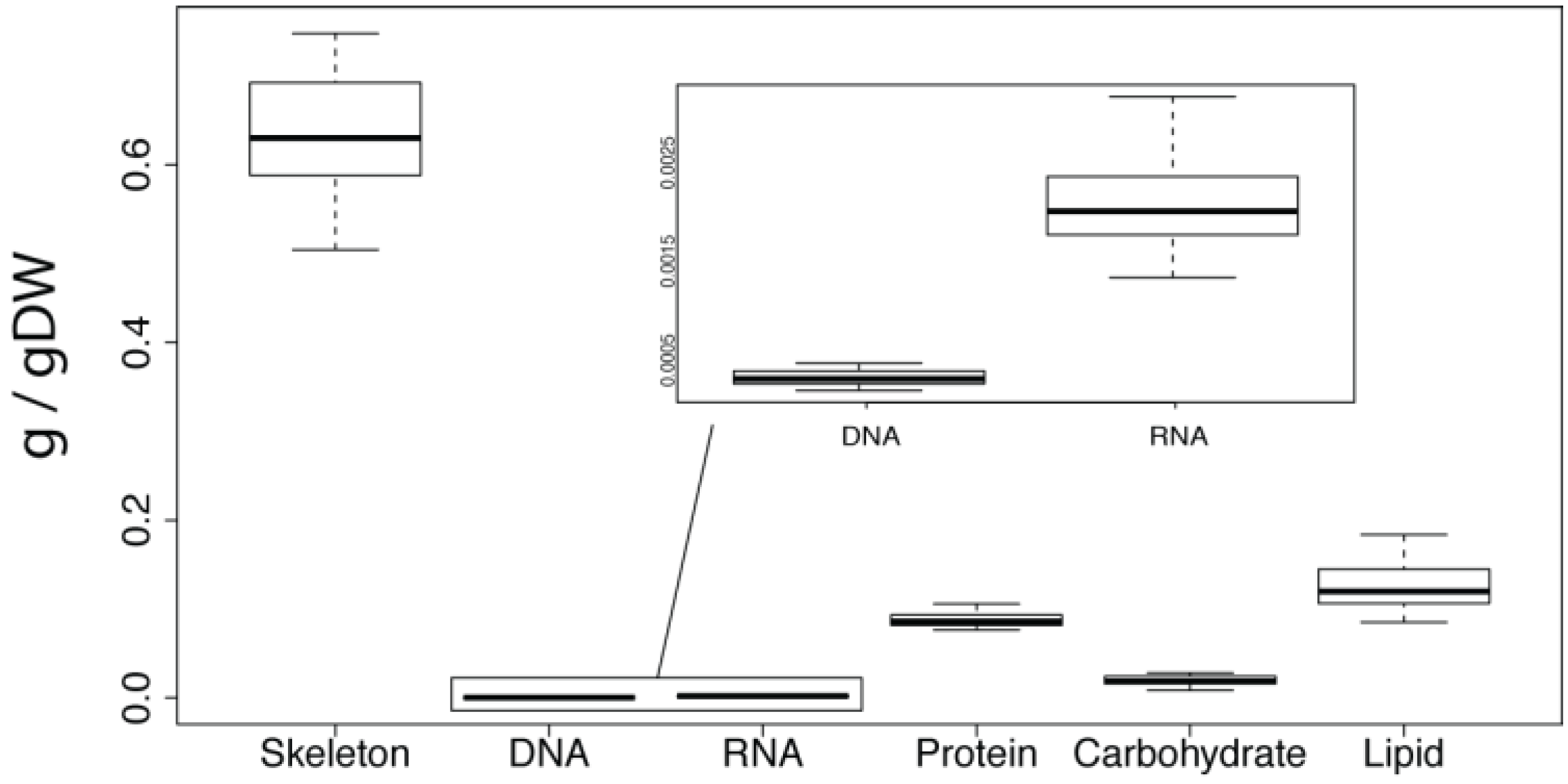
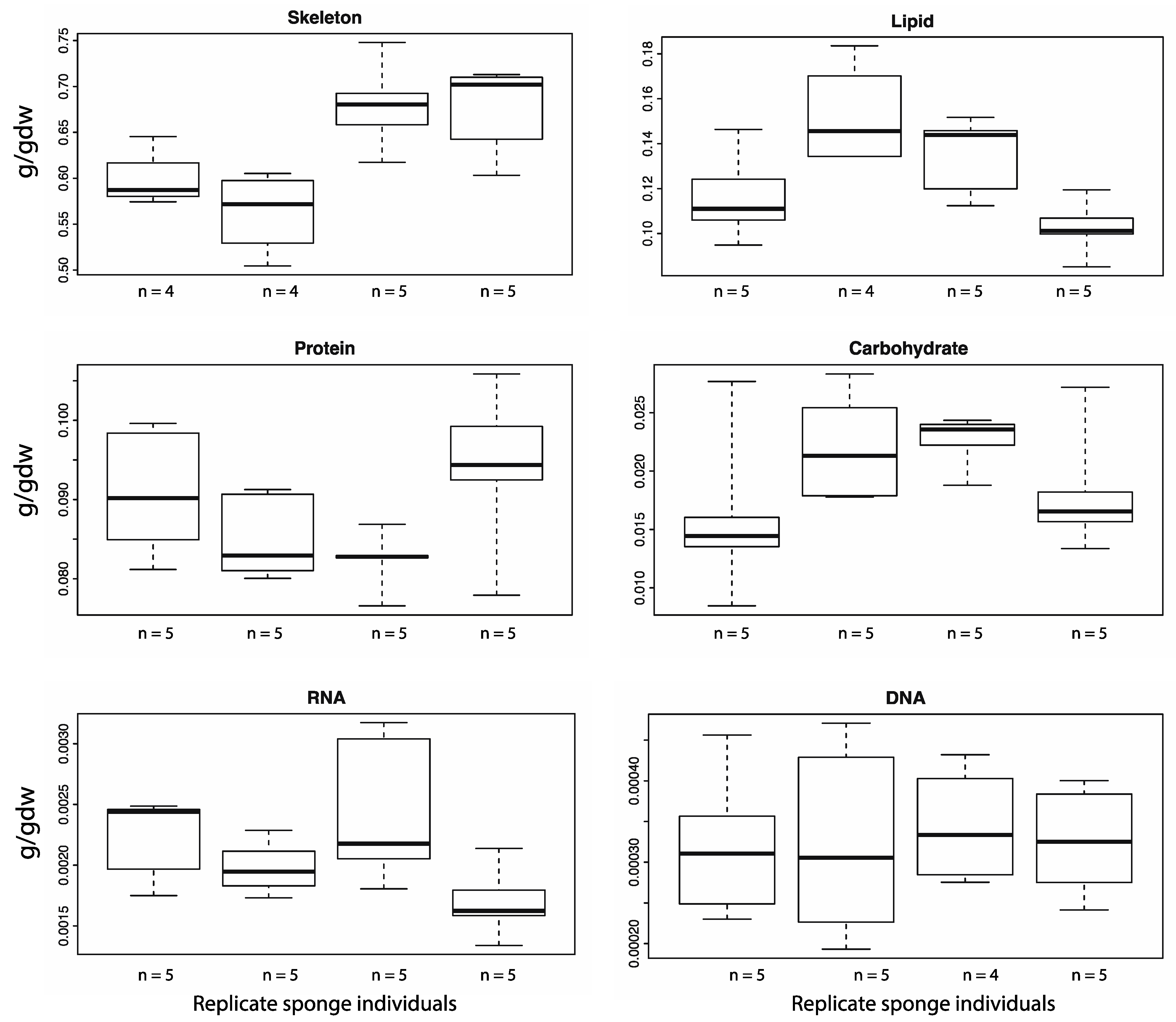
4.3. Nucleic Acids
DNA was quantified by Hoechst fluorescence assay. The lyophilised samples from 4.1 were re-hydrated with 3.0 mL of TE buffer (pH 8.0) with 0.01 g/mL lysozyme and incubated at 37 °C for 1 h on a shaker table set to 100 rpm. Re-hydrating the lyophilised biopsies caused complete lysis of the
A. queenslandica cells, but not the bacterial fraction. Lysozyme was added to ensure lysis of the bacterial fraction. After incubation, sterile tweezers were used to remove the sponge skeleton from the tube. The piece of skeleton was squeezed and rolled against the inside wall to ensure that the lysis buffer was retained. Samples were then vortexed and centrifuged for 5 min at 4000 g to pellet cellular debris. Aliquots of the supernatant were used in the Hoechst fluorescence assay. Calf thymus DNA was used to generate a standard curve [
47]. All samples and standards were measured in triplicate.
