2.1. Structure Elucidation of DAQE, DAQF, and DAQG
Following a series of chromatographic steps,
1 (
Figure 1) was obtained as red powder. The molecular formula was assigned as C
23H
28N
2O
4 on the basis of combined nuclear magnetic resonance (NMR) and high-resolution mass spectrometry (MS) experiments. This formula demanded 11 degrees of unsaturation. The unique chromophore of a fused diaza-anthracene ring system consistent with the diazaquinomycin structural class was observed in the UV spectrum of
1. Analysis of the
1H-NMR spectrum of
1 suggested the presence of an isolated aromatic hydrogen (δ
H 6.94, s, H-6). Integration of the H-6, H
3-11, H
2-12, H
2-17, H
3-16, and H
3-21 resonances in the
1H spectrum further supported a mono-normethylated DAQ core skeleton, revealing an integration value of three for the α-substituted methyl hydrogens (δ
H 2.32, H
3-11) rather than the typical integration value of 6 for this resonance in previously reported symmetric DAQs. This mono-normethylation afforded an asymmetry to
1.
Analysis of
13C-NMR data suggested the presence of two quinone carbonyls (δ
C 180.8, C-10; 173.1, C-9), two lactam carbonyls (δ
C 163.0, C-2; 162.9, C-7), one methine alkene carbon (δ
C 127.5, C-6), seven quaternary carbons (δ
C 159.7, C-5; 154.2, C-4; 137.8, C-3; 136.5, C-8a; 134.1, C-9a; 118.0, C-4a; 117.8, C-10a), eight methylene carbons (δ
C 34.9, C-17; 32.3, C-14; 31.8, C-19; 30.7, C-12; 29.6, C-18; 28.8, C-13; 22.5, C-15; 22.5, C-20), and three methyl carbons (δ
