Abstract
A series of novel cyclic marinopyrroles were designed and synthesized. Their activity to disrupt the binding of the pro-apoptotic protein, Bim, to the pro-survival proteins, Mcl-1 and Bcl-xL, was evaluated using ELISA assays. Both atropisomers of marinopyrrole A (1) show similar potency. A tetrabromo congener 9 is two-fold more potent than 1. Two novel cyclic marinopyrroles (3 and 4) are two- to seven-fold more potent than 1.
1. Introduction
Marinopyrroles were first reported to show antibiotic activity against methicillin-resistant Staphylococcus aureus (MRSA) in 2008 by the Fenical group [1]. Due to their novel molecular structures and promising biological properties, marinopyrroles have attracted considerable attention [2,3,4,5,6,7,8,9,10,11]. We reported the first total synthesis of (±)-marinopyrrole A (1) along with 12 derivatives in early 2010 [3]. Synthesis of (±)-marinopyrrole A via an intermolecular Ullman coupling as the key step to form the bispyrrole system was published by Kanakis and Sarli five months later [4]. In 2011, the Nicolaou group published a new five-step method to access marinopyrrole derivatives, (+)-1 and (–)-1 atropisomers after a chiral separation of (±)-1 using HPLC, as well as their antibiotic activities against MRSA [5]. In 2012, the Moore group published a biosynthetic approach toward marinopyrrole A via an N, C-bispyrrole homocoupling catalyzed by two flavin-dependent halogenases [8]. Last year, the total synthesis of marinopyrrole B and a review of the marinopyrroles were reported from the Clive group [9,10]. After we reported the synthesis of a novel series of “non-symmetrical” marinopyrrole derivatives and their antibiotic activities [6], we published optimization studies of the key step to avoid the formation of an oxazepine byproduct [7]. Most recently, we reported on the most potent marinopyrrole derivatives against MRSA [11]. Furthermore, we have also reported recently that (±)-marinopyrrole A antagonizes Mcl-1 and overcomes resistance of human cancer cells to the Bcl-xL antagonist, ABT-737 [12]. During preparation of this manuscript, the discovery of a novel selective Mcl-1 small-molecule inhibitor blocking pancreatic cancer growth in vitro and in vivo resulted from high throughput screening followed by structure-based chemical optimization was reported recently by Nikolovska-Coleska and co-workers [13]. Here, we report on cyclic marinopyrroles as disruptors of protein-protein interactions between the pro-apoptotic protein, Bim, and the pro-survival proteins, Bcl-xL and Mcl-1.
2. Results and Discussion
2.1. Design of Marinopyrrole Derivatives
With the success of our synthetic studies on “symmetrical” marinopyrrole derivatives [3] bearing the same substitution on both A and B phenyl rings, we focused our attention on a series of non-symmetrical derivatives. In particular, marinopyrrole F (Figure 1) reported by Hughes et al. [2] was chosen as a starting point for optimization. Marinopyrrole F adopted specific conformations that locked one aromatic ring to the bispyrrole system, due to the fused eight-membered ether linkage, as shown by crystallographic X-ray analysis [2]. We were particularly interested in exploring the structure activity relationships (SARs) of the cyclic marinopyrroles with functional groups substituted on both aromatic rings, as shown in Figure 1. The introduction of substituents in the para-position relative to the carbonyl group on both aromatic rings, such as trifluoromethanesulfonate 3, methyl ester 4 or diethyl phosphonate 5 functionality, would generate a series of compounds with the potential for a hydrogen bond (acceptor) and hydrophobic interactions with the target. Furthermore, the unmasked hydroxyl 6, carboxylic acid 7 and phosphonic acid 8 groups in the corresponding positions could serve as both a hydrogen bond donor/acceptor and a functional group to improve aqueous solubility. To evaluate the potential differences in potency between the atropisomers of 1, both (+)-1 and (–)-1 marinopyrrole A [5] were included in this study. The biological activity of brominated marinopyrrole A analog 9 [5] was also evaluated by ELISA assays.
