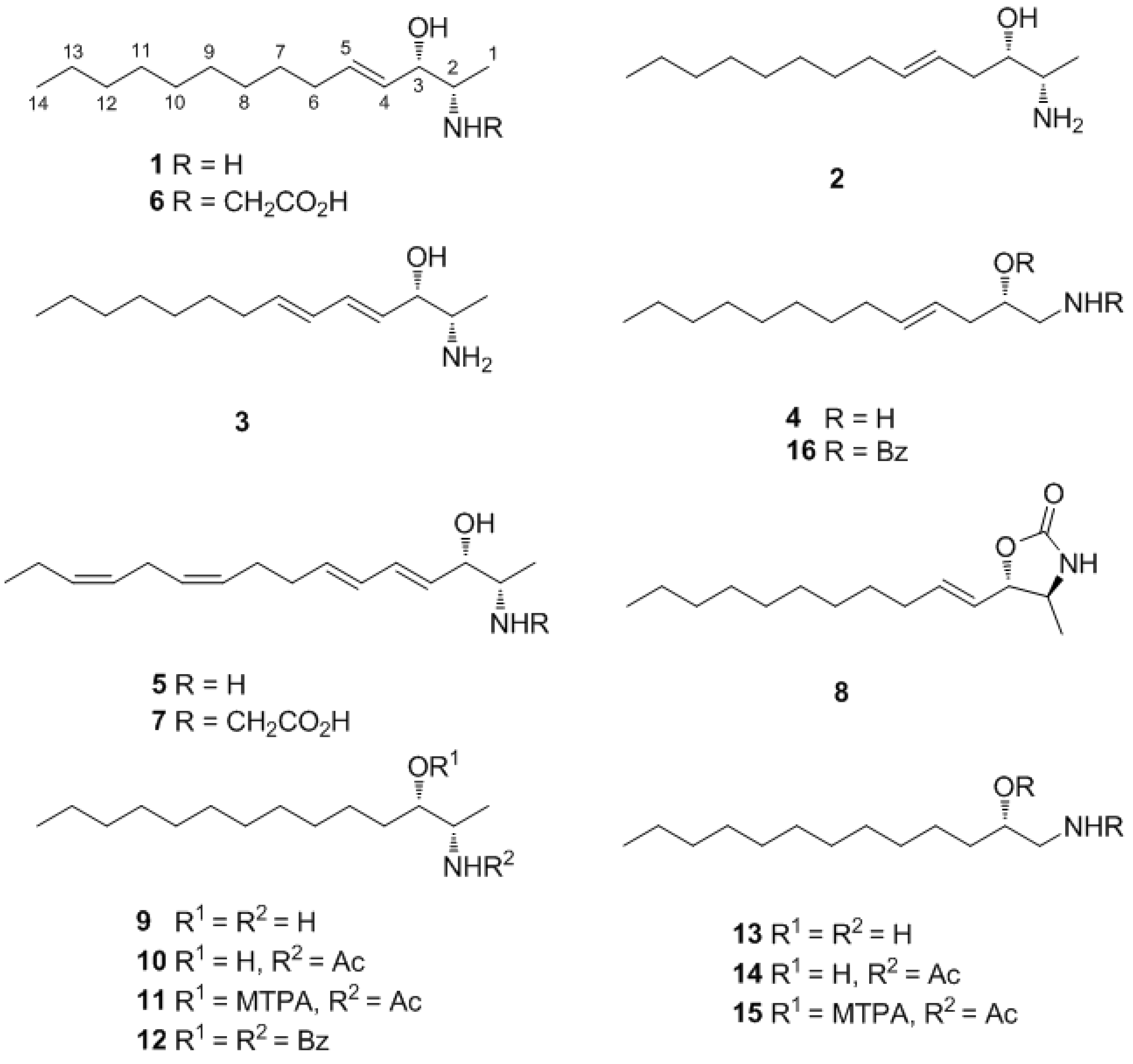
2. Results and Discussion
The ascidian specimens were lyophilized, macerated, and repeatedly extracted with MeOH and CH2Cl2. The combined extracts were separated by solvent partitioning, followed by reversed-phase flash chromatography. The polar fractions with amino alcohol mixtures, as determined by NMR analysis, were separated via reversed-phase HPLC to yield seven compounds.
Pseudoaminol A (
1) was isolated as an amorphous solid with a molecular formula of C
14H
29NO, as determined by HRFABMS analysis. The
13C NMR spectrum of this compound showed signals corresponding to two olefinic (δ
C 137.2 and 129.8), two methine (δ
C 75.0 and 53.3), and ten upfield carbons (δ
C 33.3–14.4) (
Table 1). The linear lipid nature of this compound was apparent from its lack of quaternary carbons and the presence of an oxygen and a nitrogen at the methine carbons at δ
C 75.0 and 53.3, respectively, as deduced from their chemical shifts. The sequentially correlated proton signals at δ
H 5.83, 5.42, 3.88, 3.08, and 1.22 and the corresponding carbon signals at δ
C 137.2, 129.8, 75.0, 53.3, and 15.7, respectively, were attributed to a 2-amino-3-hydroxy-alkyl moiety directly linked to a disubstituted double bond from the COSY, HSQC, and HMBC data (
Table 2). The combined 2-D NMR data also revealed that the remaining upfield protons were linearly assembled to form an eight-carbon alkyl chain attached at the previously determined double bond. The
E configuration was assigned to this double bond based on the large vicinal coupling constants between the olefinic protons (
J4,5 = 15.5 Hz), as well as the NOESY cross-peaks at H-3/H-5 and H-4/H-6. In conclusion, the planar structure of
1 was determined to be (
E)-2-aminotetradec-4-en-3-ol.
Table 1.
13C NMR (ppm, mult) Assignments for Compounds 1–7 a.
Table 1.
13C NMR (ppm, mult) Assignments for Compounds 1–7 a.
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|
| 1 | 15.7, CH3 | 15.8, CH3 | 15.7, CH3 | 45.3, CH2 | 15.7, CH3 | 12.7, CH3 | 12.9, CH3 |
| 2 | 53.3, CH | 52.7, CH | 53.3, CH | 68.7, CH | 53.6, CH | 57.8, CH | 57.8, CH |
| 3 | 75.0, CH | 73.1, CH | 74.6, CH | 39.4, CH2 | 74.6, CH | 72.2, CH | 72.1, CH |
| 4 | 129.8, CH | 38.0, CH2 | 129.7, CH | 125.6, CH | 130.1, CH | 129.4, CH | 130.4, CH |
| 5 | 137.2, CH | 125.7, CH | 135.6, CH | 135.4, CH | 135.5, CH | 134.0, CH | 132.8, CH |
| 6 | 33.3, CH2 | 135.5, CH | 130.6, CH | 33.6, CH2 | 130.9, CH | 31.2, CH2 | 129.8, CH |
| 7 | 30.1 b, CH2 | 33.7, CH2 | 137.8, CH | 30.5 b, CH2 | 137.0, CH | 28.5 b, CH2 | 134.8, CH |
| 8 | 30.3 b, CH2 | 30.6 b, CH2 | 33.6, CH2 | 30.4 b, CH2 | 33.7, CH2 | 28.5 b, CH2 | 32.1, CH2 |
| 9 | 30.4 b, CH2 | 30.6 b, CH2 | 30.3 b, CH2 | 30.3 b, CH2 | 27.9, CH2 | 28.6 b, CH2 | 26.4, CH2 |
| 10 | 30.6 b, CH2 | 30.4 b, CH2 | 30.3 b, CH2 | 30.2 b, CH2 | 129.9, CH | 28.8 b, CH2 | 129.0, CH |
| 11 | 30.7 b, CH2 | 30.3 b, CH2 | 30.3 b, CH2 | 32.9, CH2 | 129.7, CH | 28.9 b, CH2 | 128.3, CH |
| 12 | 33.1, CH2 | 33.1, CH2 | 33.0, CH2 | 23.6, CH2 | 26.4, CH2 | 31.6, CH2 | 25.2, CH2 |
| 13 | 23.7, CH2 | 23.7, CH2 | 23.7, CH2 | 14.3, CH3 | 128.3, CH | 22.1, CH2 | 127.1, CH |
| 14 | 14.4, CH2 | 14.4, CH3 | 14.4, CH3 | | 132.6, CH | 13.9, CH3 | 131.5, CH |
| 15 | | | | | 21.5, CH2 | | 20.1, CH2 |
| 16 | | | | | 14.6, CH3 | | 14.2, CH3 |
| 1′ | | | | | | 46.5, CH2 | 46.7, CH2 |
| 2′ | | | | | | 168.0, C | 168.5, C |
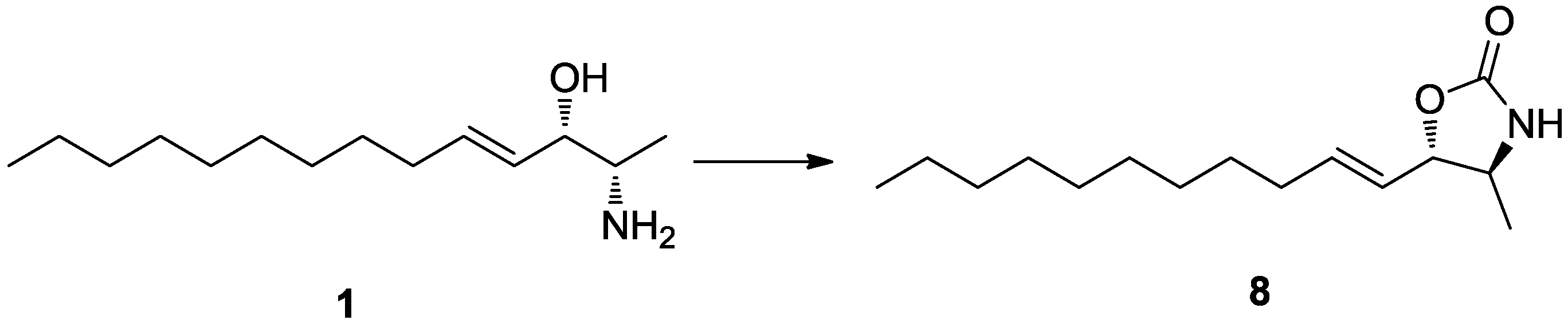
Pseudoaminol A (
1) possessed asymmetric carbon centers at C-2 and C-3. The relative configurations were determined by vicinal proton-proton coupling constants and NOESY experiments performed on the corresponding oxazolidinone derivative
8 of
1, which was prepared by treatment with 1,1′-carbonyl diimidazole (
Scheme 1). Irradiation of the H-1 and H-4 signals at δ
H 1.22 and 5.42, respectively, showed a coupling constant of
J2,3 = 7.5 Hz, which indicated a
threo configuration for C-2 and C-3. The alternative
erythro configuration would have given a
J of 4.5 Hz [
19]. This interpretation was also confirmed by the cross-peaks at H-1/H-3 and H-2/H-5 in the NOESY data.