C 14.1, C-16; 14.1, C-21; 13.0, C-11) (
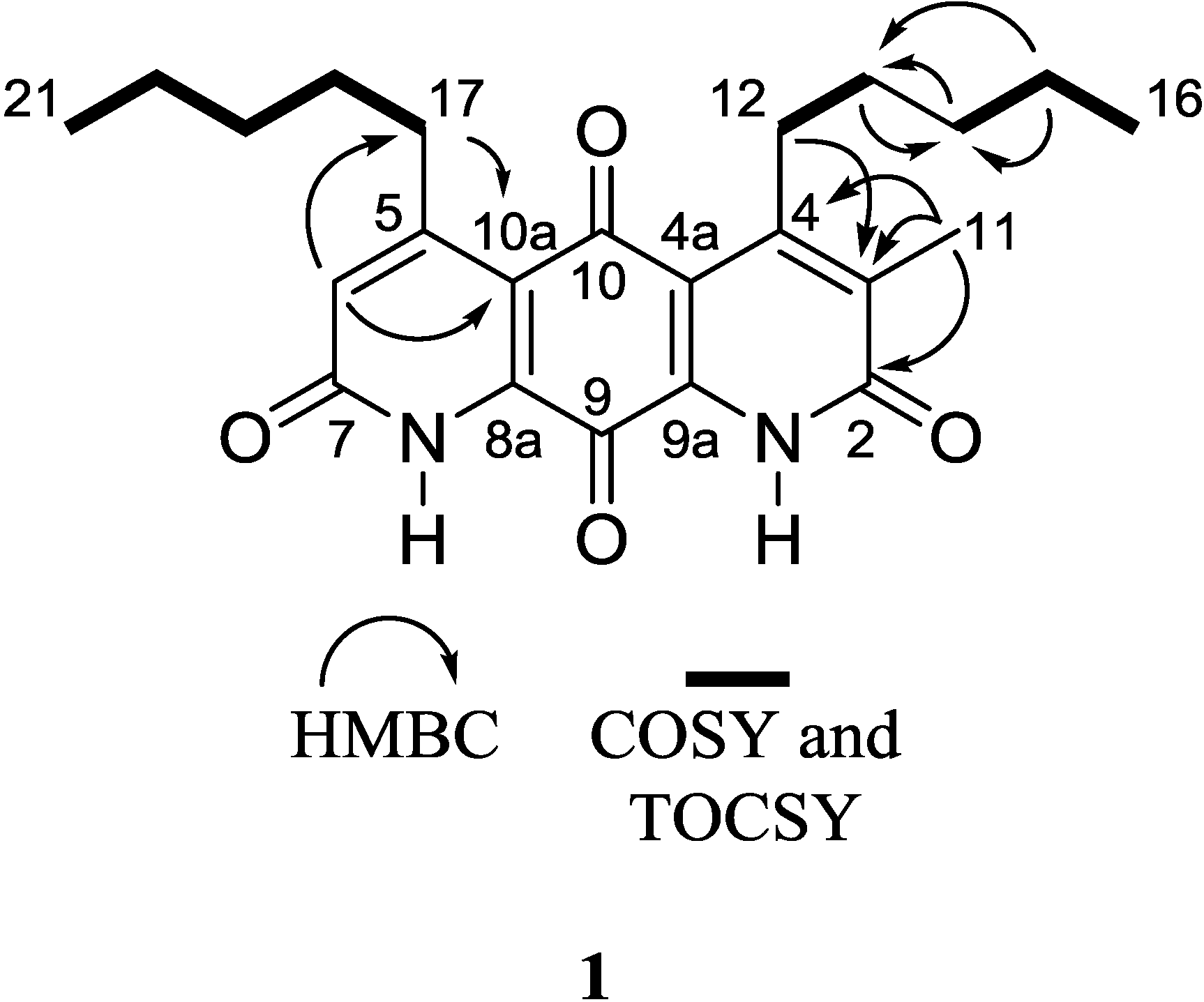
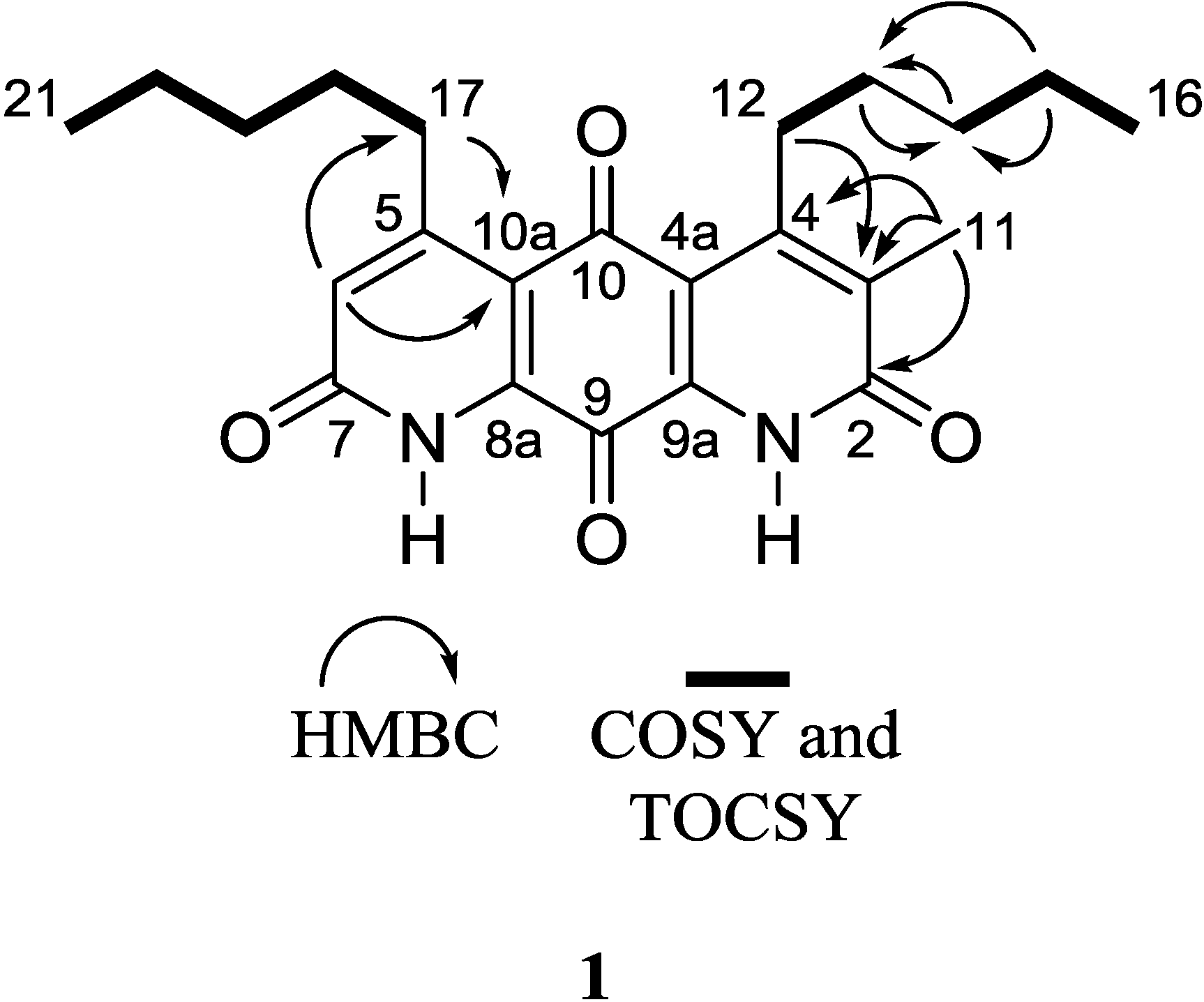
Table 1). Given that the molecular formula afforded 11 degrees of unsaturation and the molecule contained four carbonyls and eight quaternary alkene carbons, the remaining degrees were satisfied by the fused ring system. Key HMBC, COSY, and 1D-TOCSY correlations are given in
Figure 2. Since the molecule is asymmetric, we were able to employ proton-based spectroscopic experiments to distinguish between resonances of the β-substituted alkyl groups. Interpretation of COSY and 1D-TOCSY data defined two distinct spin systems, which were then connected to the core ring system using HMBC correlations (
Figure 3 and
Supplementary Figures S4–S8).
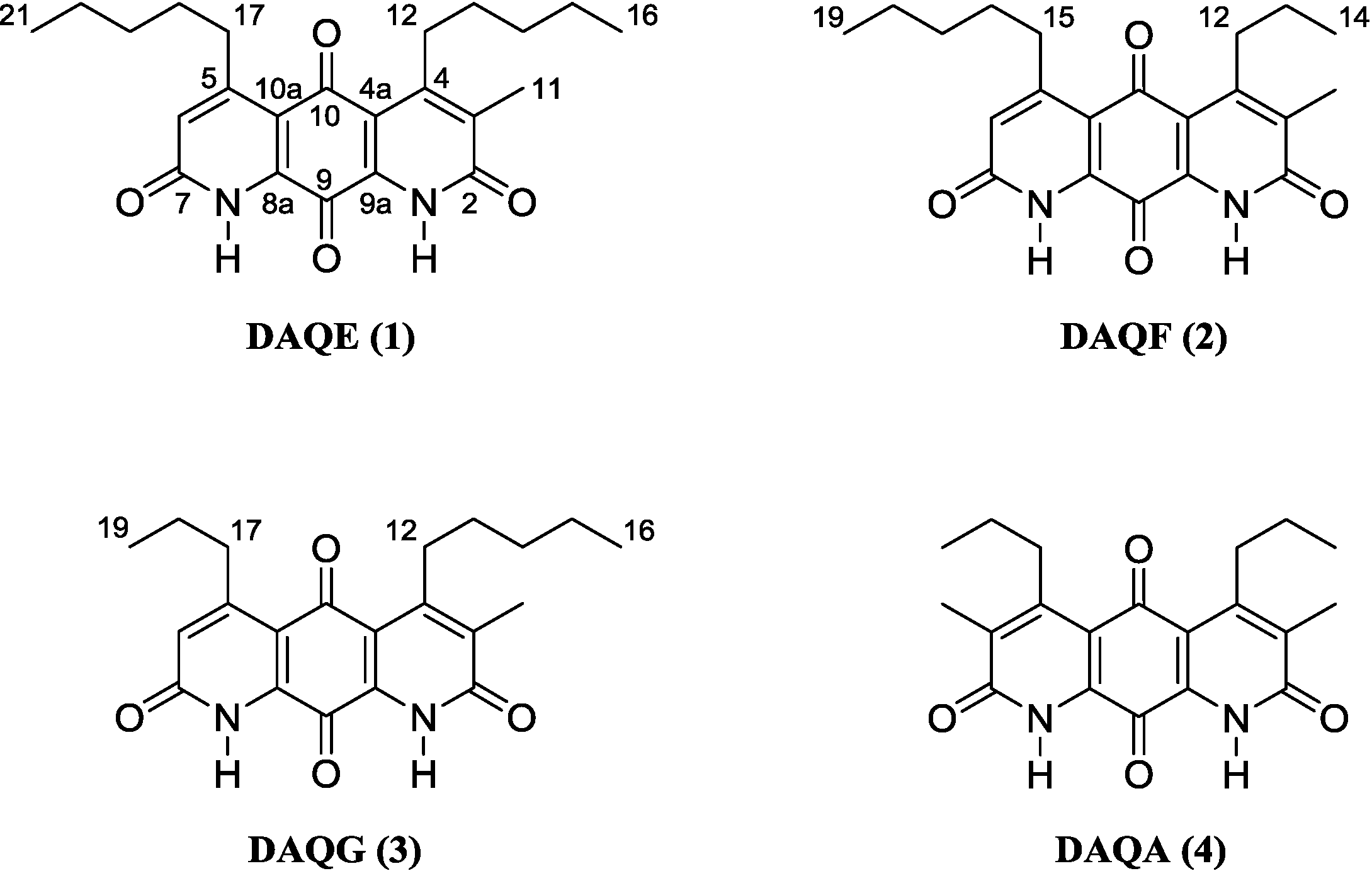
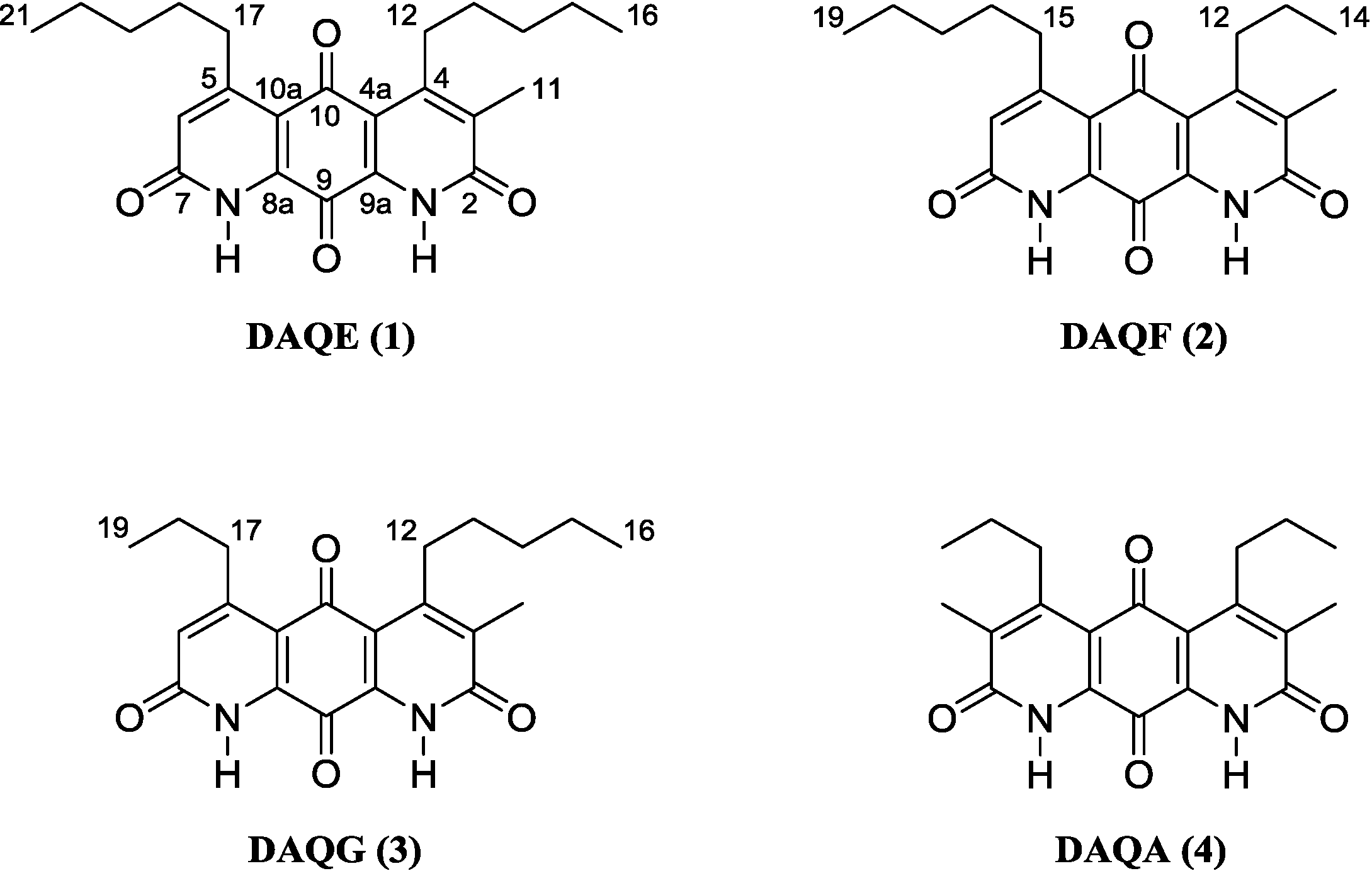
Figure 1.
Structure of diazaquinomycins E (1), F (2), G (3), and A (4).
Figure 1.
Structure of diazaquinomycins E (1), F (2), G (3), and A (4).
Table 1.
1H and 13C-NMR data (CDCl3/1% CF3CO2D) of 1.
Table 1.
1H and 13C-NMR data (CDCl3/1% CF3CO2D) of 1.
| Position | 13C a | 1H mult. (J, Hz) b |
|---|
| 2 | 163.0 c | |
| 3 | 137.8 | |
| 4 | 154.2 | |
| 4a | 118.0 | |
| 5 | 159.7 | |
| 6 | 127.5 | 6.94 s |
| 7 | 162.9c | |
| 8a | 136.5 | |
| 9 | 173.1 | |
| 9a | 134.1 | |
| 10 | 180.8 | |
| 10a | 117.8 | |
| 11 | 13.0 | 2.32 s |
| 12 | 30.7 | 3.12 bt (6.4) |
| 13 | 28.8 | 1.52 m |
| 14 | 32.3 | 1.51 m |
| 15 | 22.5 | 1.42 m |
| 16 | 14.1 | 0.96 t (7.3) |
| 17 | 34.9 | 3.11 t (7.7) |