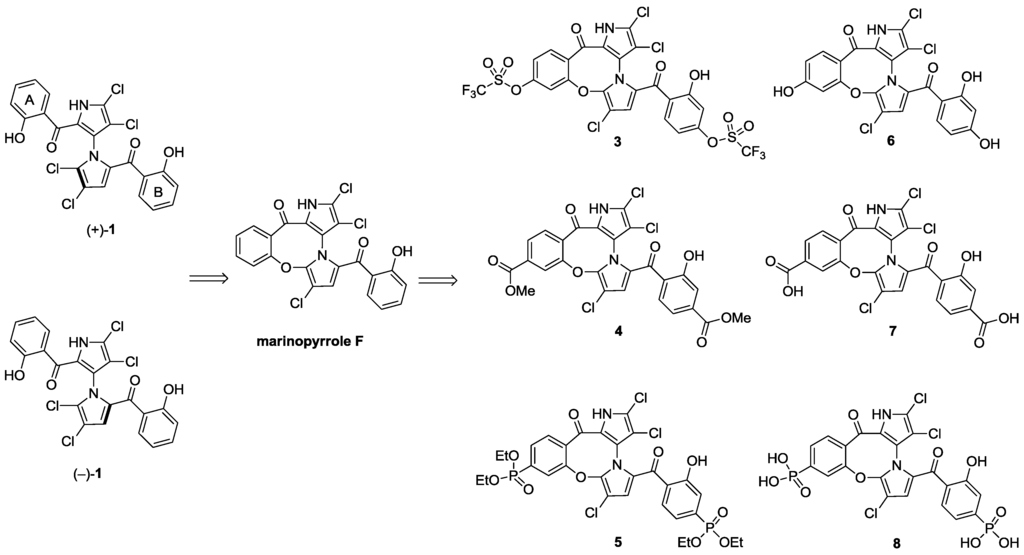
Figure 1.
Structure of marinopyrrole A (1) and cyclic marinopyrroles 3–8.
2.2. Synthesis of Marinopyrrole Derivatives
Starting from our previously reported compound 2 [11], macrocycle 3 was obtained in an 80% yield after heating 2 in dimethylformamide (DMF) at 110 °C (Scheme 1). Removal of the trifluoromethanesulfonic groups by saponification in methanolic tetrahydrofuran (THF) gave phenol 6 in an 81% yield. Palladium-mediated carbonylation [14] of 2 provided symmetrical marinopyrrole 4a and cyclic marinopyrrole 4 in a 25% and 22% yield, respectively. Further heating of compound 4a at 80 °C generated 4, presumably by spontaneous cyclization of 8-OH with 5′-Cl. Saponification of 4 and 4a yielded the corresponding carboxylic acid derivatives, 7 and 7a, respectively (Scheme 2). Palladium-catalyzed phosphorylation [15] of 2 with HPO(OEt)2 furnished a mixture of symmetrical marinopyrrole 5a in a 43% yield, as well as cyclized 5 in a 54% yield (Scheme 3). Intramolecular cyclization of 5a could also occur upon heating at 81–82 °C. Finally, upon treatment with Me3SiBr, 5 and 5a could be smoothly converted to the corresponding bisphosphonic acids, 8 and 8a [16].
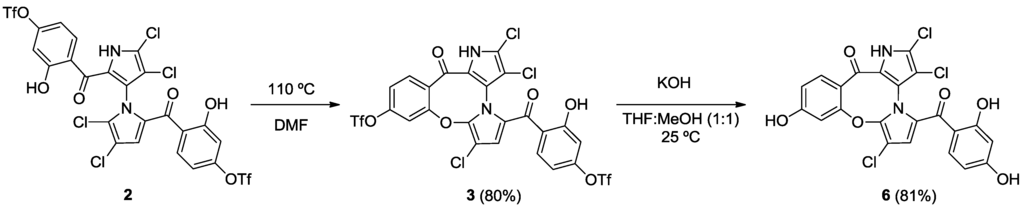
Scheme 1.
Synthesis of cyclic marinopyrroles 3 and 6.
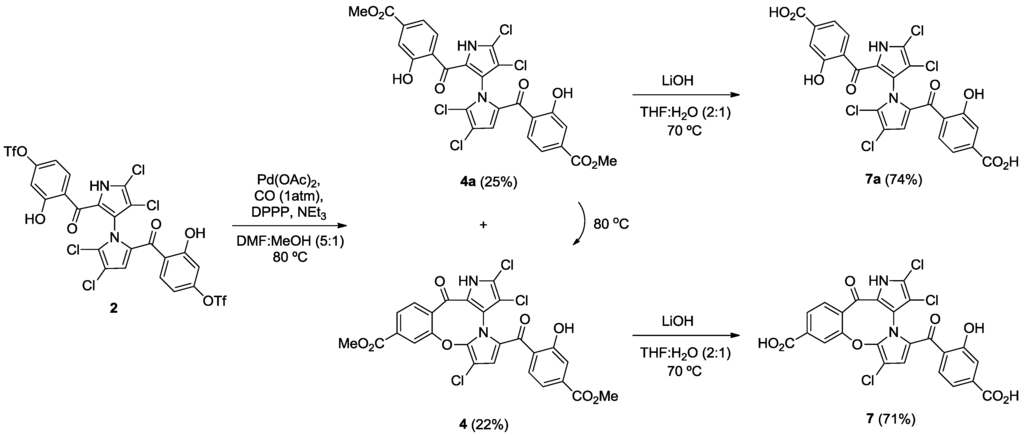
Scheme 2.