RNA content was quantified by digesting the lyophilised samples from 4.1 with 3.0 mL of 0.3 M KOH for 1 h at 37 °C [
48]. The reaction was then acidified with the addition of 1.0 mL of 3 M perchloric acid. [
49]. Following centrifugation to remove DNA and protein, the RNA-containing supernatant was quantified in triplicate by UV absorbance at 260 nm [
48].
The nucleotide composition of both the DNA and RNA was calculated using the GC content of the genome and an adult transcriptome. The calculations assumed that the DNA and RNA is all sponge derived as the bacterial genomic and transcriptomic sequence data, in addition to bacterial diversity and abundance data is not yet available. The GC content of the
A. queenslandica genome and adult transcriptome are 31.14% [
18] and 39.76% [
20], respectively. The percentage contribution of each nucleotide was calculated, assuming an even guanine-cytosine and thymine-adenine/thymine-uracil ratios. Next the average molecular weight of one nucleotide was calculated. This was done by multiplying the abundance of each nucleotide by its molecular weight. These values were then added together to give the average molecular weight. This was multiplied by the mean total amount of DNA or RNA (g/gDW) to give the number of mols of average nucleotide per gram of dry weight. The amount of each nucleotide (mols) was then calculated using this value and the guanine-cytosine to thymine-adenine/thymine-uracil ratios. Values were converted to micromoles for reporting.
4.7. Fatty Acids and Sterols
A fatty acid methyl ester (FAME) analysis was performed on the extracted lipids as previously described [
54]. The lipid solid was dissolved in hexane and 5.1 μg of nondecanoic acid (Sigma-Aldrich, St. Louis, MO, USA) was used as an internal standard (ISTD) to a final concentration of 3.6 μg/mL. Saponification, methylation and GC/MS analysis of the lipid mass fraction followed the protocol described in [
54] The software program AMDIS (version 2.64, National Institute of Standards and Technology (NIST), Gaithersburg, MD, USA) was used for the analysis of the fatty acid and sterols. A minimum match factor of 80 and signal threshold of 700 was set for identifying compounds. A FAME library was built using the target ions and retention times of thirty-two known fatty acid standards (Sigma-Aldrich, St. Louis, MO, USA). An alkane (C
7-30) standard was used as a retention time index. To define compounds important for biomass formation, we used a cut-off of 0.05% of the largest peak in the MS analysis, which means that peaks below those signal intensities would not be reported. For unknown compounds (
i.e., that were not identified using the standard library), their fragmentation patterns were compared to the NIST database library (version 2.0, NIST, Gaithersburg, MD, USA). For a range of peaks both the target library as well as the NIST library did not provide accurate hits. Based on manual interpretation of fragmentation patterns, unknowns that were found consistently in the majority (>75%) of the samples were classified into sterols or fatty acids. From the quantified lipid fraction (g/gDW) the estimated weight of glycerol and phosphate groups was subtracted assuming a glycerolipid to phospholipid ratio of 20:80. It was then assumed that the reminder of the lipid fraction would generate a signal in GC-MS and that the MS response and ionization efficiency for all FA and sterol ions was comparable (due to the unknown character of many FA’s this cannot be tested, but individual errors for individual compounds will not impact dramatically on the later modelling). All peak areas were normalized to the total signal generated for that sample. Using the molecular weight of the knowns and an average molecular weight (calculated from all identified FA’s and sterols) for the unknowns the individual concentrations were calculated in mol/gDW. A total of 23 separate fractions of biomass were analyzed and the standard deviation was calculated.