| 18 | 29.6 | 1.60 p (7.6) |
| 19 | 31.8 | 1.43 m |
| 20 | 22.5 | 1.40 m |
| 21 | 14.1 | 0.94 t (7.3) |
Figure 2.
Key 2D NMR correlations of 1.
Figure 2.
Key 2D NMR correlations of 1.
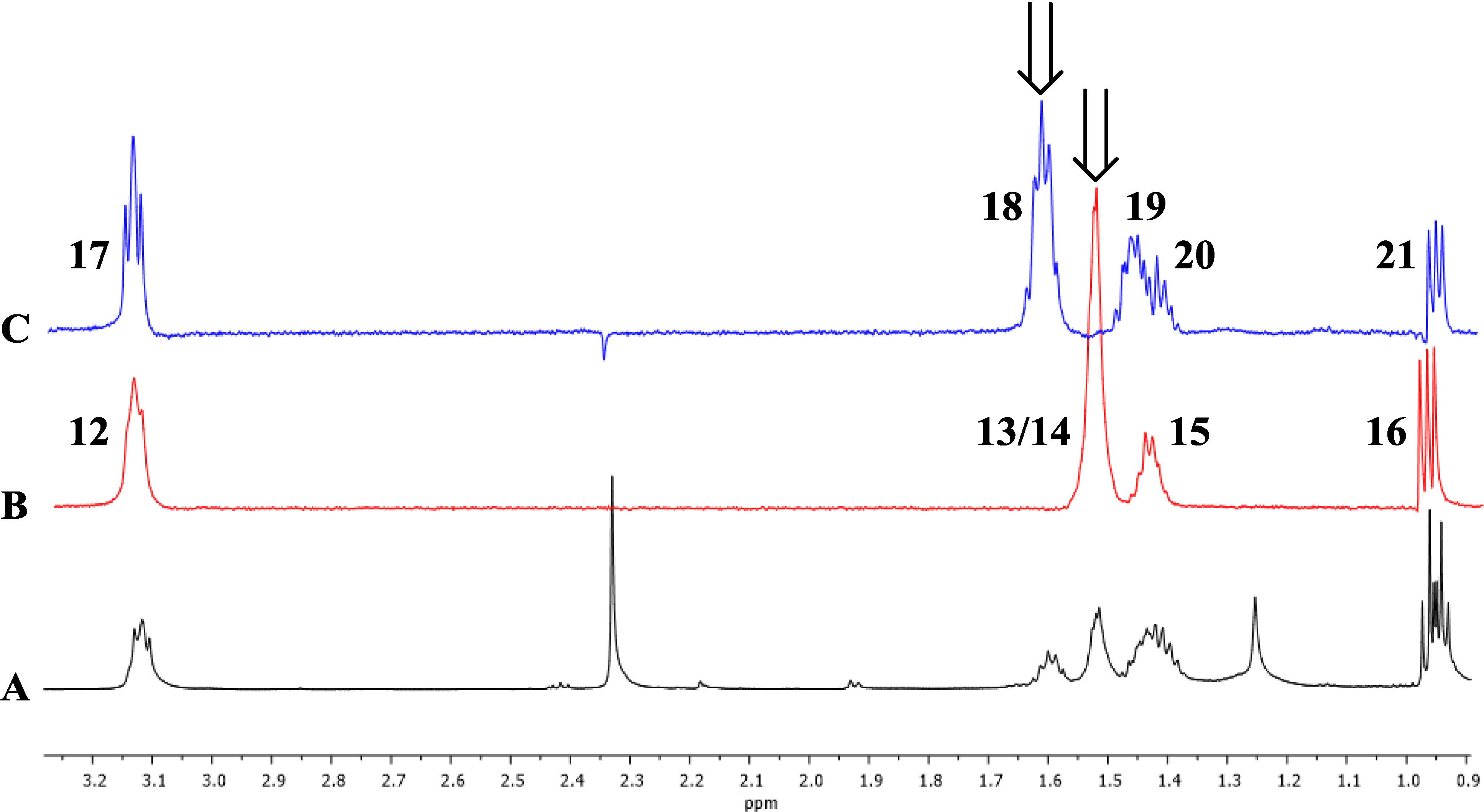
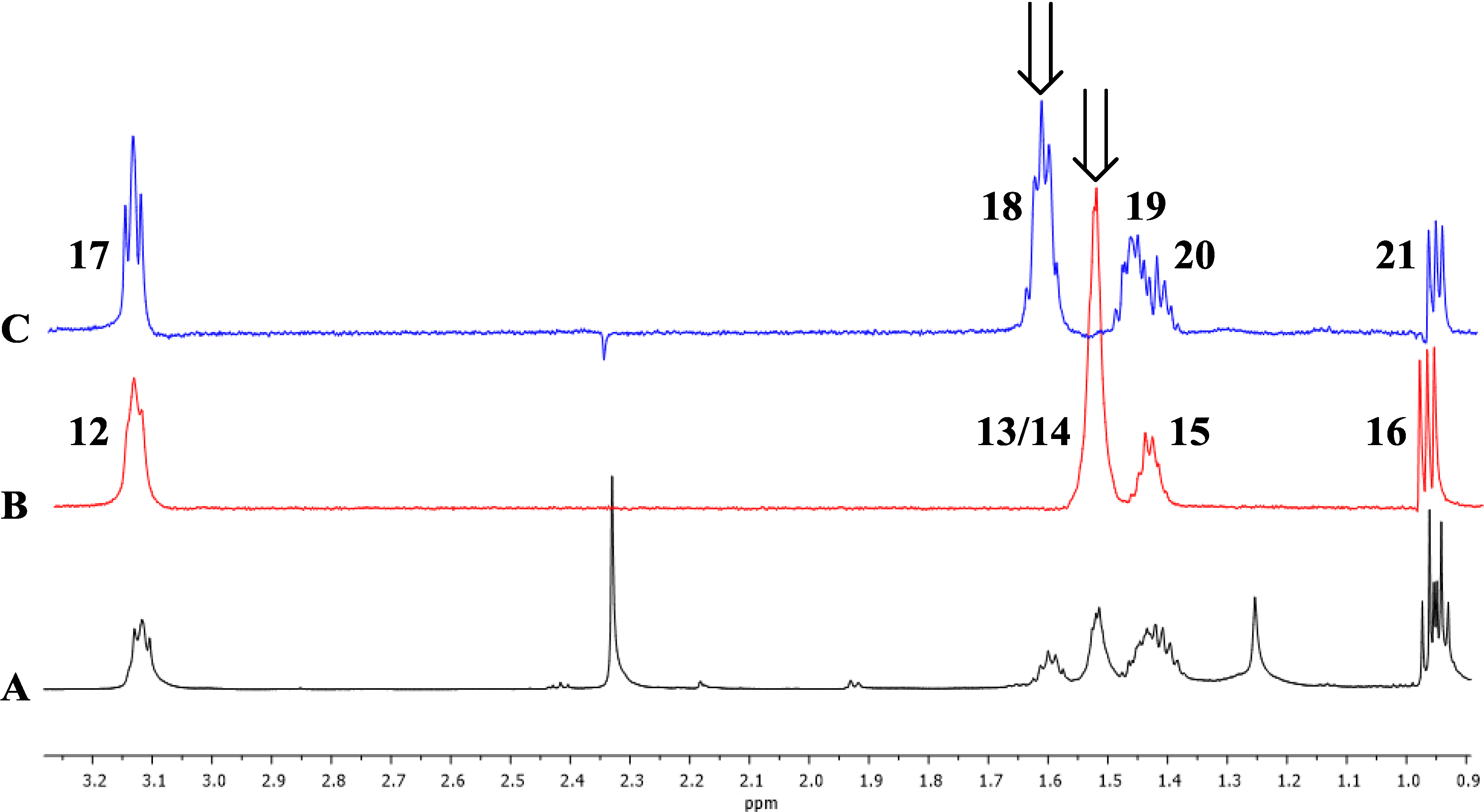
Figure 3.
1D TOCSY correlations of 1. Arrows indicate irradiated resonances. (A) Expansion of 1H-NMR spectrum (600 MHz) of 1; (B) Expansion of 1D TOCSY spectrum of 1 (irradiation of 1.51 ppm); (C) Expansion of 1D TOCSY spectrum of 1 (irradiation of 1.62 ppm).
Figure 3.
1D TOCSY correlations of 1. Arrows indicate irradiated resonances. (A) Expansion of 1H-NMR spectrum (600 MHz) of 1; (B) Expansion of 1D TOCSY spectrum of 1 (irradiation of 1.51 ppm); (C) Expansion of 1D TOCSY spectrum of 1 (irradiation of 1.62 ppm).
An HMBC correlation from H
2-17 to C-10a, and of H-6 to C-17 and C-10a, positioned the spin system of C-17–C-18–C-19–C-20–C-21 on the normethyl half of the diaza-anthracene core. Similarly, an HMBC correlation from H
2-12 to C-3 positioned the spin system of C-12–C-13–C-14–C-15–C-16 on the α-methylated ring of the diaza-anthracene core. Two lactam carbonyl resonances were observed in the
13C DEPTQ spectrum; due to overlap it was not possible to distinguish between them in an HMBC experiment (
Table 1). The remaining quinone carbons C-8a, C-9, C-9a, and C-10 were assigned based on comparison with reported values of other diazaquinomycin analogs [
5,
6]; our assignments were highly consistent with reported values. Therefore, the structure of