Synthesis of cyclic and symmetrical marinopyrroles.
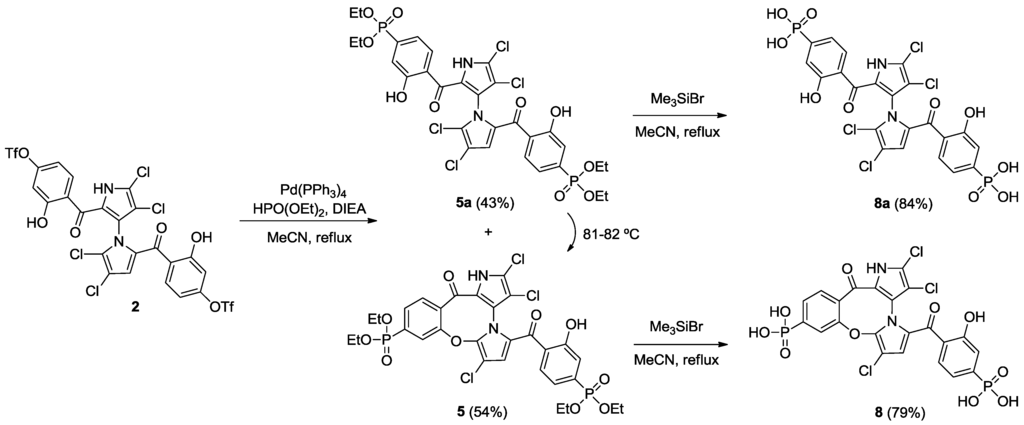
Scheme 3.
Synthesis of cyclic and symmetrical marinopyrroles.
2.3. Physicochemical Properties and SAR of the Marinopyrroles
Consistent with our previous report [12], the IC50 value of racemic marinopyrrole A to disrupt the binding of Mcl-1 to Bim was 8.9 μM. Although the activity of racemic marinopyrrole A against Bcl-xL/Bim binding was lower than we reported previously [12], this reflects the four times lower Bcl-xL concentration utilized in our present assay. We observed no significant activity difference between atropisomers (+)-1 and (–)-1, as both exhibited similar potencies against Mcl-1/Bim and Bcl-xL/Bim (Figure 2). Symmetrically para-substituted marinopyrroles with a carboxy methyl ester, 4a, and diethyl phosphonate 5a showed activity against Mcl-1/Bim, but were inactive against Bcl-xL/Bim (IC50 > 100 μM). Furthermore, substitution in the para-position of the carbonyl group with carboxylic acid 7a showed lower activity than 1 against Mcl-1/Bim and little activity against Bcl-xL/Bim. Bisphosphonic acid marinopyrrole 8a was slightly less potent than 1 against Mcl-1-Bim, but not Bcl-xL/Bim. Interestingly, the brominated marinopyrrole congener 9 [5] is two-fold more potent than 1 against both Mcl-1/Bim and Bcl-xL/Bim.
Both pKa and log p values were calculated using ChemAxon Software Version 5.12.3 [17,18]. The pKa values of marinopyrrole A (1) are predicted to be 7.8 (pKa 1) and 8.4 (pKa 2), respectively (Figure 2). As reported previously [11], the difference in pKa values for the hydroxyl group in ring A and ring B is presumably due to the axially chiral environment. The pKa values of 1 are 1.6–2.2 log units lower than that of phenol (pKa = 9.98 [19]). An equilibrium may exist between open conformations and closed conformations in 1, similar to those observed in a recent report of intramolecular hydrogen bonding [20]. The Fenical group reported the X-ray structure of marinopyrrole B (3′-Br analogue of 1) that indicated the preference for the formation of intramolecular hydrogen bonds between the peri-hydroxyl and the carbonyl group [1]. These intramolecular hydrogen bond interactions contribute to increasing not only compound acidity, but also its lipophilicity [20]. The calculated log p value of 1 is 5.6, which marginally violates the Rule of Five (RO5), drug-like properties formulated by Lipinski [21]. The calculated pKa 1 and pKa 2 values of marinopyrroles in Figure 2 range from 6.8 to 8.4. Compound 7a has pKa 3 (3.8) and pKa 4 (3.2) values, due to the carboxylic acid, while 8a has a pKa 3 (0.7–5.5) and pKa 4 (1.0–5.8) range of values corresponding to the phosphonic acid functional group. Clog p values of both compounds 7a (4.6) and 8a (2.4) reside within the suggested range for drug-like compounds. Despite the expected improvement in aqueous solubility of 7a and 8a over 1, both were found to be less active against Mcl-1/Bim and Bcl-xL/Bim, perhaps due to unfavorable ionic and/or hydrogen bond interactions with the targets.