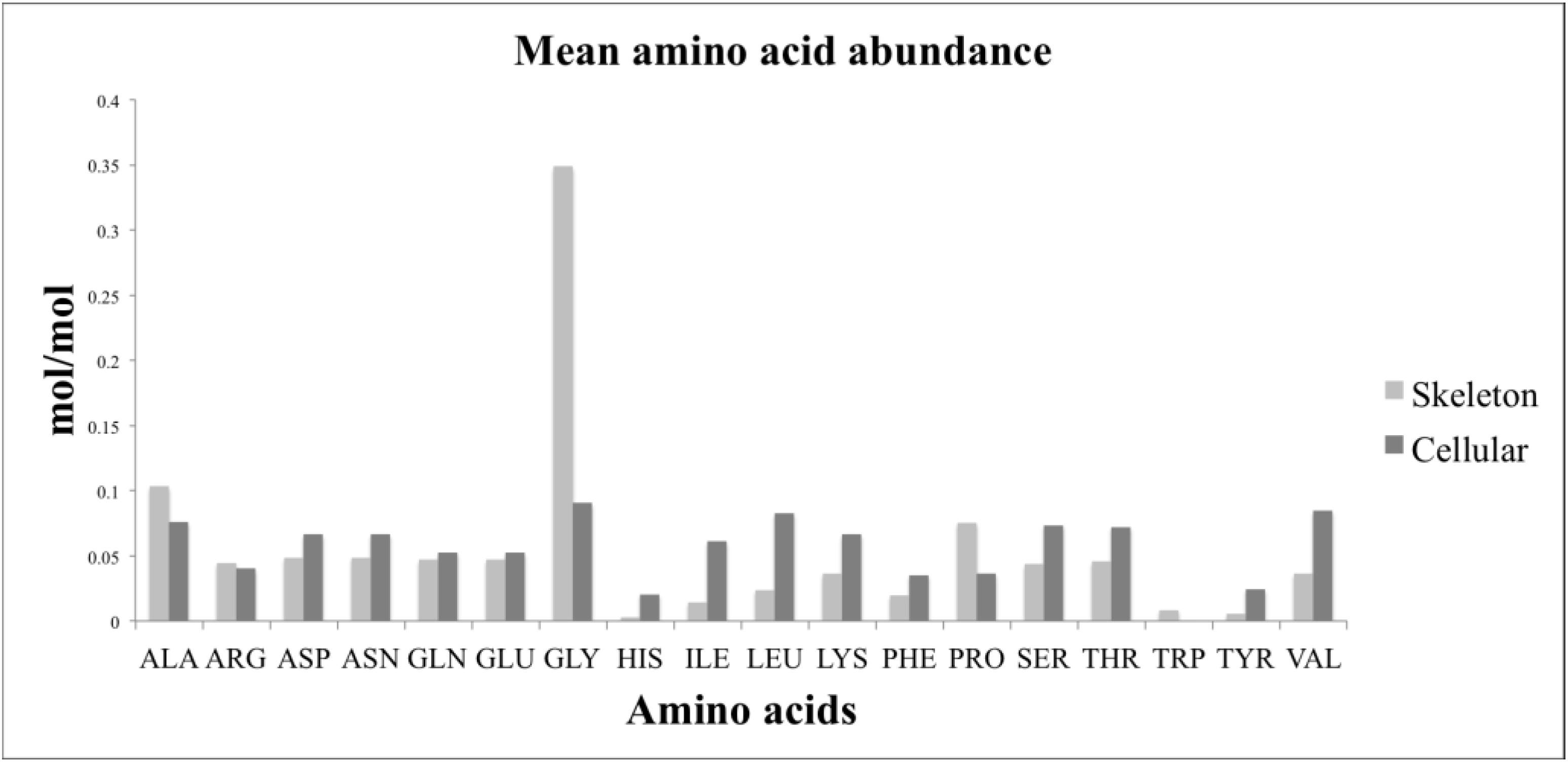
4.8. Amino Acids
Amino acid analysis was performed by reversed phase high performance liquid chromatography. 20 samples containing cellular and skeleton components were analysed in addition to 20 samples of skeleton, prepared as above. 5 mL of 6 M HCl was added to each sample and incubated at 105 °C. Hydrolysis times were first optimized and found to be 54 h for the complete biomass samples and 48 h for skeleton samples. Optimization monitored the concentration increase of all amino acids over the time course and 54/48 h was found to be a time at which most amino acids reached the peak concentration. Longer times may favor some compounds but will lead to degradation of others. This compromise was necessary since otherwise individual experiments need to be conducted for all 20 amino acids. Samples were allowed to cool, mixed, and then a 500 μL aliquot was taken and filtered with a 0.22 μm Ultrafree-MC GV centrifugal filter (Merck Millipore, Darmstadt, Germany). The HCl was evaporated by heating to 40 °C under a constant flow of nitrogen. The solid pellet was stored at −80 °C. Derivatisation was performed in a high-performance autosampler (Agilent HiP-ALS SL, G1367C, Santa Clara, CA, USA). 0.5 μL of sample containing 250 μM of internal standards, sarcosine and 2-aminobutyric acid, was added into 2.5 μL of borate buffer (0.4 N, pH 10.2, Agilent PN: 5061-3339, Santa Clara, CA, USA), mixed and incubated for 20 s at 4 °C. 1 μL of OPA reagent (10 mg o-pthalaldehyde/mL in 3-mercaptopropionic acid, Agilent PN: 5061-3335, Santa Clara, CA, USA) was then added to initially derivatise primary