1 was determined as shown and named diazaquinomycin E.
Following a series of chromatographic steps, the co-eluting isomeric mixture of
2 and
3 (
Figure 1) was obtained as red powder. The molecular formula for the mixture of constitutional isomers was assigned as C
21H
24N
2O
4 on the basis of combined NMR and high-resolution MS experiments. This formula demanded 11 degrees of unsaturation. A chromophore of a fused diaza-anthracene ring system consistent with
1 was observed in the UV spectrum of the compound mixture. The diaza-anthracene core of DAQF and DAQG was determined to be mono-methylated as was previously described for
1. This was evidenced by the presence of an aromatic hydrogen in the
1H-NMR spectrum (δ
H 6.98, s, H-6 of
2 or
3). Evidence for a second minor isomer was observed adjacent to the resonance at δ
H 6.98 (δ
H 6.95, s, H-6 of
2 or
3) (
Supplementary Information, Figures S11 and S14). Integration of the major aromatic hydrogen and the n-pentyl and n-propyl group resonances in the
1H spectrum further supported a mono-normethylated DAQ core skeleton. Distinguishing separate, complete sets of
13C shifts from DEPTQ and HMBC experiments was not possible due to the structural similarity and inability to separate
2 and
3. Partial carbon shift data shared by the isomers was extracted from an HSQC experiment (
Supplementary Information, Table S1). Interpretation of COSY data defined two distinct spin systems, one n-pentyl group and one n-propyl group.