Table 2.
1H NMR (ppm, mult) Assignments for Compounds 1–3 a.
Table 2.
1H NMR (ppm, mult) Assignments for Compounds 1–3 a.
| Position | 1 | 2 | 3 |
|---|
| 1 | 1.22, d (7.0) | 1.26, d (7.0) | 1.22, d (7.0) |
| 2 | 3.08, dq (7.5, 7.0) | 3.11, dq (3.0, 7.0) | 3.10, dq (7.5, 7.0) |
| 3 | 3.88, dd (7.0, 7.0) | 3.50, dd (5.0, 7.0, 7.0) | 3.90, dd (7.5, 7.5) |
| 4 | 5.42, dd (7.5, 15.5) | 2.33, ddd (7.0, 7.0, 14.5); 2.17, ddd (7.0, 7.0, 14.5) | 5.51, dd (7.5, 15.0) |
| 5 | 5.83, td (7.0, 15.5) | 5.48, dt (7.0, 7.0, 15.0) | 6.33, dd (11.0, 15.0) |
| 6 | 2.09, td (7.5, 7.0) | 5.57, dt (7.0, 7.0, 15.0) | 6.00, dd (11.0, 15.0) |
| 7 | 1.34, tt (7.5, 7.5) | 2.03, dt (7.0, 7.0) | 5.78, dd (7.5, 15.0) |
| 8 | 1.23–1.27, m | 1.26–1.32, m | 2.09, dt (7.0, 7.0) |
| 9 | 1.23–1.27, m | 1.26–1.32, m | 1.38, tt (7.0, 7.0) |
| 10 | 1.23–1.27, m | 1.26–1.32, m | 1.26–1.32, m |
| 11 | 1.23–1.27, m | 1.26–1.32, m | 1.26–1.32, m |
| 12 | 1.23–1.27, m | 1.26–1.32, m | 1.26–1.32, m |
| 13 | 1.23–1.27, m | 1.26–1.32, m | 1.26–1.32, m |
| 14 | 0.89, t (7.0) | 0.89, t (7.0) | 0.89, t (7.0) |
Scheme 1.
Reagents and Conditions: 1,1′-carbonyldiimidazole, CH2Cl2, rt, 2 h.
Scheme 1.
Reagents and Conditions: 1,1′-carbonyldiimidazole, CH2Cl2, rt, 2 h.
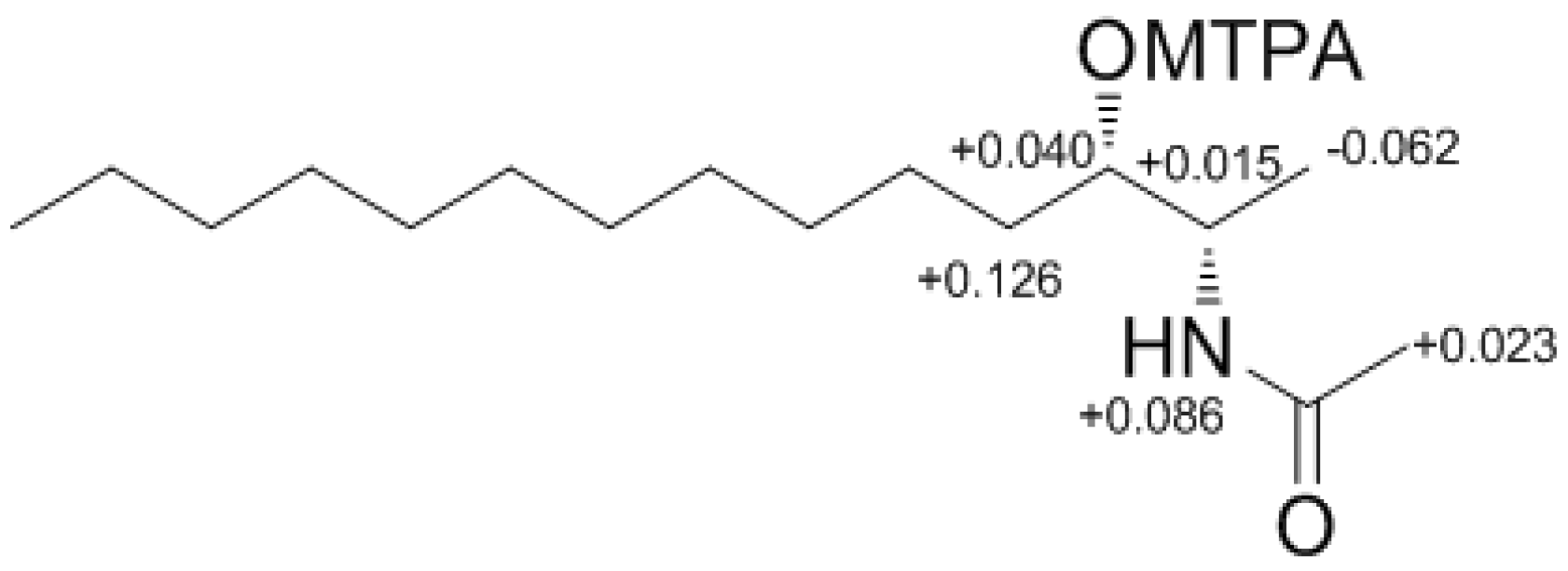
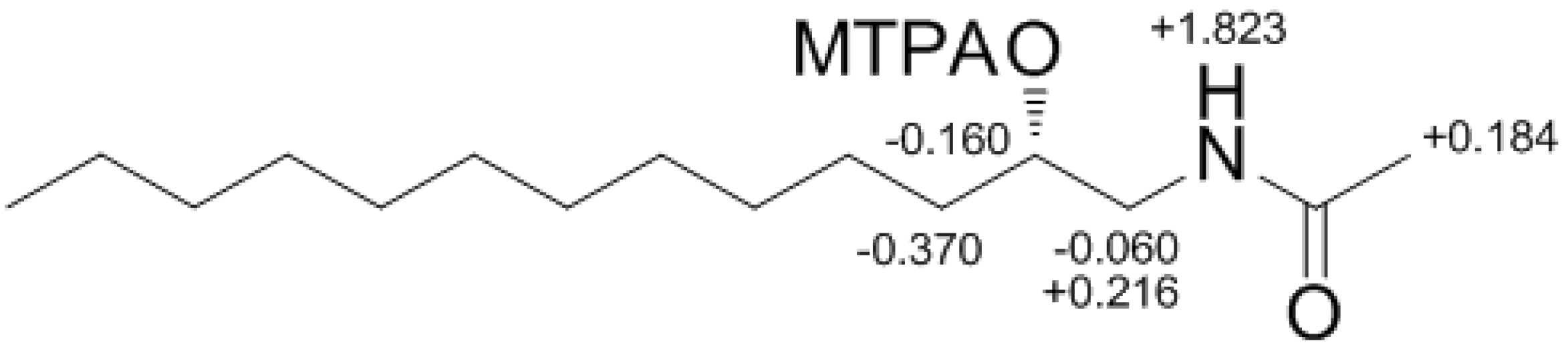
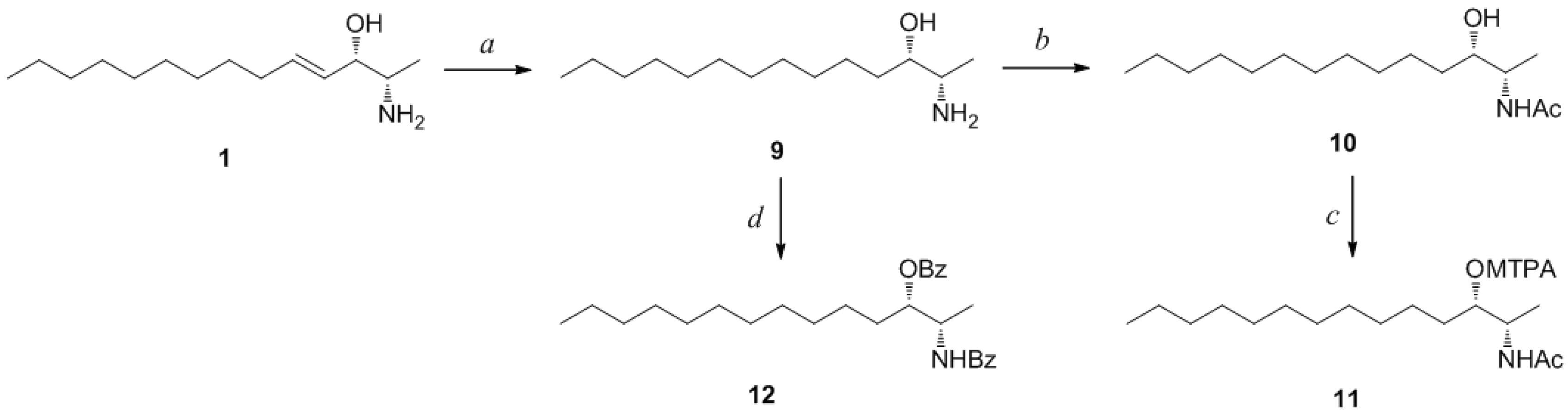
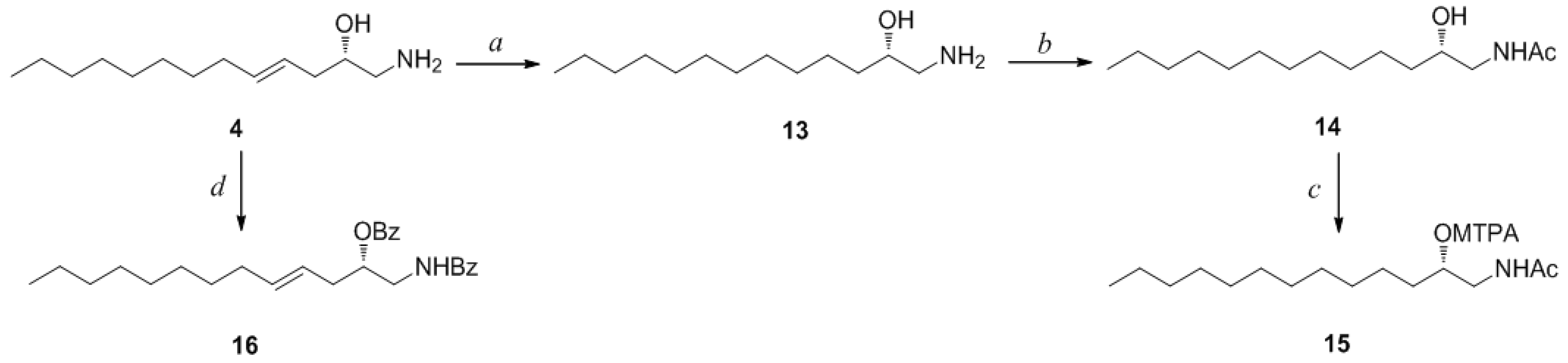
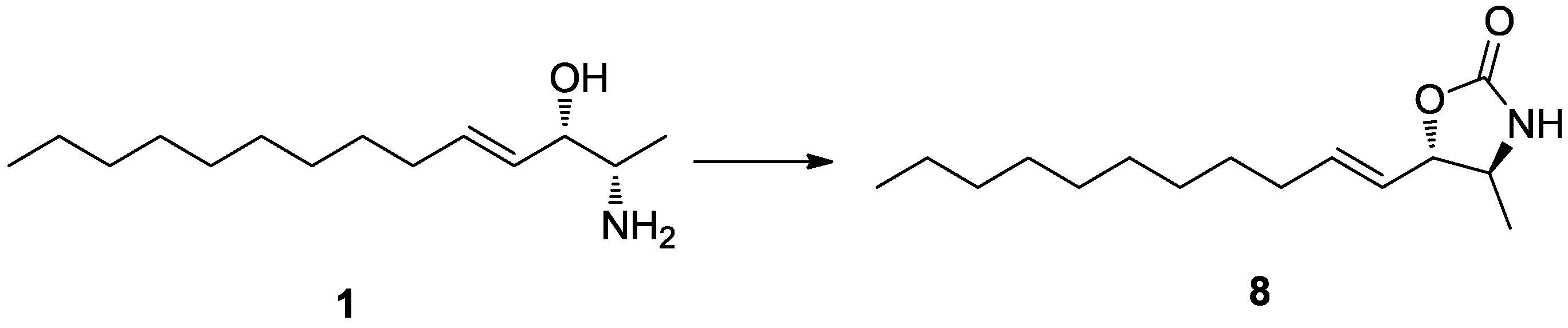
Determination of the absolute configuration of
1 was initially attempted by application of the Mosher method, which involved MTPA-Cl esterification of the C-3 hydroxyl group. To avoid possible steric hindrance by the neighboring C-4 double bond and concomitant MTPA esterification at the C-2 amine group,
1 was sequentially hydrogenated (
9) and acetylated (
10) prior to MTPA esterification (
Scheme 2). However, the MTPA esters of compound
10 exhibited irregular diamagnetic proton shifts, possibly due to the spatial crowding between C-2 and C-3 (
Figure 2). These results contradict previous determinations of absolute configurations using the Mosher method for similar amino alcohols [
6,
10,
14].
Scheme 2.