amino acids. The reaction was mixed and incubated for 20 s at 4 °C. Then 0.4 μL of FMOC reagent (2.5 mg 9-fluorenylmethyl chloroformate/mL in acetonitrile, Agilent PN:5061-3337, Santa Clara, CA, USA) was added, mixed and incubated for 20 s at 4 °C to derivatise the secondary amines proline and sarcosine. 45.6 μL of Buffer A (40 mM Na2HPO4, 0.02% NaN3, pH 7.8) was added to lower the pH of the reaction prior to injecting the 50 μL reaction onto an Agilent Zorbax Extend C-18 column (3.5 μm, 4.6 × 150 mm, Agilent PN: 763953-902, Santa Clara, CA, USA) with a guard column (SecurityGuard Gemini C18, Phenomenex PN: AJO-7597, Santa Clara, CA, USA). Column temperature was kept at 39 °C in a thermostatted column compartment (Agilent TCC, G1316B, Santa Clara, CA, USA). Chromatography was performed using an Agilent 1200-SL HPLC system, equipped with an active seal wash and a degasser (Agilent Degasser, G1379B, Santa Clara, CA, USA). The HPLC gradient was 2%–45% buffer B (45% acetonitrile, 45% methanol and 10% water) from 0 to 18 min, 50%–60% buffer B from 18.1 to 20 min. 100% buffer B from 20.1 to 24 min, and 2% buffer B from 24.1 to 27 min—using a binary pump (Agilent Bin Pump SL, G1312B, Santa Clara, CA, USA). Flow rate was 2 mL/min. Derivatised amino acids were monitored using a fluorescence detector (Agilent FLD, G1321A, Santa Clara, CA, USA). OPA-derivatised amino acids were detected at 340ex and 450em nm from 1 to 18 min, and FMOC-derivatised amino acids at 266ex and 305em nm from 18 to 27 min. Chromatograms were integrated using ChemStation (Rev B.03.02, Agilent, Santa Clara, CA, USA).



{kind=link}
{kind=link}
{kind=link}