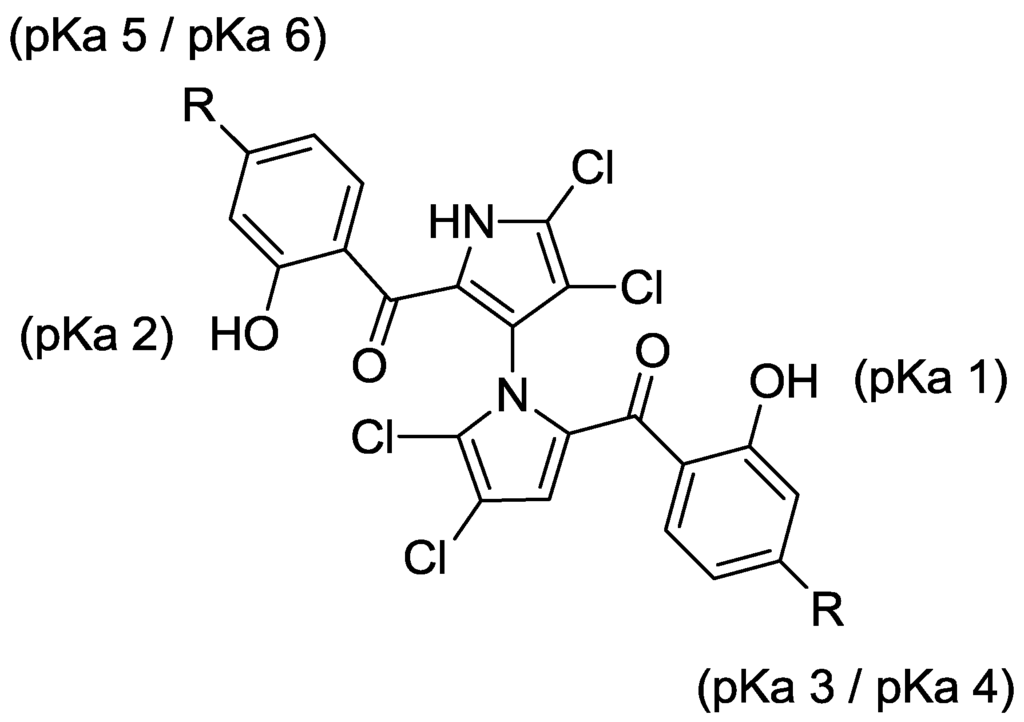
Figure 2.
ELISA and physicochemical properties of 1 and symmetrical marinopyrroles.
Compared to the SARs of the symmetrical marinopyrroles described (vide supra), cyclic marinopyrroles behaved similarly. Compounds containing functional groups with potential ionic and/or hydrogen bond interactions (6–8) reduce both anti-Mcl-1/Bim and Bcl-xL/Bim activity, as the cyclic marinopyrroles, phosphonic acid 8 and ester 5, lack activity against both Mcl-1/Bim and Bcl-xL/Bim (IC50 > 100 μM in Figure 3). Conversely, methyl ester 4 is two-fold more potent than 1 against Mcl-1/Bim and seven-fold more potent against Bcl-xL/Bim. Interestingly, trifluoromethanesulfonate 3 is the most potent cyclic marinopyrrole, showing six- and seven-fold higher potency than 1 against Mcl-1/Bim and Bcl-xL/Bim, respectively. Compound 4 has a Clog p value of 4.7, while the most potent compound, 3, has a Clog p value outside the advised range of RO5. The Clog p value for Compound 5 is marginally higher than the range of RO5, while the rest of the compounds (6–8) have Clog p values all within the recommended range for RO5. This series of cyclic marinopyrroles, which adopt constrained molecular geometries, due to the locked ring system [2], displayed an enhanced ability to disrupt the binding of Bim to Mcl-1 and Bcl-xL.