Reagents and Conditions: (a) H2, Pd/C, MeOH, rt, 2 h; (b) acetic anhydride, pyridine, rt, 1 h; (c) MTPA-Cl, pyridine, DMAP, rt, 1 h; (d) benzoyl chloride, pyridine, DMAP, rt, 3 h.
Scheme 2.
Reagents and Conditions: (a) H2, Pd/C, MeOH, rt, 2 h; (b) acetic anhydride, pyridine, rt, 1 h; (c) MTPA-Cl, pyridine, DMAP, rt, 1 h; (d) benzoyl chloride, pyridine, DMAP, rt, 3 h.
Figure 2.
The Result (∆δ11S−11R) of MTPA esterification for compound 11.
Figure 2.
The Result (∆δ11S−11R) of MTPA esterification for compound 11.
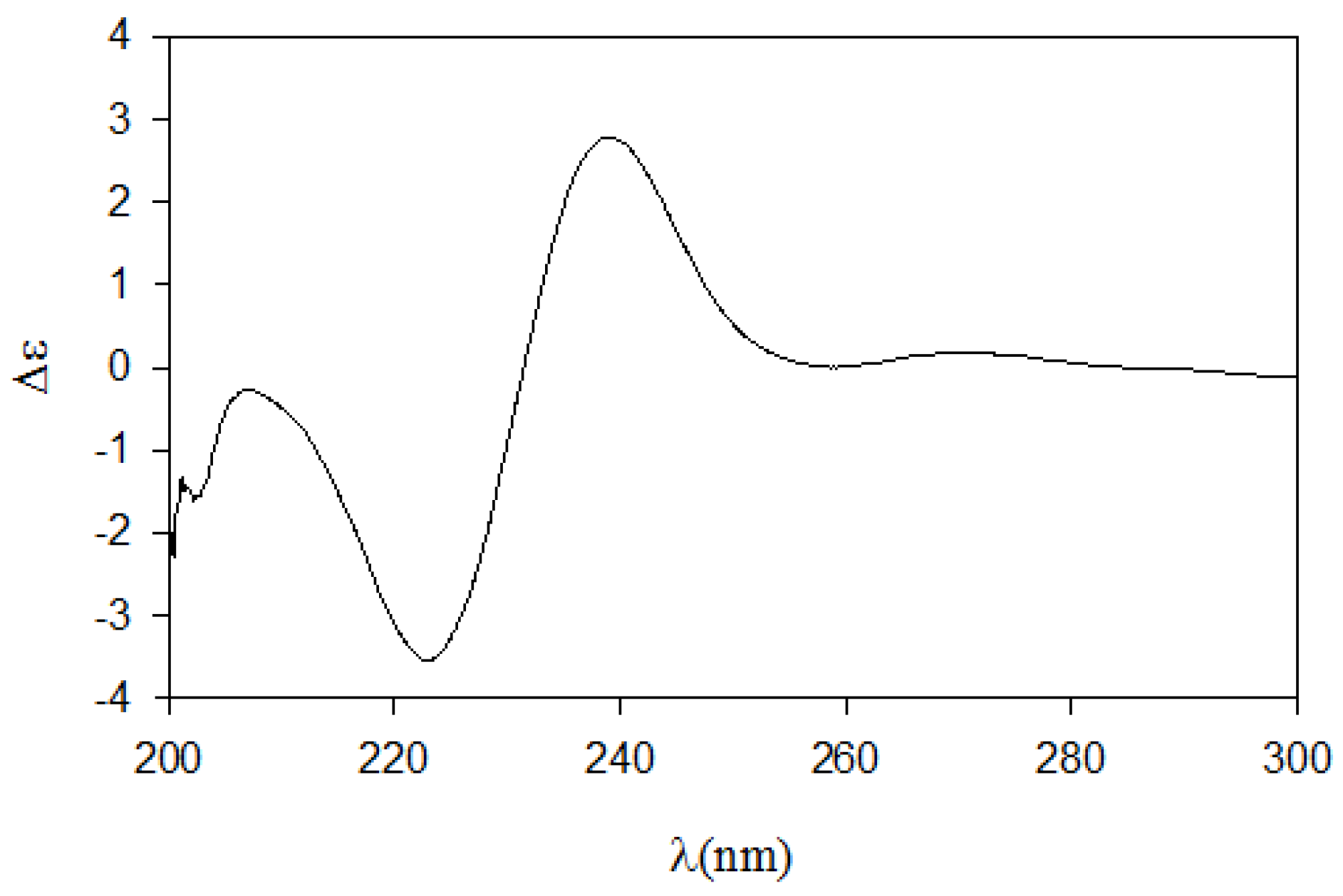
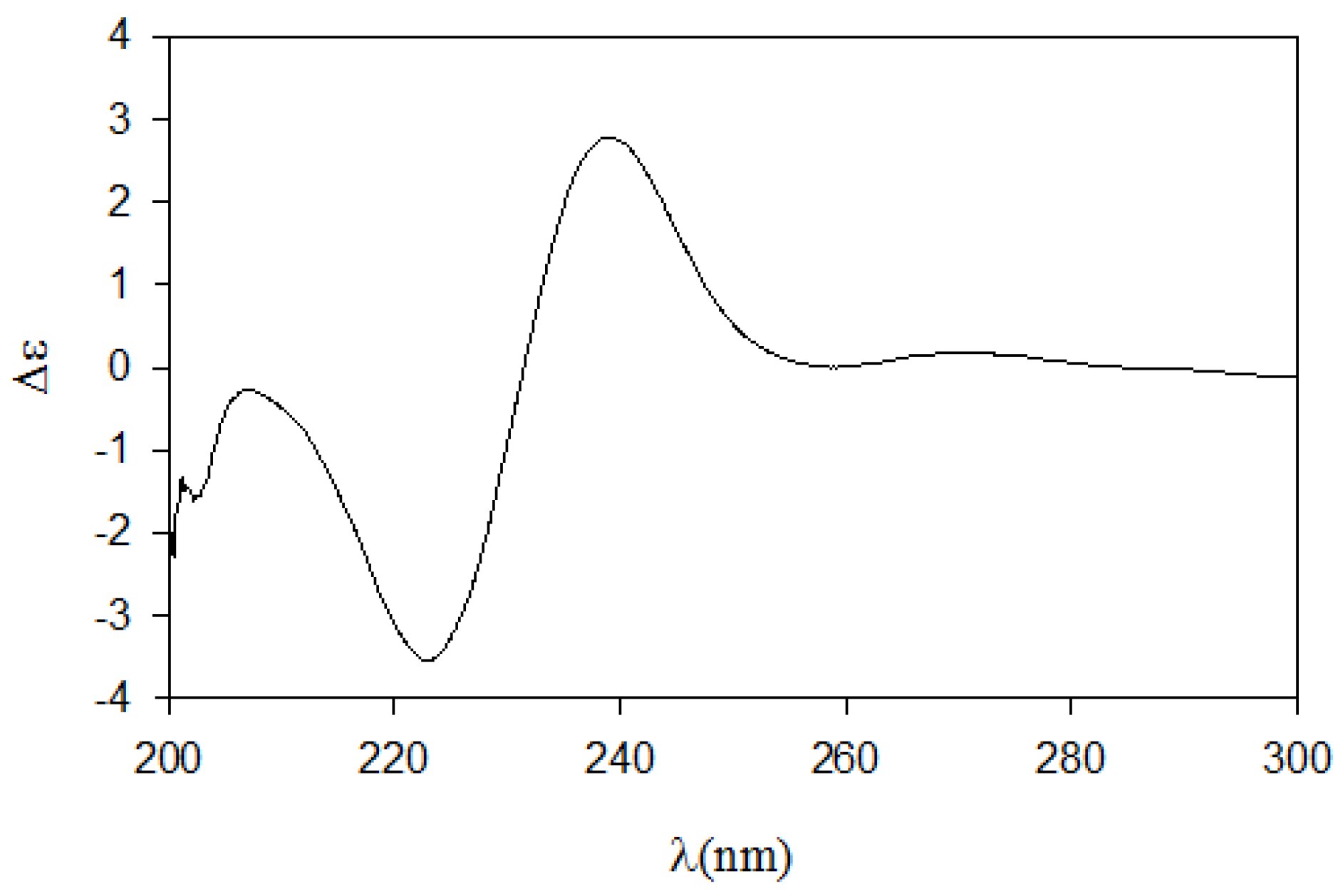
Next, we employed the recently proposed CD method, which has been used to unambiguously assign both relative and absolute configurations of diastereomeric
N,
O-dibenzoyl-2-amino-3-alkanols [
7]. The CD spectrum of the dibenzoyl derivative
11 obtained by treatment of
8 with benzoyl chloride (
Scheme 2) closely matched the (2
S,3
S)-
threo model because it exhibited the same sign for the negative and positive cotton effects at approximately 220 nm and 240 nm, respectively, and because it exhibited the same band magnitude (
Figure 3). Thus, the absolute configurations of pseudoaminol A (
1) were determined to be 2
S and 3
S.
Figure 3.
The CD spectrum of compound 12.
Figure 3.
The CD spectrum of compound 12.
The molecular formula of pseudoaminol B (
2) was deduced as C
14H
29NO by HRFABMS analysis, which was the same as that of
1. The NMR data of this compound were also similar to those of
1; the shifts of a methylene resonance (δ
C 38.0 and δ
H 2.17 and 2.33) were the most noticeable differences between the spectra of
1 and
2 (
Table 1 and
Table 2). On the basis of the results of combined 2-D NMR experiments, pseudoaminol B (
2) was determined to be a derivative of
1, with migration of the C-4 double bond to C-5. The
E configuration was assigned to this double bond on the basis of the large proton coupling constants (
J4,5 = 15.5 Hz).
Despite their structural similarity, the specific rotation of
2 was opposite that of
1 ( ![Marinedrugs 12 03754 i001]()
−4.4 and +5.1 for
1 and
2, respectively), which led us to conduct a detailed investigation. Thus,
2 was hydrogenated and converted to the dibenzoyl derivative, as shown in