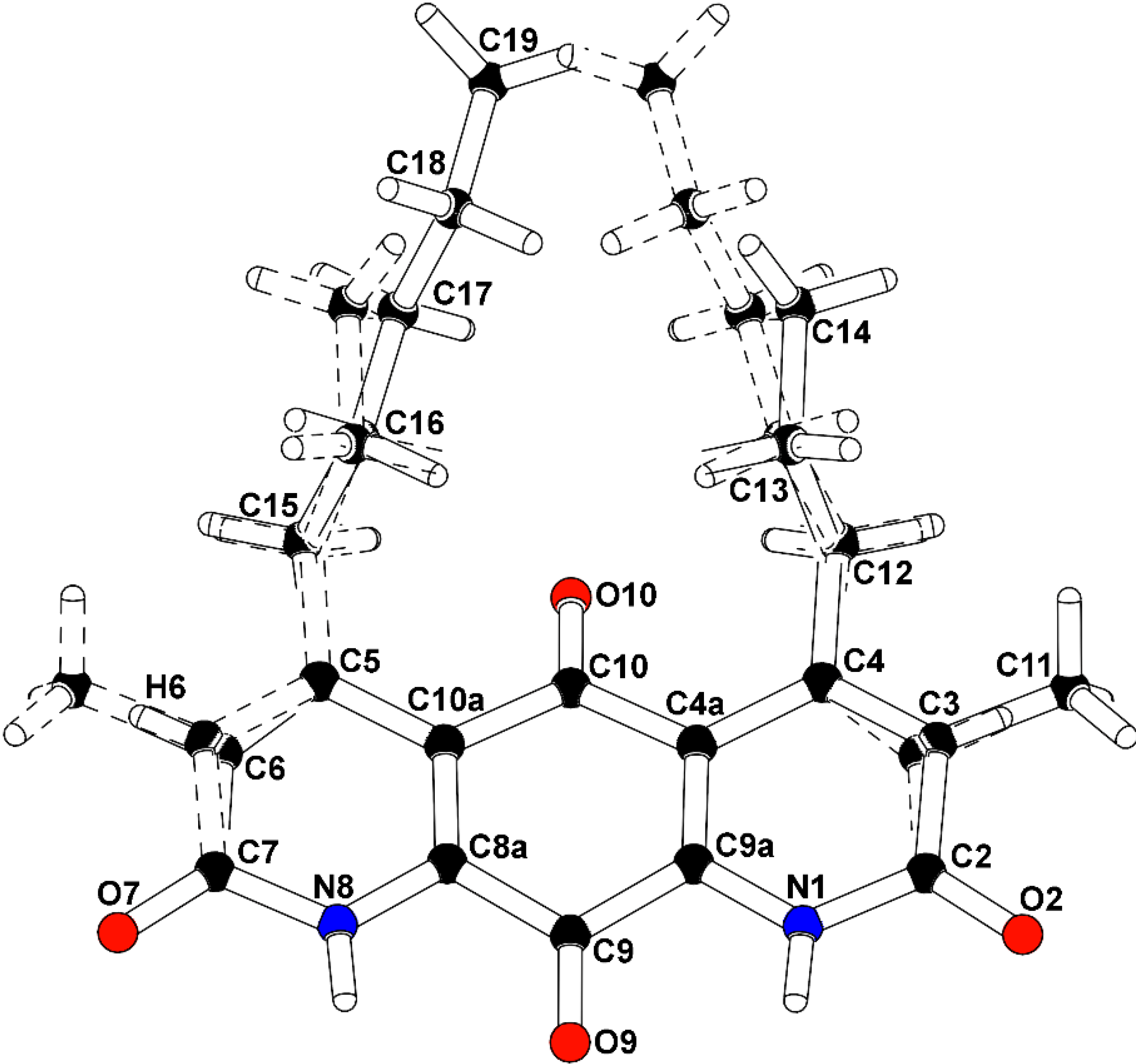
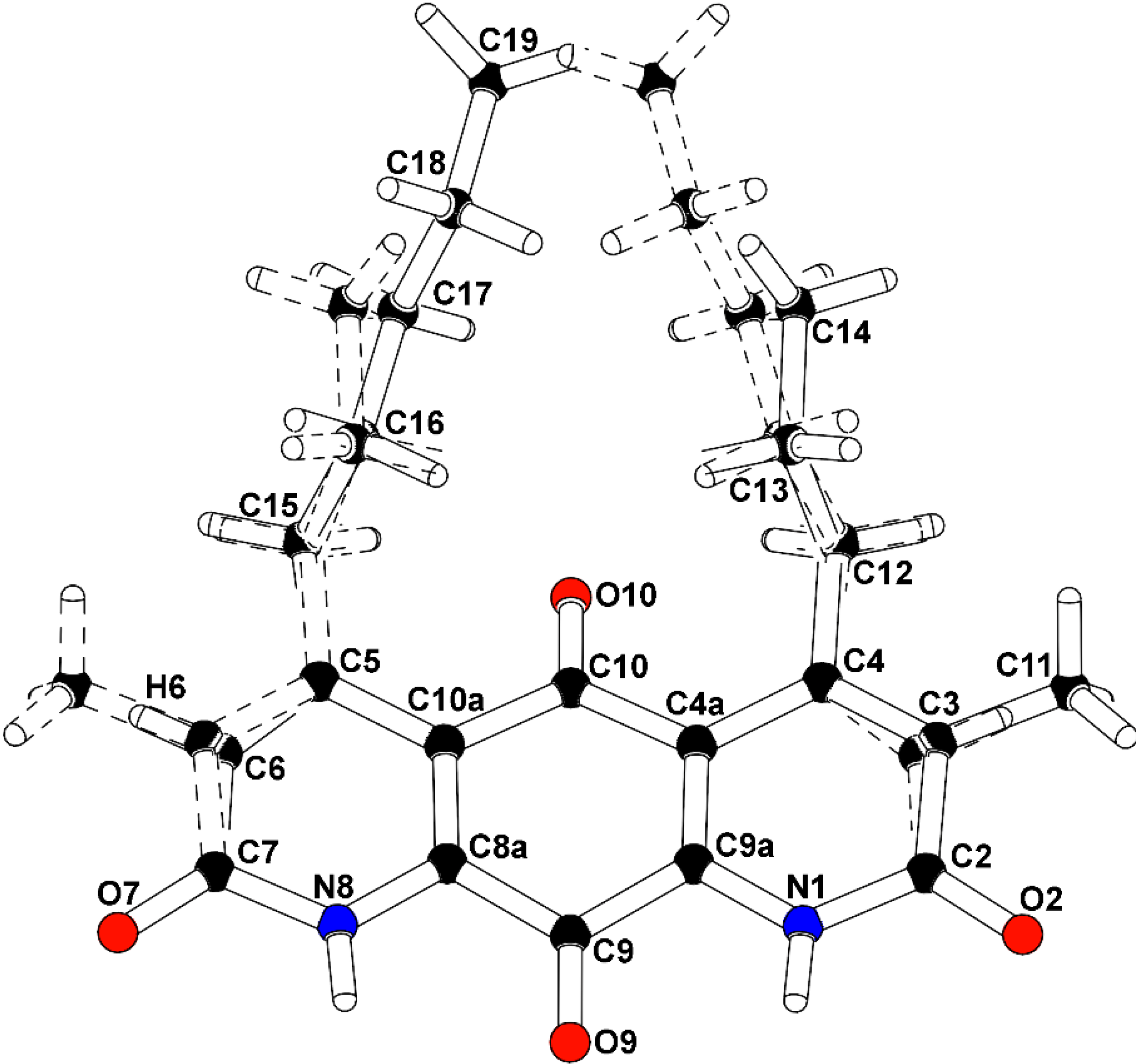
To confirm the structural features observable by NMR analysis and to determine the remaining connectivity of the structures, an X-ray structure determination was attempted. The mixture of
2 and
3 co-crystallized from methanol using a slow evaporation technique. Compounds
2 and
3 occupied the same molecular site, crystallizing in the monoclinic space group P2
1/m (No. 11), with a mirror plane bisecting the molecules through the central carbonyl atoms C9-O9 and C10-O10. The major isomer (
2), present as 52.6% of the crystal, is defined with a methyl group carbon atom, C11, on atom C3 adjacent to the propyl-substituted C4 atom, and a methine group carbon atom, C6, adjacent to the pentyl-substituted C5 atom. A constitutional isomer (
3), present as 47.4% of the crystal, is modeled with the methyl group carbon atom, C11, on atom C3 adjacent to the pentyl-substituted C4 atom, and a methine group carbon atom, C6, adjacent to the propyl-substituted C5 atom. Partially occupied water molecules are evident from the electron density maps, and near the methyl groups. The C3(methyl)-C6(H):C3(
H)-C6(methyl) fragment occupancy was refined to a ratio of 0.704(8):0.296(8), and the C4(
n-propyl):C4(
n-pentyl) fragment occupancy ratio was 0.566(9):0.434(9). The molecules are packed in the crystal through hydrogen bonding with the amide hydrogen atoms and adjacent carbonyls. The intensity data was collected at experimental station 21-ID-D, Life Sciences-Collaborative Access Team (LS-CAT), at Advanced Photon Source, Argonne National Laboratory, and processed with XDS [
7]. The structure was solved and refined with SHELX [
8]. The final R-factor was 0.0799 for 1525 intensities greater than 2σ, and 0.0961 for all 2030 unique data. The X-ray analysis supports the proposed structures of