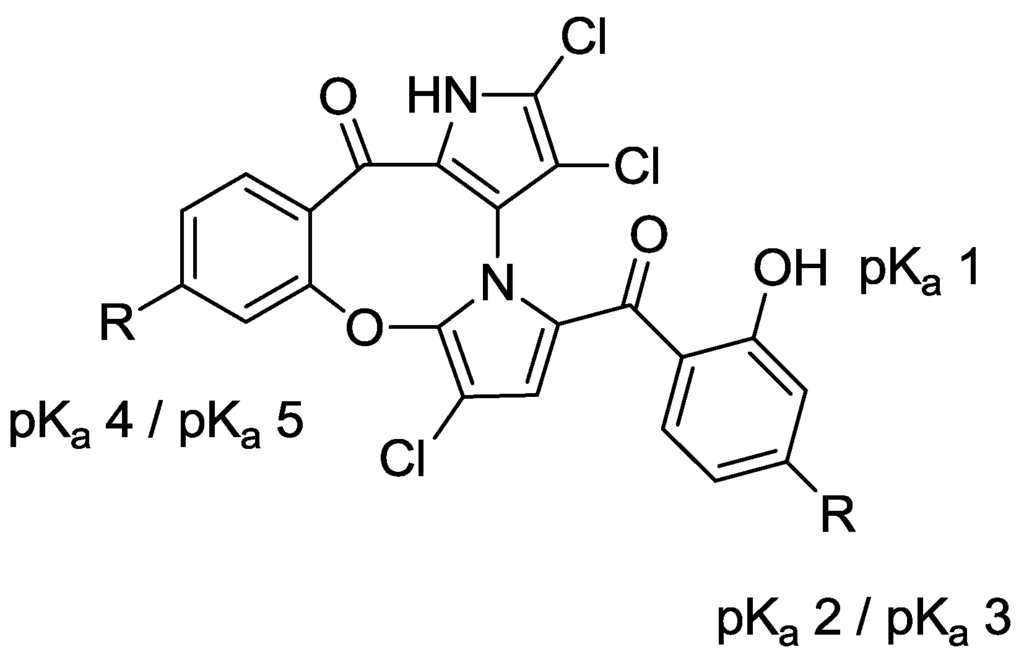
Figure 3.
ELISA and physicochemical properties of cyclic marinopyrroles.
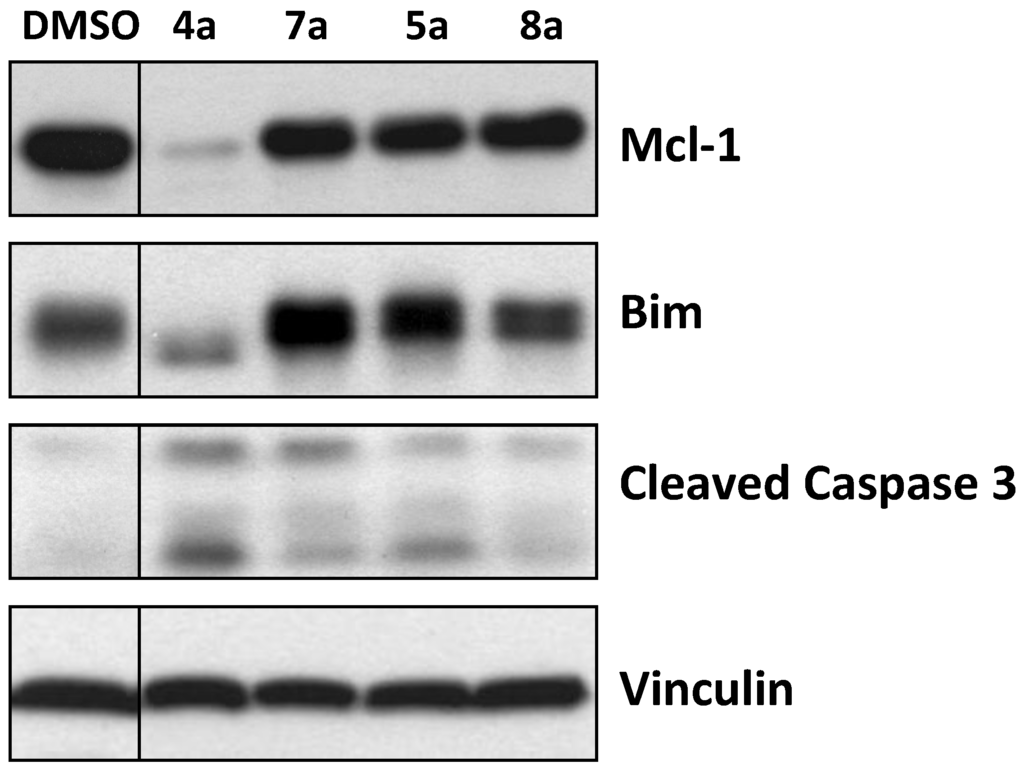
Figure 4.