Scheme 2. After each step, the specific rotations for the corresponding derivatives of
1 and
2 were compared. The specific rotations of the hydrogenated derivatives (
![Marinedrugs 12 03754 i001]()
−6.9 and −7.1, respectively) and dibenzoyl analogs (
![Marinedrugs 12 03754 i001]()
−12.3 and −12.4, respectively) were virtually identical, regardless of their origin. The CD spectra of the dibenzoyl derivatives of
1 and
2 were also identical (See
Supplementary Information, Figure S1). In addition to similar
13C and
1H NMR chemical shifts, chemical derivatization allowed unambiguous assignment of the same absolute configuration to
1 and
2. The significant difference in the specific optical rotation between these compounds is a result of the direct influence of the neighboring electron-rich double bond.
Pseudoaminol C (3) exhibited a molecular formula of C14H27NO, as determined by HRFABMS analysis, indicating that it possessed two fewer protons than 1. The NMR data for this compound showed four olefinic signals (δC 137.8, 135.6, 130.6, and 129.7; δH 6.33, 6.00, 5.78, and 5.51), which were assigned to a conjugated diene moiety on the basis of the COSY and HSQC analyses. Aided by the HMBC data, the structure of 3 was determined to be a linear amino alcohol derivative with a conjugated diene at C-4. The configurations of both double bonds were assigned as E on the basis of the large coupling constants between the olefinic protons (J4,5 = J6,7 = 15.0 Hz).
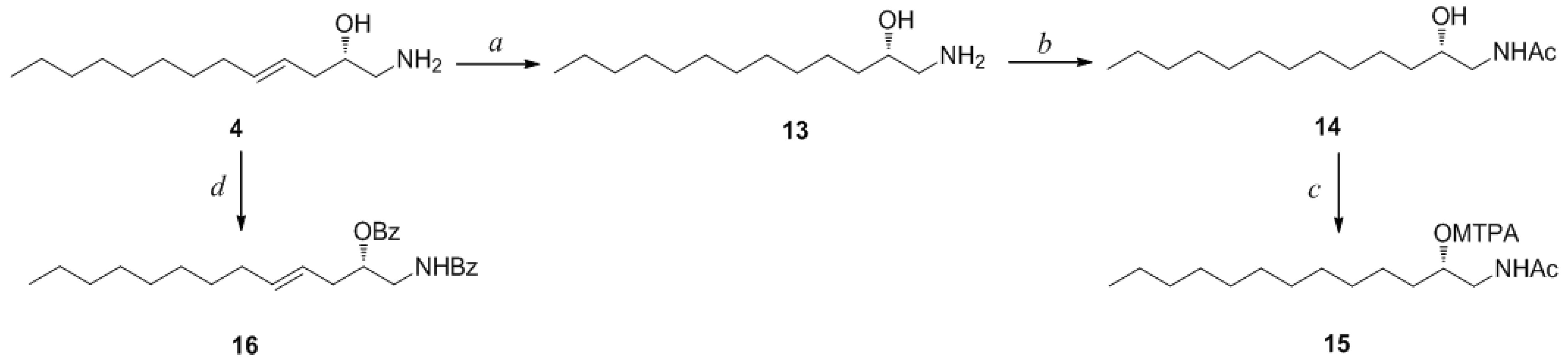
The molecular formula of pseudoaminol E (
4) was determined to be C
13H
27NO by HRFABMS analysis. The most conspicuous difference between the NMR results for
4 and
2 was the absence of the C-1 terminal methyl signal (δ
C 15.8 and δ
H 1.26), which coincided with the loss of a carbon on the basis of the MS analysis. In addition, signals of nitrogenous terminal methylene protons (δ
H 2.74 and 3.00) were present in the
1H NMR spectrum (
Table 3). Based on the combined 2-D NMR analyses, the structure of
4 was determined to be a linear C
13 amino alcohol derivative with a terminal amino group. The geometry of the double bond was assigned as
E on the basis of the proton coupling constants (
J4,5 = 15.0 Hz).