2 and
3 (
Figure 4,
Supplementary Figures S17 and S18). Therefore,
2 and
3 were determined as shown and named diazaquinomycin F and diazaquinomycin G, respectively. Crystallographic data for the structure of
2 and
3 were deposited under accession number CCDC 996646 and can be obtained free of charge from The Cambridge Crystallographic Data Centre. The ratio of
2 and
3 in the co-crystal does not necessarily reflect the weight percentage in the compound mixture, as evidenced by uneven H-6 resonances in the
1H-NMR spectrum (
Supplementary Figures S11 and S14).
Figure 4.
Co-crystal structure of diazaquinomycin F (2) and diazaquinomycin G (3).
Figure 4.
Co-crystal structure of diazaquinomycin F (2) and diazaquinomycin G (3).
Structure numbered to emphasize DAQF occupancy in the co-crystal.
The known metabolite DAQA (
4) was isolated and characterized based on comparison of
1H-NMR and HRMS data with those appearing in literature [
5,
9].
2.2. DAQA (4) Induces DNA Damage, Cell Cycle Arrest, and Apoptosis through Cleaved-PARP
Compounds
1 and
4 were tested for
in vitro cytotoxicity against the ovarian cancer cell line OVCAR5. Dose response analysis of the isolated compounds revealed an LC
50 of 9.0 μM for
1 and 8.8 μM for
4 after treatment of cells for 96 h (
Supplementary Figure S1).
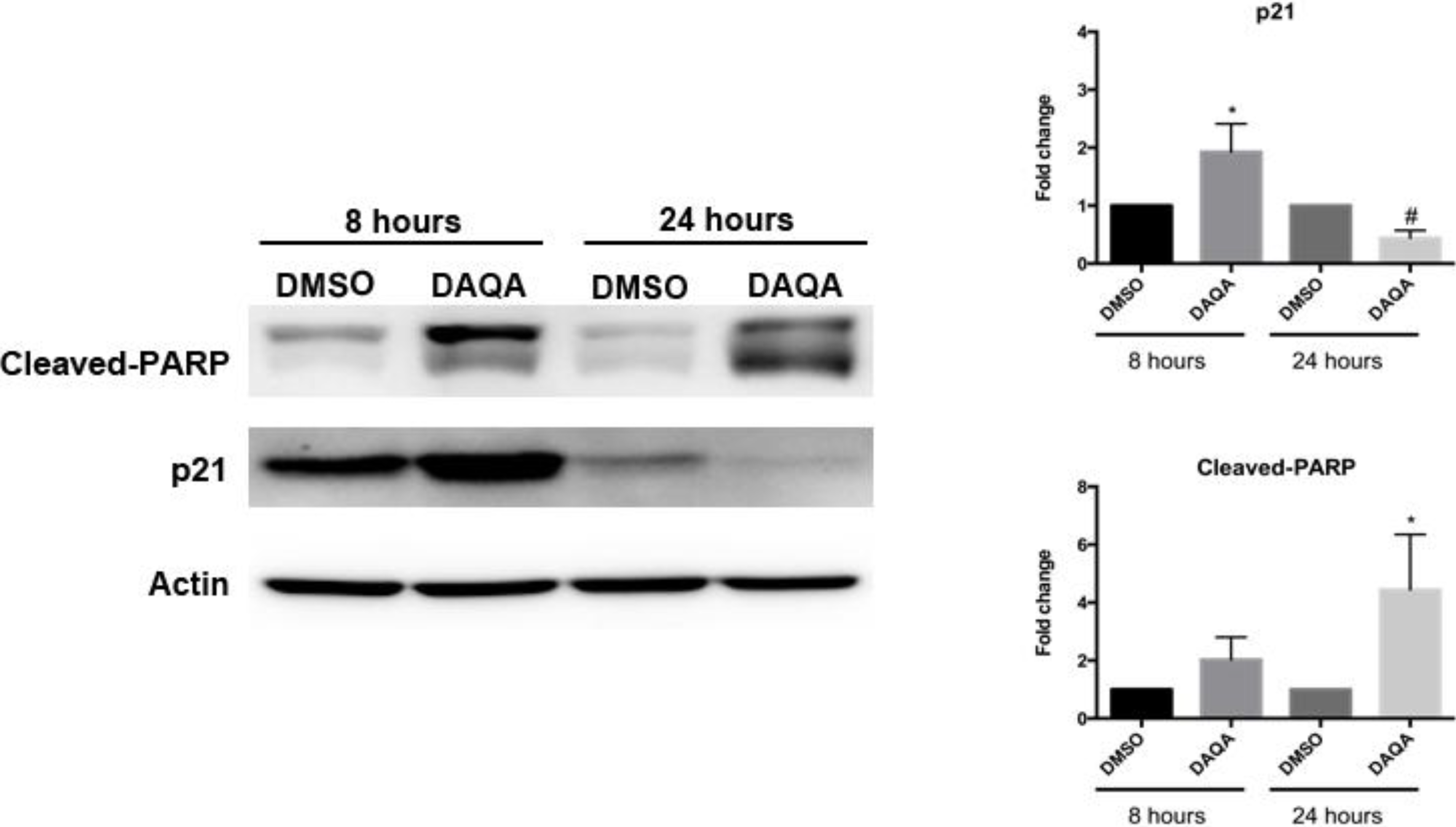
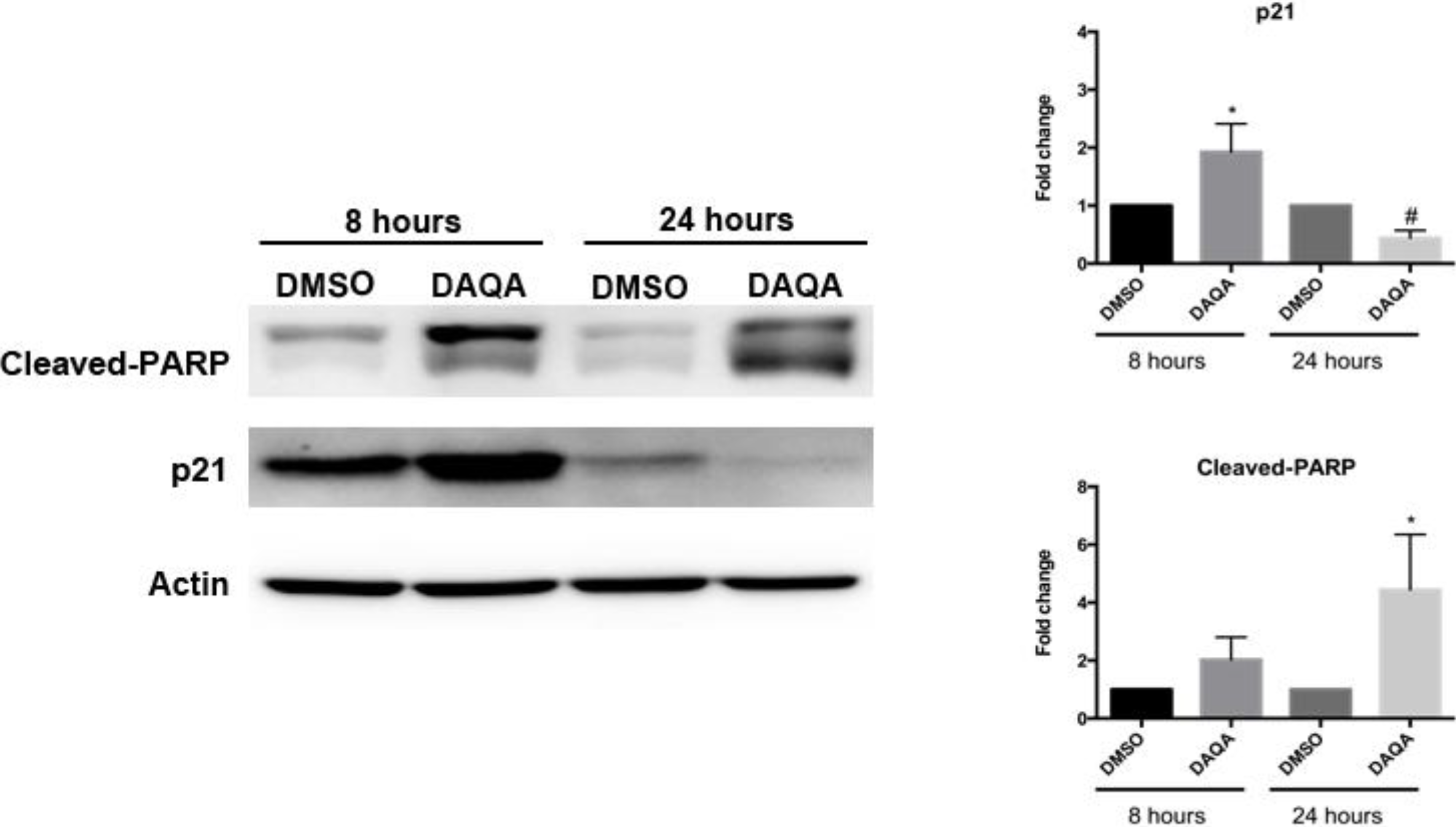
Further cell-based experiments were performed on
4 due to its high yield. Western blot analysis of OVCAR5 cells treated with 17.6 μM (LC
100) of
4 showed increased levels of p21 (a cell cycle inhibitor) after 8 h (
Figure 4). Interestingly, levels of p21 decreased after 24 h when compared to solvent control (