Effect of marinopyrroles on Mcl-1, Bim and caspase 3 in human breast cancer cells.
2.4. Activity in Intact Human Breast Cancer Cells
To determine if the marinopyrroles are active in intact cells, the human breast cancer MDA-MB-468 cells were treated with the marinopyrrole derivatives (10 μM for 16 h). The cells were then processed for Western blotting exactly as described by us previously [22]. Figure 4 shows that treatment of the cells with 4a resulted in a significant decrease in the levels of Mcl-1 and Bim, and cleavage of caspase 3. Compound 7a, the free carboxylic acid analogue of 4a, did not decrease Mcl-1 and Bim and resulted in little caspase 3 cleavage. The phosphate 8a and its corresponding ethyl ester 5a had little effect on Mcl-1, Bim or caspase 3 (Figure 4). The (±)-marinopyrrole A (1) as reported by us previously [12] and its atropisomers, (+)-1 and (–)-1, as well as 9, tetrabromo-(±)-1, were able to decrease Mcl-1 and Bim and to cleave caspase 3 (data not shown). However, none of the cyclic marinopyrroles were active in intact cells (data not shown).
3. Experimental Section
3.1. Synthesis of Marinopyrrole Derivatives
All chemicals were purchased from commercial suppliers and used without further purification. All solvents were dried and distilled before use. Tetrahydrofuran was distilled from sodium/benzophenone. Dichloromethane and acetonitrile were distilled over calcium hydride. Flash column chromatography was performed with silica gel (200–300 mesh). 1H NMR spectra were recorded at either 400 MHz or 600 MHz at ambient temperature. 13C NMR spectra were recorded at either 100 or 150 MHz at ambient temperature. Infrared spectra were recorded on a Perkin-Elmer Spectrum 100 spectrometer. Copies of the NMR spectra of all the described compounds are provided in an Electronic Supporting Information (ESI) document. Melting points were determined with a melting point apparatus (Fukai X-4). High resolution mass spectra were performed by electrospray ionization (ESI) on an Agilent ESI-TOF LC-MS 6200 system. Analytical HPLC was performed on an Agilent 1100 series instrument with diode array detectors and auto samplers. All tested compounds possessed a purity of not less than 95%. 1H and 13C NMR spectra, and HPLC trace of the final compounds can be found in Supplementary Information (Figures S1–S30).
3-Hydroxy-4-(2,3,7-trichloro-13-oxo-10-(((trifluoromethyl)sulfonyl)oxy)-1,13 dihydrobenzo[g]di-pyrrolo[2,1-b:3′,2′-d][1,3]oxazocine-5-carbonyl)phenyl trifluoromethanesulfonate (3). Under N2, (4,4′,5,5′-tetrachloro-1′H-1,3′-bipyrrole-2,2′-diyl)bis(((2-hydroxy-4-hydroxytrifluoromethanesulfonate)-phenyl)methanone) (2) [11] (150 mg, 0.19 mmol) and NaI (120 mg, 0.75 mmol) were dissolved in DMF (5 mL). The mixture was heated to 110 °C and stirred for about 24 h. The reaction was quenched by the addition of saturated aqueous Na2S2O3 (20 mL) and extracted with EtOAc (ethyl acetate; 15 mL × 3). The suspension was filtered, and the filtrate was concentrated in vacuum. The residue was purified by flash column chromatography (16% EtOAc/petroleum ether, Rf = 0.2) to give 3 (115 mg, 80%) as a light yellow solid. mp 94.8–96.4 °C; 1H NMR (400 MHz, CDCl3) δ 5.72 (s, 1H), 6.89 (dd, J = 8.8, 2.0 Hz, 1H), 6.98 (d, J = 2.4 Hz, 1H), 7.39 (dd, J = 8.8, 2.0 Hz, 1H), 7.73 (d, J = 2.4 Hz, 1H), 7.92 (d, J = 8.8 Hz, 1H), 8.14 (d, J = 8.8 Hz, 1H), 9.72 (br s, 1H), 11.55 (s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 101.68, 106.22, 111.40, 112.60, 116.88, 117.00, 119.02, 120.19, 120.98, 121.12, 121.28, 122.85, 124.18, 125.33, 129.73, 134.45, 135.18, 144.90, 152.81, 154.62, 157.16, 164.28, 174.98, 186.66 ppm; HRMS (M + H+) calcd. for C24H10Cl3F6N2O10S2 768.8747, found 768.8809; IR (KBr): 3423, 3244, 2960, 2922, 2852, 1626, 1604, 1580, 1462, 1426, 1217, 1139, 1095, 965, 790 cm−1. HPLC purity, 99.1% (flow rate: 1 mL/min; column: Agilent ZORBAX 300SB-C8, 5 μm, 150 × 4.6 mm; wavelength: UV 254 nm; temperature: 25 °C; mobile phase: MeOH:H2O = 80:20; tR = 28.9 min).