Table 3.
1H NMR (ppm, mult) Assignments for Compounds 4–7 a.
Table 3.
1H NMR (ppm, mult) Assignments for Compounds 4–7 a.
| Position | 4 | 5 | 6 | 7 |
|---|
| 1 | 2.74, dd (10.0, 13.0); 3.00, dd (3.0, 13.0) | 1.23, d (7.0) | 1.10, d (7.0) | 1.22, d (7.0) |
| 2 | 3.76, dddd (3.0, 7.0, 7.0, 10.0) | 3.11, dq (7.5, 7.0) | 2.98, dq (7.5, 7.0) | 2.99, dq (7.5, 7.0) |
| 3 | 2.24, ddd (7.0, 7.0, 14.0); 2.19, ddd (7.0, 7.0, 14.0) | 3.96, dd (7.5, 7.5) | 3.88, dd (7.5, 7.5) | 4.02, dd (7.5, 7.5) |
| 4 | 5.44, ddd (7.0, 7.0, 15.0) | 5.53, dd (7.5, 15.0) | 5.37, dd (15.0, 7.5) | 5.53, dd (15.0, 7.5) |
| 5 | 5.56, dt (15.0, 7.0) | 6.32, dd (10.5, 15.0) | 5.68, ddd (7.0, 7.0, 15.0) | 6.21, dd (11.0, 15.0) |
| 6 | 2.02, dt (7.0, 7.0) | 6.11, dd (10.5, 15.0) | 2.00, ddd (7.0, 7.0, 15.0) | 6.06, dd (11.0, 15.0) |
| 7 | 1.35, tt (7.0, 7.0) | 5.78, dt (15.0, 7.0) | 1.33, m | 5.73, dd (7.5, 7.5) |
| 8 | 1.25–1.33, m | 2.16, s | 1.23–1.28, m | 2.11, br s |
| 9 | 1.25–1.33, m | 2.16, s | 1.23–1.28, m | 2.11, br s |
| 10 | 1.25–1.33, m | 5.35, m | 1.23–1.28, m | 5.33, m |
| 11 | 1.25–1.33, m | 5.35, m | 1.23–1.28, m | 5.33, m |
| 12 | 1.25–1.33, m | 2.77, dd (6.0, 6.0) | 1.23–1.28, m | 2.73, dd (6.0, 6.0) |
| 13 | 0.89, t (7.0) | 5.31, m | 1.23–1.28, m | 5.25, dt (10.0, 6.0) |
| 14 | | 5.35, m | 0.84, t (7.0) | 5.33, m |
| 15 | | 2.06, dq (7.0, 7.0) | | 2.02, dq (7.0, 7.0) |
| 16 | | 0.96, t (7.0) | | 0.91, t (7.0) |
| 1′ | | | 3.35, d (15.0) | 3.34, d (15.0) |
| | | | 3.23, d (15.0) | 3.23, d (15.0) |
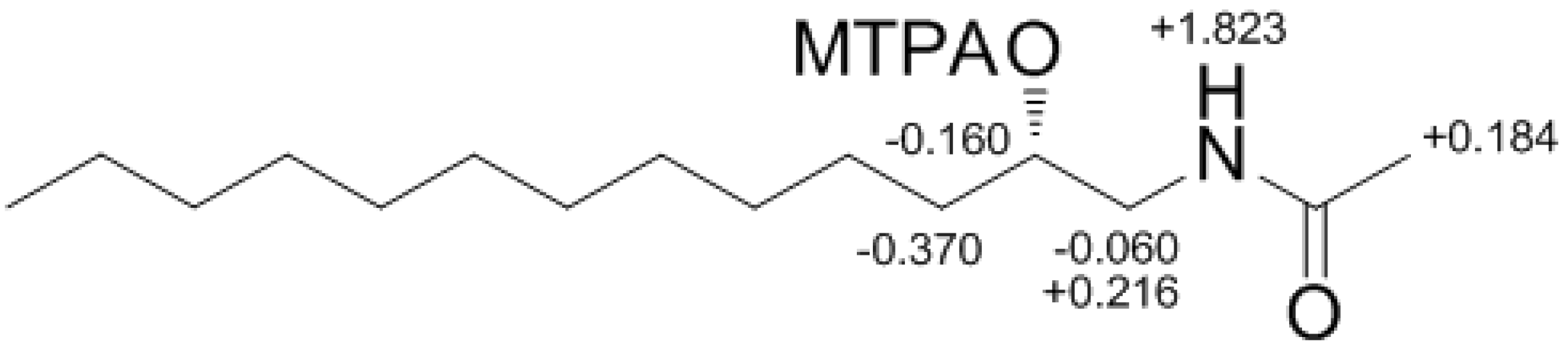
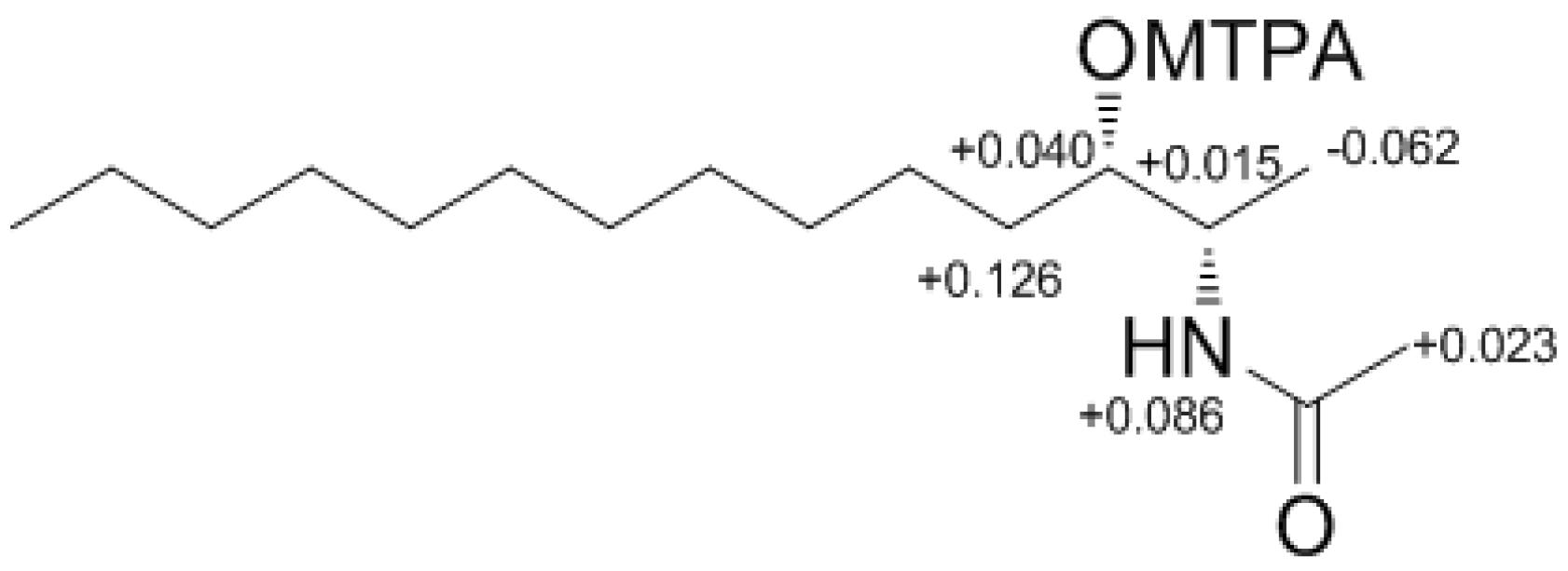
Compound
4 contained an asymmetric carbon center at the C-2 oxymethine. The Mosher method was used to determine the absolute configuration of this carbon. Similar to the case of
1, compound
4 was sequentially hydrogenated (
13) and acetylated (
14) prior to MTPA esterification in order to avoid potential steric hindrance from the neighboring C-4 double bond and additional MTPA esterification at the C-1 amine group (
Scheme 3). The ∆δ values of the signals of the protons near the hydroxyl group at C-2 were negative to the left and positive to the right, suggesting the 2
S configuration, with the exception of one of the H-1 methylene protons (
Figure 4). The absolute configuration was confirmed by CD method on a synthetic derivative. The CD spectrum of the dibenzoyl derivative (
16) obtained by treatment of
4 with benzoyl chloride (
Scheme 3) showed a positive cotton effect (222 nm (∆ε −5.8) and 239 nm (∆ε +7.1)), which was opposite to the previously reported 2
R model [
20] (See
Supplementary Information, Figure S2). Overall, the absolute configuration was determined to be
S in accord with the configurations of other amino alcohols at the oxymethine position.