Figure 4). Reduction of p21 protein after 24 h correlated with an increase in cleaved-PARP, an indication of apoptosis (
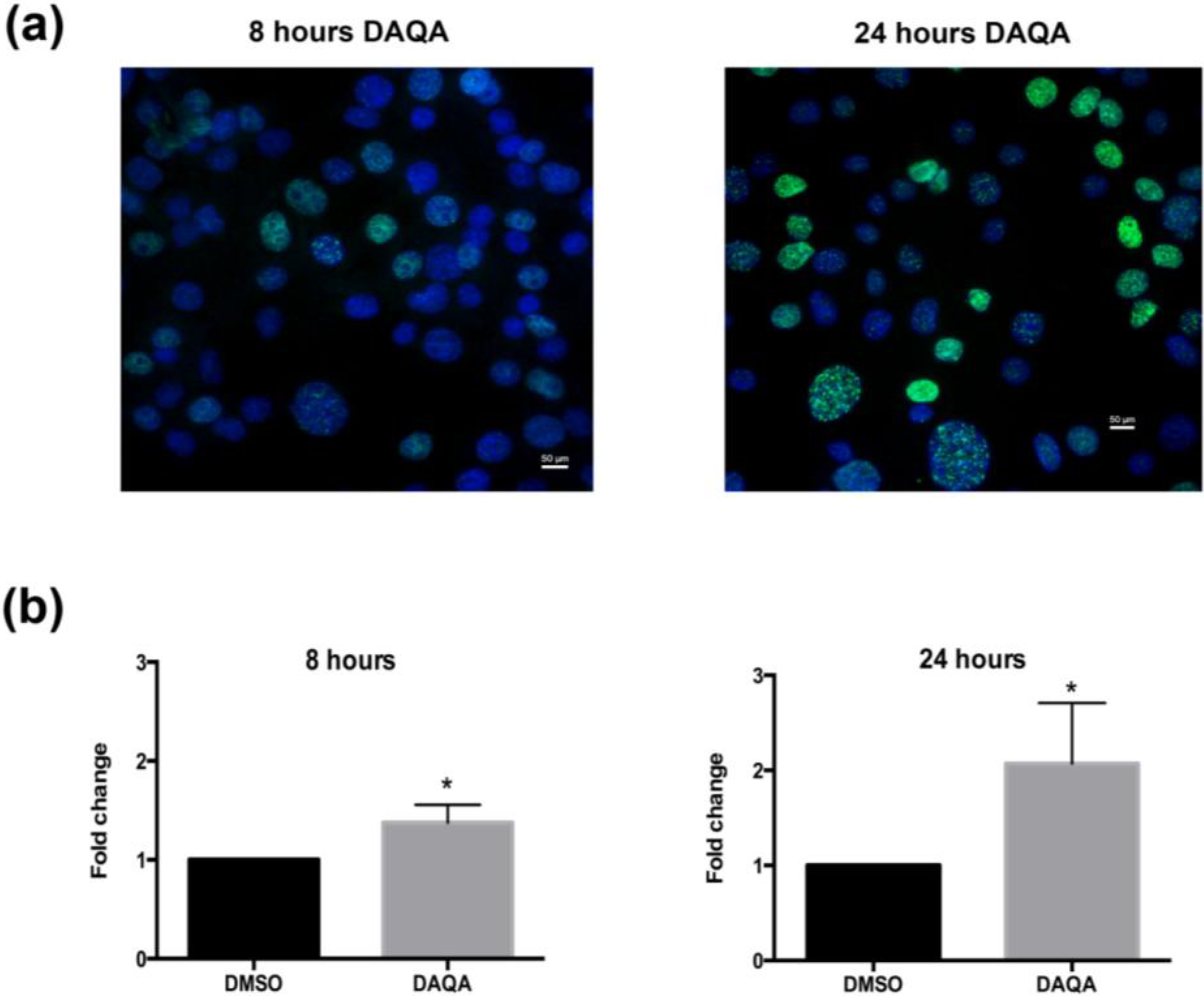
Figure 5). The induction of cell cycle arrest, leading to apoptosis suggests significant DNA damage. To address this possibility, immunofluorescence for phospho-histone H2A.X was performed on OVCAR5 cells treated with
4 at 17.6 μM. Enhanced DNA damage, as monitored by increased phospho-histone H2A.X staining, was seen after 8 h and 24 h treatment with 17.6 μM of
4 when compared to solvent control (
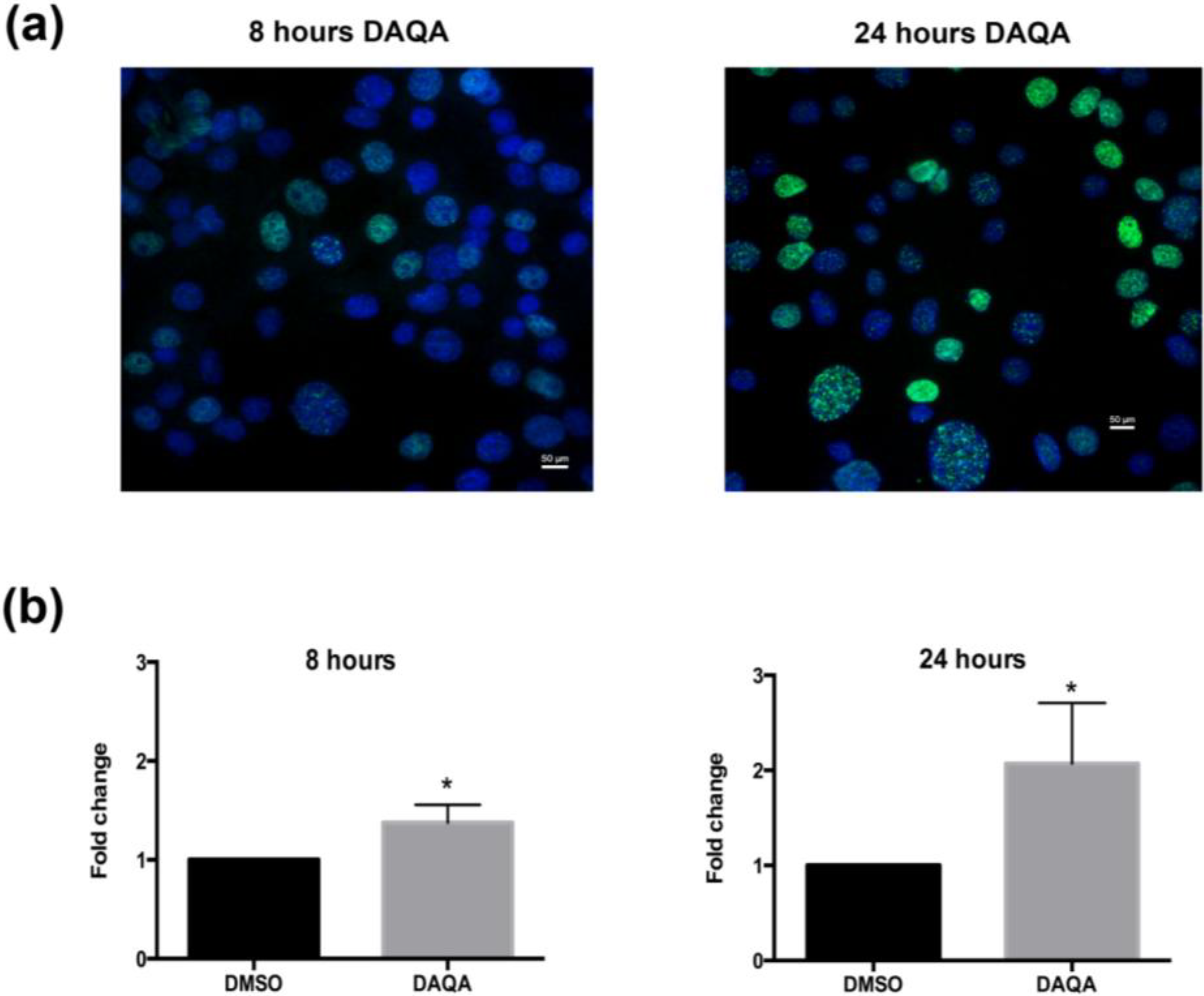
Figure 6a,b).
Figure 5.
Compound 4 induces cell cycle arrest followed by apoptosis.
Figure 5.
Compound 4 induces cell cycle arrest followed by apoptosis.
OVCAR5 cells treated with 17.6 μM of 4 showed induction of cell cycle arrest through increased p21 after 8 h, and induction of apoptosis through increased cleaved-PARP after 24 h. Densitometry of westerns was performed from three replicates. Statistical significance is donated by * for increased expression, and # for decreased expression. Student t-test was performed for all statistics and are represented as SEM +/−, * and # p ≤ 0.05.
Figure 6.
Compound 4 induces DNA damage in OVCAR5 cells. (a) H2A.X foci images taken after treatment of OVCAR5 cells with DAQA at 17.6 μM 8 h and 24 h; (b) Quantification of phospho-histone H2A.X foci as a fold increase over DMSO solvent control. Statistical significance is donated by * using student t-test. * p ≤ 0.05.
Figure 6.
Compound 4 induces DNA damage in OVCAR5 cells. (a) H2A.X foci images taken after treatment of OVCAR5 cells with DAQA at 17.6 μM 8 h and 24 h; (b) Quantification of phospho-histone H2A.X foci as a fold increase over DMSO solvent control. Statistical significance is donated by * using student t-test. * p ≤ 0.05.