2,3,7-Trichloro-5-(2,4-dihydroxybenzoyl)-10-hydroxybenzo[g]dipyrrolo[2,1-b:3′,2′-d][1,3]oxazo-cin-13(1H)-one (6). To a solution of 3 (65 mg, 0.08 mmol) in a mixture of MeOH/THF (1:1, 4 mL) was added KOH (47 mg, 0.80 mmol) at room temperature. The mixture was heated to 70 °C and stirred for 10 h. The reaction mixture was adjusted to pH 7.0 with 0.5 N HCl and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by flash column chromatography (33% EtOAc/petroleum ether, Rf = 0.3) to yield 6 (34 mg, 81%) as a yellow solid. mp 274.7–276.0 °C; 1H NMR (400 MHz, acetone-d6) δ 6.37 (s, 1H), 6.84 (d, J = 2.0 Hz, 1H), 6.95 (br s, 1H), 7.44 (dd, J = 8.8, 2.4 Hz, 1H), 7.72 (d, J = 2.0 Hz, 1H), 8.17 (dd, J = 8.4, 3.2 Hz, 1H), 8.38 (d, J = 8.4 Hz, 1H) ppm; 13C NMR (CD3OD, 100 MHz) δ 91.20, 94.31, 96.54, 100.56, 100.90, 105.20, 106.97, 109.25, 113.22, 113.86, 115.03, 115.39, 115.68, 125.60, 125.62, 127.17, 150.97, 156.11, 157.43, 158.23, 168.02 178.12 ppm; HRMS (M + K+) calcd. for C22H11Cl3KN2O6 542.9320, found 542.9297; IR (KBr): 3415, 3251, 2962, 2924, 1619, 1581, 1547, 1476, 1456, 1310, 1090, 796 cm−1. HPLC purity, 98.9% (flow rate: 1 mL/min; column: Agilent ZORBAX 300SB-C8, 5 μm, 150 × 4.6 mm; wavelength: UV 254 nm; temperature: 25 °C; mobile phase: MeOH:H2O = 75:25; tR = 7.5 min).
Methyl-2,3,7-trichloro-5-(2-hydroxy-4-(methoxycarbonyl)benzoyl)-13-oxo-1,13-dihydrobenzo[g]-dipyrrolo[2,1-b:3′,2′-d][1,3]oxazocine-10-carboxylate (4) and dimethyl-4,4′-(4,4′,5,5′-tetrachloro-1′H-[1,3′-bipyrrole]-2,2′-dicarbonyl)bis(3-hydroxybenzoate) (4a). Under CO (1 atm), 2 (400 mg, 0.50 mmol), DPPP (bis(diphenylphosphino)propane; 26 mg, 0.10 mmol), Pd(OAc)2 (11 mg, 0.05 mmol) and Et3N (251 mg, 2.50 mmol) were dissolved in a mixture of DMF/MeOH (5:1, 5 mL). The reaction was heated to 80 °C and stirred for 3 h. The reaction mixture was quenched with water (10 mL) and extracted with EtOAc (15 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by flash column chromatography (50% EtOAc/petroleum ether, Rf = 0.2) to give 4 (70 mg, 22%) and 4a (80 mg, 25%) as a pale yellow solid.
4: mp 135.7–137.0 °C; 1H NMR (400 MHz, CDCl3) δ 3.96 (s, 3H), 3.98 (s, 3H), 5.72 (s, 1H), 7.56 (d, J = 8.0 Hz, 1H), 7.69 (s, 1H), 7.84 (d, J = 6.8 Hz, 1H), 8.07 (s, 2H), 8.40 (s, 1H), 9.88 (s, 1H), 11.22 (s, 1H) ppm; 13C NMR (acetone-d6, 100 MHz) δ 52.81, 53.16, 107.56, 118.58, 118.60, 120.76, 124.61, 128.87, 132.99, 132.99, 133.71, 134.02, 134.26, 136.82, 136.99, 149.00, 157.40, 157.51, 164.91, 165.33, 166.12, 167.90, 174.87, 176.18, 178.01, 183.00 ppm; HRMS (M + H+) calcd. for C26H16Cl3N2O8 588.9972, found 588.9967; IR (KBr): 3416, 3236, 2954, 2852, 1730, 1609, 1580, 1461, 1414, 1288, 1207, 1090, 988, 806 cm−1. HPLC purity, 95.6% (flow rate: 1 mL/min; column: Waters C18, 5 μm, 150 × 4.6 mm; wavelength: UV 254 nm; temperature: 25 °C; mobile phase: MeOH:H2O = 90:10; tR = 4.6 min).