Scheme 3.
Reagents and Conditions: (a) H2, Pd/C, MeOH, rt, 2 h; (b) acetic anhydride, pyridine, rt, 1 h; (c) MTPA-Cl, pyridine, DMAP, rt, 1 h; (d) benzoyl chloride, pyridine, DMAP, rt, 3 h.
Scheme 3.
Reagents and Conditions: (a) H2, Pd/C, MeOH, rt, 2 h; (b) acetic anhydride, pyridine, rt, 1 h; (c) MTPA-Cl, pyridine, DMAP, rt, 1 h; (d) benzoyl chloride, pyridine, DMAP, rt, 3 h.
Figure 4.
The Result (∆δ15S-15R) of MTPA esterification for compound 15.
Figure 4.
The Result (∆δ15S-15R) of MTPA esterification for compound 15.
The molecular formula of pseudoaminol E (
5) was deduced as C
16H
28NO by HRFABMS analysis. The
13C NMR spectrum of this compound showed signals corresponding to eight olefinic (δ
C 137.0–128.3), two methine (δ
C 53.6 and 74.6), and six upfield carbons (δ
C 33.7–14.6). The presence of eight methylene signals at δ
H 2.77–2.06 in the
1H NMR spectrum indicated the presence of allylic and
bis-allylic methylenes and provided insight into the double-bond system. On the basis of the combined results of 2-D NMR analyses, the conjugated double bond was located at C-4 and the isolated double bonds were located at C-10 and C-13. Overall, the structure of compound
5 was determined to be a highly unsaturated, linear amino alcohol possessing a conjugated diene moiety. The configurations of the four double bonds were assigned on the basis of the proton coupling constants and carbon chemical shifts.
E configuration was assigned to the C-4 and C-6 double bonds based on the large vicinal proton coupling constants (
J4,5 =
J6,7 = 15.0 Hz). In contrast, the
Z configuration of the C-10 and C-13 double bonds was determined on the basis of the upfield chemical shifts of the allylic and
bis-allylic carbons (δ
C 27.9, 26.4, and 21.5 for C-9, C-12, and C-15, respectively) in the
13C NMR spectrum [
21]. The severely overlapping olefinic proton signals hindered both the accurate measurement of the proton coupling constants and NOESY analysis.
In addition to the linear amino alcohols, structurally related metabolites with unusual functionalities were isolated and analyzed. The molecular formula of pseudoaminol F (
6) was established as C
16H
31NO
3 by HRFABMS analysis. The
13C NMR spectrum of this compound was very similar to that of
1; the presence of a carbonyl and a methylene carbon at δ
C 168.0 and 46.5, respectively, were the most noticeable differences. A difference was also observed in the
1H NMR spectrum, as proton signals of additional methylenes were observed at δ
H 3.35 (1H, d,
J = 15.0 Hz) and 3.23 (1H, d,
J = 15.0 Hz) (
Table 3). A strong absorption band was also present at 1747 cm
−1 in the IR spectrum; this result, in conjunction with the MS results, indicated that the carbonyl group was a carboxylic acid. The combined 2-D NMR experiments of
6 revealed the presence of the same linear carbon framework as
1. The new functional group was observed to be a carboxymethyl group directly attached to the 2-amino group on the basis of the long-range carbon-proton correlations at H-2/C-1′, H-1′/C-2, and H-1′/C-2′ in the HMBC spectra. To the best of our knowledge, this
N-carboxymethyl group possibly derived from amino acid glycine, is the first example of its type among amino alcohols.
Related compound 7 was determined to have a molecular formula of C18H29NO3 by HRESIMS analysis. Other than the signals from the N-carboxymethyl group (δC 168.5, δH 3.34 and 3.23), the 1H and 13C NMR spectra of this compound were very similar to those of 5. The combined 2-D NMR analyses confirmed this similarity and 7, designated pseudoaminol G, was determined to be a N-carboxymethyl-substituted amino alcohol.
The marine-derived amino alcohols and related metabolites exhibited cytotoxic [
5,
6,
8,
9,
16] and antimicrobial activities [
2,
7,
10,
12,
13,
15,
17]. In our cytotoxicity assays of the natural and synthetic derivatives, compounds
1–
5,
8–
10, and
12 showed cytotoxicity against the A549 and K562 cell lines that was substantially lower than that of doxorubicin, whereas the other compounds were inactive (
Table 4). The significant cytotoxicity of the crude extract (LC
50 14.6 μg/mL against the A549 cell line) from this specimen suggests either the presence of minor but significantly cytotoxic constituents or the existence of synergic effects between these amino alcohols that may require further investigation.
In antibacterial assays against Gram-positive and Gram-negative strains, compounds 1, 2, 8, and 9 exhibited moderate inhibition, whereas other compounds with more elaborate structures were inactive. These compounds were also tested against microbial enzymes, isocitrate lyase, sortase A, and Na+/K+-ATPase. Weak inhibition was observed for 1, 2, 8, and 9 against enzyme Na+/K+-ATPase, whereas no other enzyme-inhibitory activities were observed for the amino alcohols. These results suggest that fewer double bonds and free amino and hydroxy groups are important for the antibacterial or related enzyme-inhibitory activity of the compounds. The absence of bioactivity for 6 and 7, which possess the unprecedented N-carboxymethyl group, emphasizes the role the free amino group plays, via either an electronic or steric effect, in determining the bioactivity.
Table 4.
The Result of Bioactivity test.
Table 4.
The Result of Bioactivity test.
| | LC50 (μM) | MIC (μg/mL) | IC50 (μM) |
|---|
| | K562 | A549 | Gram(+) Bacterium | Gram(−) Bacterium | Na+/K+-ATPase |
|---|
| Compound | | | A | B | C | D | E | F | |
|---|
| 1 | 13.6 | 13.8 | 12.5 | 25 | 12.5 | 6.25 | 25 | >100 | 62.0 |
| 2 | 12.6 | 13.1 | 12.5 | 25 | 12.5 | 12.5 | 25 | >100 | 78.4 |
| 3 | 12.4 | 12.4 | 50 | 100 | 100 | 50 | 100 | >100 | >200 |
| 4 | 11.9 | 13.2 | 100 | >100 | 100 | 50 | >100 | >100 | 190.3 |
| 5 | >100 | 12.3 | 100 | >100 | 100 | 50 | >100 | >100 | 101.2 |
| 6 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >200 |
| 7 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >200 |
| 8 | 12.7 | 10.5 | 50 | 50 | 25 | 12.5 | 50 | >100 | 71.4 |
| 9 | >100 | 14.0 | 12.5 | 25 | 12.5 | 6.25 | 25 | >100 | 46.8 |
| 10 | 17.3 | 11.7 | 50 | 100 | 100 | 50 | 100 | >100 | 118.1 |
| 12 | 5.9 | 8.0 | >100 | >100 | >100 | >100 | >100 | >100 | 108.2 |
| 13 | >100 | >100 | 100 | >100 | 100 | 100 | >100 | >100 | 130.3 |
| 14 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >200 |
| Doxorubicin | 1.1 | 0.9 | | | | | | | |
| Ouabain | | | | | | | | | 6.1 |
| Ampicillin | | | 0.4 | 0.4 | 0.4 | 0.4 | 1.6 | 6.3 | |
3. Experimental Section
3.1. General Experimental Procedures
Optical rotations were measured on a JASCO P-1020 polarimeter (Jasco, Tokyo, Japan) using a 1 cm cell. UV spectra were acquired with a Hitachi U-3010 spectrophotometer (Hitachi High-Technologies, Tokyo, Japan). CD spectra were obtained on a JASCO J-715 (Jasco, Tokyo, Japan) using a 0.2 mm cell. IR spectra were recorded on a JASCO 4200 FT-IR spectrometer (Jasco, Tokyo, Japan) using a ZnSe cell. NMR spectra were recorded in DMSO-
d6 and MeOH-
d4 solutions containing Me
4Si as an internal standard on Bruker Avance 600 and 500 spectrometers (Bruker, Massachusetts, MA, USA). Proton and carbon NMR spectra were measured at 600 and 150 MHz (
1,
3,
4,
6, and
7) or 500 and 125 MHz (
2 and
5), respectively (See
Supplementary Information, Figures S3–S37). High-resolution FAB mass spectrometric data were obtained at the Korea Basic Science Institute (Daegu, Korea) and were acquired using a JEOL JMS 700 mass spectrometer (Jeol, Tokyo, Japan) with
meta-nitrobenzyl alcohol (NBA) as a matrix for the FABMS. High-resolution ESIMS data were obtained at the National Instrumentation Center for Environmental Management (Seoul, Korea) using a Thermo-Finnigan LTQ-Orbitrap instrument (Thermo, Waltham, MA, USA) equipped with a Dionex U-3000 HPLC system (Thermo, Waltham, MA, USA). Low-resolution ESIMS data were recorded on an Agilent Technologies 6130 quadrupole mass spectrometer (Santa Clara, CA, USA) coupled to an Agilent Technologies 1200 series HPLC (Santa Clara, CA, USA). Semi-preparative HPLC was performed on a Spectrasystem p2000 (Thermo, Waltham, MA, USA) equipped with a refractive-index detector (Spectrasystem RI-150) and a YMC ODS-A column (10 × 250 mm). All solvents used were spectroscopic grade or were distilled prior to use.