Diazaquinomycins A and B were originally isolated from a
Streptomyces sp. after exhibiting moderate inhibitory activity against four Gram-positive bacteria (three
Staphylococcus aureus and one
Streptococcus faecium IFO 3181 strains) in agar-based assays [
5,
9]. A follow-up publication by the same group reported that DAQA exhibited cytotoxicity against Vero and Raji cell lines, while also inhibiting thymidylate synthase in Ehrlich ascites carcinoma [
10]. Additional studies were carried out in order to increase the solubility and bioactivity of DAQA, and some success was achieved through modification of the C-3 and C-6 methyl groups to short chain ester and ether derivatives [
11]. Nearly 15 years later, DAQC was isolated from a
Streptomyces sp.; this report was also the first mention of DAQD, though the metabolite was only observed through (−)-ESI MS experiments and was never fully characterized [
6]. The observation of an aromatic resonance in the
1H spectrum (H-6) of
1 is a feature unique among existing diazaquinomycin analogs; previously reported structures of this class contain a methyl group at this position [
5,
6,
9]. In the current study, though four secondary metabolites were isolated and characterized from strain F001, we observed ten additional diazaquinomycin analogs (based on the presence of characteristic UV spectra and MS
2 fragmentation patterns) in LCMS data of our bioactive fractions. Positive ion values ranged from
m/
z 355.1 [M + H]
+ to 425.2 [M + H]
+, and given that UV spectra remained consistent among derivatives, structural modifications likely occurred in the α-substituted methyl and β-substituted alkyl groups. Finally,
1 and
4 exhibited moderate cytotoxicity toward OVCAR5 cells by inducing apoptosis and enhancing DNA damage.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}