3.2. Animal Materials
Specimens of Pseudodistoma sp. (sample number 12CH-24) were collected by hand using scuba equipment at a depth of 20 m off the coast of Chuja-do, Korea, on 10 October 2012. The colony has conical heads on short, thick, and wrinkled cylindrical stalks. Nine stalks (16–29 mm long) were on a basal test mass up to 62 mm wide. The colony was reddish-orange in live form and was yellowish-beige in ethanol. The zooids were reddish-orange in color during the tests. The zooids were 10.2–32.0 mm in length; the thorax was 3.5–4.2 mm, and the abdomen was 6.7–27.8 mm. These morphological features indicated that the specimen belongs to the genus Pseudodistoma, and in particular, was very similar to P. antinboja Tokioka; however, the lack of gonads and larvae prevented adequate species identification. The voucher specimens were deposited at the Natural History Museum, Ehwa Womans University, under the curatorship of B.J.R.
3.3. Extraction and Isolation
Freshly collected specimens were immediately frozen and stored at −25 °C until use. Lyophilized specimens were macerated and repeatedly extracted with MeOH (3 L × 3) and CH2Cl2 (3 L × 2). The combined extracts (25.71 g) were successively partitioned between H2O (18.67 g) and n-BuOH (7.05 g); the latter fraction was repartitioned between H2O–MeOH (15:85) (4.50 g) and n-hexane (2.55 g). The former layer was separated by C18 reversed-phase flash chromatography using sequential mixtures of MeOH and H2O as the eluents (six fractions in the gradient, H2O–MeOH, from 50:50 to 0:100), followed by acetone and finally EtOAc.
On the basis of the 1H NMR results and cytotoxicity analyses, the fractions eluted with 40:60 H2O–MeOH (0.88 g) and 20:80 H2O–MeOH (0.22 g) were chosen for separation. The 40:60 H2O–MeOH fraction (0.88 g) was separated by reversed-phase semi-preparative HPLC (H2O–MeOH, 50:50), yielding five peaks rich with secondary metabolites. The five peaks provided, in order of elution, compounds 5, 3, 4, 1, and 2; the products were highly pure and required no further purification.
The 20:80 H2O–MeOH fraction (0.22 g) was separated by reversed-phase semi-preparative HPLC (H2O–MeOH, 30:70) to yield, in order of elution, compounds 6 and 7. The purified metabolites were isolated in the following amounts: 87.0, 75.3, 6.3, 3.3, 35.0, 3.9, and 4.1 mg of 1–7, respectively.
Pseudoaminol A (
1): Yellow amorphous solid;
![Marinedrugs 12 03754 i001]()
−4.4 (
c 0.40, MeOH); IR (ZnSe) ν
max 3352, 2924, 1655 cm
−1;
1H and
13C NMR data, see
Table 1 and
Table 2, respectively; HRFABMS
m/
z 228.2329 [M + H]
+ (calcd for C
14H
30NO, 228.2327).
Pseudoaminol B (
2): Yellow amorphous solid;
![Marinedrugs 12 03754 i001]()
+5.1 (
c 0.50, MeOH); IR (ZnSe) ν
max 3336, 2925, 1671 cm
−1;
1H and
13C NMR data, see
Table 1 and
Table 2, respectively; HRFABMS
m/
z 228.2330 [M + H]
+ (calcd for C
14H
30NO, 228.2327).
Pseudoaminol C (
3): Yellow amorphous solid;
![Marinedrugs 12 03754 i001]()
+2.7 (
c 0.50, MeOH); UV (MeOH) λ
max (log ε) 204 (2.58), 228 (2.48) nm; IR (ZnSe) ν
max 3389, 2925, 1670 cm
−1;
1H and
13C NMR data, see
Table 1 and
Table 2, respectively; HRFABMS
m/
z 226.2175 [M + H]
+ (calcd for C
14H
28NO, 226.2171).
Pseudoaminol D (
4): Yellow amorphous solid;
![Marinedrugs 12 03754 i001]()
+15.9 (
c 0.65, MeOH); IR (ZnSe) ν
max 3336, 2924, 1716, 1651 cm
−1;
1H and
13C NMR data, see
Table 1 and
Table 3, respectively; HRFABMS
m/
z 214.2171 [M + H]
+ (calcd for C
13H
28NO, 214.2171).
Pseudoaminol E (
5): Yellow amorphous solid;
![Marinedrugs 12 03754 i001]()
+2.4 (
c 0.45, MeOH); UV (MeOH) λ
max (log ε) 205 (2.48), 230 (2.49), 278 (1.69) nm; IR (ZnSe) ν
max 3340, 2928, 1670 cm
−1;
1H and
13C NMR data, see
Table 1 and
Table 3, respectively; HRFABMS
m/
z 250.2174 [M + H]
+ (calcd for C
16H
28NO, 250.2171).
Pseudoaminol F (
6): White amorphous solid;
![Marinedrugs 12 03754 i001]()
+3.4 (
c 0.50, MeOH); IR (ZnSe) ν
max 3386, 2923, 1747, 1640 cm
−1;
1H and
13C NMR data, see
Table 1 and
Table 3, respectively; HRFABMS
m/
z 286.2386 [M + H]
+ (calcd for C
16H
32NO
3, 286.2382).
Pseudoaminol G (
7): White amorphous solid;
![Marinedrugs 12 03754 i001]()
+1.4 (
c 0.50, MeOH); UV (MeOH) λ
max (log ε) 204 (2.48), 232 (2.34), 275 (1.58) nm; IR (ZnSe) ν
max 3381, 2925, 1736, 1646 cm
−1;
1H and
13C NMR data, see
Table 1 and
Table 3, respectively; HRESIMS
m/
z 308.2220 [M + H]
+ (calcd for C
18H
30NO
3, 308.2222).
3.4. Preparation of Oxazolidinone Derivative
To a stirred solution of 3.0 mg (0.013 mmol) of compound 1 in 2.0 mL of dry CH2Cl2 was added 15.0 mg of 1,1′-carbonyldiimidazole. The mixture was stirred at room temperature for 2 h, concentrated under reduced pressure, and purified by HPLC (YMC ODS-A column, 10 × 250 mm; H2O–MeOH, 20:80) to yield pure compound 8 (2.5 mg, 75%).
Compound 8 (8): 1H NMR (MeOH-d4) δ 5.86 (1H, dt, J = 15.0, 7.0 Hz, H-5), 5.53 (1H, dd, J = 15.0, 7.5 Hz, H-4), 4.45 (1H, dd, J = 7.5, 7.5 Hz, H-3), 3.59 (1H, dq, J = 7.5, 6.2 Hz, H-2), 2.08 (2H, dt, J = 7.0, 7.0 Hz, H-6), 1.24–1.39 (14H, m, H-7–H-13), 0.88 (3H, t, J = 7.0 Hz, H-14); LRESIMS m/z 254.2 [M + H]+ (calcd for C15H28NO2, 254.2).
3.5. Hydrogenation of Amino Alcohols
To a stirred mixture of 8.7 mg (0.038 mmol) of 1 in 1.0 mL of dry MeOH was added 10.0 mg of Pd/C. The mixture was stirred and hydrogenated using H2 at room temperature for 2 h. The mixture was filtered through Celite under vacuum. The residue was concentrated under reduced pressure and purified by HPLC (YMC ODS-A column, 10 × 250 mm; H2O–MeOH, 50:50) to yield pure compound 9 (7.1 mg, 81%). Compound 4 (2.8 mg, 0.013 mmol) was hydrogenated to afford pure compound 13 (2.3 mg, 81%) via the same procedure as that for 1.
Compound 9 (9): 1H NMR (MeOH-d4) δ 3.20 (1H, m, H-3), 2.60 (1H, dq, J = 7.0, 7.0 Hz, H-2), 1.49 (2H, m, H-4), 1.23–1.38 (18H, m, H-5–H-13), 1.04 (3H, d, J = 7.0 Hz, H-1), 0.89 (3H, t, J = 7.0 Hz, H-14); LRESIMS m/z 230.3 [M + H]+ (calcd for C14H32NO, 230.3).
Compound 13 (13): 1H NMR (MeOH-d4) δ 3.62 (1H, m, H-2), 2.85 (1H, m, H-1a), 2.63 (1H, m, H-1b), 1.44 (2H, m, H-3), 1.22–1.36 (12H, m, H-4–H-12), 0.88 (3H, t, J = 7, H-13); LRESIMS m/z 215.2 [M + H]+ (calcd for C13H30NO, 215.2).
3.6. Preparation of N-Acetyl Derivatives
To a stirred mixture of 2.9 mg (0.013 mmol) of 9 in 1.0 mL of dry pyridine was added 5.0 mg of acetic anhydride. The mixture was stirred at room temperature for 1 h, concentrated under reduced pressure, and purified by HPLC (YMC ODS-A column, 10 × 250 mm; H2O–MeOH, 20:80) to yield pure compound 10 (2.3 mg, 71%). Compound 13 (2.3 mg, 0.011 mmol) was acetylated to afford pure compound 14 (1.9 mg, 77%) via the same procedure as that for 9.
Compound 10 (10): 1H NMR (CDCl3) δ 5.86 (1H, s, NH), 3.70 (1H, m, H-2), 3.51 (1H, m, H-1), 3.10 (1H, m, H-1), 2.04 (3H, s, H-NHCOMe-Me), 1.44 (2H, m, H-3), 1.24–1.33 (12H, m, H-7–H-12), 0.88 (3H, t, J = 7, H-13); LRESI-MS m/z 258.2 [M + H]+ (calcd for C15H32NO2, 258.2).
Compound 14 (14): 1H NMR (CDCl3) δ 5.83 (1H, s, NH), 3.69 (1H, m, H-2), 3.52 (1H, m, H-1), 3.13 (1H, m, H-1), 2.01 (3H, s, H-NHCOMe-Me), 1.43 (1H, m, H-3), 1.24–1.30 (18H, m, H-4–H-12), 0.88 (3H, t, J = 7.5, H-13); LRESI-MS m/z 256.3 [M + H]+ (calcd for C15H30NO2, 256.2).
3.7. Esterification with of (–)-(R)-α-Methoxy-α-(trifluoromethyl)phenylacetic (MTPA) Chloride
To a stirred mixture of 1.0 mg (0.004 mmol) of 9 and 0.1 mg of DMAP in 1.0 mL of dry pyridine was added 20 μL of (–)-(R)-MTPA-chloride. The mixture was stirred at room temperature for 1 h. The mixture was concentrated under reduced pressure and purified by HPLC (YMC ODS-A column, 10 × 250 mm, H2O–MeOH, 10:90) to give 0.7 mg (32%) of (S)-MTPA ester 11S. The corresponding (R)-MTPA ester 11R (0.8 mg, 36%) was also obtained from a similar esterification reaction of 9 (1.0 mg, 0.004 mmol) with (+)-(S)-MTPA-chloride. Compound 14 (1.0 mg, 0.004 mmol) was esterified to afford pure compounds 15S (0.6 mg, 32%) and 15R (0.6 mg, 32%) via the same procedure as that for 9.
Compound 11S (11S): 1H NMR (CDCl3) δ 7.508 (2H, dd, J = 7.6, 1.8 Hz, MTPA-Ar), 7.396–7.421 (3H, m, MTPA-Ar), 6.916 (1H, d, J = 9.0 Hz, NH), 4.920 (1H, dt, J = 5.0, 7.0 Hz, H-3), 4.254 (1H, ddq, J = 9.0, 5.0, 7.0 Hz, H-2), 3.408 (3H, s, MTPA-OMe), 2.092 (3H, s, COMe), 1.561 (2H, dt, J = 7.0, 7.0 Hz, H-4), 1.205–1.314 (18H, m, H-5–H-13), 1.119 (3H, d, J = 7.0 Hz, H-1), 0.880 (3H, t, J = 7.0 Hz, H-14); LRESIMS m/z 510.3 [M + Na]+ (calcd for C26H40NO4F3Na, 510.3).
Compound 11R (11R): 1H NMR (CDCl3) δ 7.545 (2H, dd, J = 7.5, 2.0 Hz, MTPA-Ar), 7.348–7.442 (3H, m, MTPA-Ar), 6.830 (1H, d, J = 9.0 Hz, NH), 4.880 (1H, dt, J = 5.0, 7.0 Hz, H-3), 4.239 (1H, ddq, J = 9.0, 5.0, 7.0 Hz, H-2), 3.437 (3H, s, MTPA-OMe), 1.969 (3H, s, COMe), 1.435 (2H, dt, J = 7.0, 7.0 Hz, H-4), 1.202–1.316 (18H, m, H-5–H-13), 1.171 (3H, d, J = 7.0 Hz, H-1), 0.880 (3H, t, J = 7.0 Hz, H-14); LRESIMS m/z 510.3 [M + Na]+ (calcd for C26H40NO4F3Na, 510.3).
Compound 15S (15S): 1H NMR (CDCl3) δ 7.497–7.540 (2H, m, MTPA-Ar), 7.377–7.418 (3H, m, MTPA-Ar), 7.035 (1H, dd, J = 5.5, 7.0 Hz, NH), 4.961 (1H, ddt, J = 3.5, 7.0, 7.0 Hz, H-2), 3.542 (1H, ddd, J = 3.5, 5.5, 13.5 Hz, H-1), 3.462 (1H, ddd, J = 7.0, 7.0, 13.5 Hz, H-1), 3.395 (3H, s, MTPA-OMe), 2.023 (3H, s, COMe), 1.299 (2H, m, H-3), 1.215–1.304 (18H, m, H-4–H-12), 0.880 (3H, t, J = 7.0 Hz, H-13); LRESIMS m/z 474.2 [M + H]+ (calcd for C25H39NO4F3, 474.3).
Compound 15R (15R): 1H NMR (CDCl3) δ 7.510–7.564 (2H, m, MTPA-Ar), 7.385–7.451 (3H, m, MTPA-Ar), 5.212 (1H, dd, J = 5.5, 7.0 Hz, NH), 5.121 (1H, ddt, J = 3.5, 7.0, 7.0 Hz, H-2), 3.602 (1H, ddd, J = 3.5, 5.5, 13.5 Hz, H-1), 3.566 (3H, s, MTPA-OMe), 3.246 (1H, ddd, J = 7.0, 7.0, 13.5 Hz, H-1), 1.839 (3H, s, COMe), 1.669 (2H, m, H-3), 1.214–1.315 (18H, m, H-4–H-12), 0.880 (3H, t, J = 7.0 Hz, H-13); LRESIMS m/z 474.2 [M + H]+ (calcd for C25H39NO4F3, 474.3).
3.8. Preparation of Dibenzoyl Derivatives
To a stirred mixture of 2.7 mg (0.012 mmol) of 9 and 0.1 mg of DMAP in 1.0 mL of dry pyridine was added 20.0 mg of benzoyl chloride. The mixture was stirred at room temperature for 3 h, concentrated under reduced pressure, and separated by HPLC (YMC ODS-A column, 10 × 250 mm; H2O–MeOH, 10:90) to yield compound 12 (2.1 mg, 41%). Compound 4 (2.5 mg, 0.012 mmol) was reacted to afford pure compound 16 (2.3 mg, 46%) via the same procedure as that for 9.
Compound 12 (12): 1H NMR (CDCl3) δ 8.03 (2H, d, J = 7.8 Hz, Ar), 7.72 (2H, d, J = 7.8 Hz, Ar), 7.54 (1H, t, J = 7.8 Hz, Ar), 7.45 (5H, m, Ar), 6.38 (1H, d, J = 9.2 Hz, NH), 5.22 (1H, dt, J = 7.5, 5.5 Hz, H-3), 4.54 (1H, ddq, J = 7.5, 9.2, 7.0 Hz, H-2), 1.78 (1H, dddd, J = 7.5, 7.5, 7.5, 15.0 Hz, H-4), 1.78 (1H, dddd, J = 5.5, 7.5, 7.5, 15.0 Hz, H-4), 1.41 (2H, tt, J = 7.5, 7.5 Hz, H-5), 1.29 (3H, d, J = 7.0 Hz, H-1), 1.19–1.25 (16 H, m, H-6–H-13), 0.87 (3H, t, J = 7.0 Hz, H-14); LRESIMS m/z 438.3 [M + H]+ (calcd for C28H40NO3, 438.3).
Compound 16 (16): 1H NMR (CDCl3) δ 8.05 (2H, d, J = 7.9 Hz, Ar), 7.74 (2H, d, J = 7.9 Hz, Ar), 7.58 (1H, t, J = 7.9 Hz, Ar), 7.45 (5H, m, Ar), 6.69 (1H, dd, J = 5.5, 5.5 Hz, NH), 5.60 (1H, ddd, J = 14.5, 7.5, 7.5 Hz, H-5), 5.45 (1H, ddd, J = 14.5, 7.5, 7.5 Hz, H-4), 5.29 (1H, dddd, J = 7.5, 7.5, 7.5, 3.0 Hz, H-2), 3.83 (1H, ddd, J = 13.5, 5.5, 3.0 Hz, H-1), 3.72 (1H, ddd, J = 13.5, 7.5, 5.5 Hz, H-1), 2.50 (2H, dd, J = 7.5, 7.5 Hz, H-3), 1.98 (2H, dt, J = 7.5, 7.5 Hz, H-6), 1.20–1.31 (12H, m, H-7–H-12), 0.87 (3H, t, J = 7.0 Hz, H-13); LRESIMS m/z 422.2 [M + H]+ (calcd for C27H36NO3, 422.3).
3.9. Biological Assays
Antimicrobial assays were performed according to the method described previously [
22]. Cytotoxicity assays were performed in accord with literature protocols [
23]. Isocitrate lyase, sortase A, and Na
+/K
+-ATPase inhibition assays were performed according to previously described methods [
24,
25,
26].



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

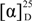 −4.4 and +5.1 for 1 and 2, respectively), which led us to conduct a detailed investigation. Thus, 2 was hydrogenated and converted to the dibenzoyl derivative, as shown in Scheme 2. After each step, the specific rotations for the corresponding derivatives of 1 and 2 were compared. The specific rotations of the hydrogenated derivatives (
−4.4 and +5.1 for 1 and 2, respectively), which led us to conduct a detailed investigation. Thus, 2 was hydrogenated and converted to the dibenzoyl derivative, as shown in Scheme 2. After each step, the specific rotations for the corresponding derivatives of 1 and 2 were compared. The specific rotations of the hydrogenated derivatives (