4a: mp 99.4–101.0 °C; 1H NMR (400 MHz, acetone-d6) δ 3.84 (s, 3H), 3.86 (s, 3H), 6.18 (s, 1H), 7.27 (dd, J = 8.4, 1.6 Hz, 1H), 7.42–7.44 (m, 3H), 7.87 (d, J = 8.4 Hz, 1H), 8.04 (d, J = 8.0 Hz, 1H) ppm; 13C NMR (acetone-d6, 100 MHz) δ 52.68, 52.75, 110.16, 118.28, 118.51, 119.94, 120.60, 122.61, 123.20, 124.43, 126.43, 126.59, 127.10, 128.72, 130.72, 133.06, 135.43, 136.51, 158.90, 159.95, 166.17, 166.17, 184.78, 185.08, 185.60, 186.13 ppm; HRMS (M + H+) calcd. for C26H17Cl4N2O8 624.9739, found 624.9736; IR (KBr): 3,245, 2,954, 1,727, 1,632, 1,599, 1,441, 1,291, 1,223, 1,093, 884, 759, 672 cm−1. HPLC purity, 96.3% (flow rate: 1 mL/min; column: Agilent ZORBAX 300SB-C8, 5 μm, 150 × 4.6 mm; wavelength: UV 254 nm; temperature: 25 °C; mobile phase: MeOH:H2O = 80:20; tR = 6.6 min).
5-(4-Carboxy-2-hydroxybenzoyl)-2,3,7-trichloro-13-oxo-1,13-dihydrobenzo[g]dipyrrolo[2,1-b:3′,2′-d][1,3]oxazocine-10-carboxylic acid (7). To a solution of 4 (44 mg, 0.07 mmol) in a mixture of H2O/THF (1:2, 5 mL) was added LiOH (27 mg, 1.1 mmol) at room temperature. The reaction was heated to 70 °C and stirred for 10 h. The reaction mixture was adjusted to pH 5.0 with 0.5 N HCl and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by reverse-phase flash column chromatography (6% AcOH, 23% H2O, 71% MeOH, Rf = 0.2) to give 7 (30 mg, 71%) as a light yellow solid. mp 215.5–217.0 °C; 1H NMR (400 MHz, DMSO-d6) δ 6.03 (s, 1H), 7.40 (m, 3H), 8.03 (m, 2H), 8.14 (s, 1H) ppm; 13C NMR (DMSO-d6, 100 MHz) δ 100.08, 107.72, 117.52, 120.03, 120.62, 123.74, 124.50, 124.73, 125.18, 128.83, 130.17, 130.78, 132.98, 133.43, 136.00, 139.29, 145.72, 156.81, 156.81, 166.57, 167.44, 173.04, 175.98, 183.02 ppm; HRMS (M + H+) calcd. for C24H12Cl3N2O8 560.9659, found 560.9669; IR (KBr): 3420, 3240, 3127, 2925, 2600, 1710, 1604, 1580, 1462, 1413, 1311, 1210, 1025, 996, 906, 799, 761 cm−1. HPLC purity, 99.3% (flow rate: 1 mL/min; column: Waters C18, 5 μm, 150 × 4.6 mm; wavelength: UV 254 nm; temperature: 25 °C; mobile phase: MeOH:H2O = 55:45; tR = 6.7 min).
4,4′-(4,4′,5,5′-Tetrachloro-1′H-[1,3′-bipyrrole]-2,2′-dicarbonyl)bis(3-hydroxybenzoic acid) (7a). To a solution of 4a (27 mg, 0.04 mmol) in a mixture of H2O/THF (1:2, 3 mL) was added LiOH (16 mg, 0.65 mmol) at room temperature. The reaction was heated to 70 °C and stirred for 10 h. The reaction mixture was adjusted to pH 5.0 with 0.5 N HCl and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by reverse-phase flash column chromatography (6% AcOH, 30% H2O, 64% MeOH, Rf = 0.2) to give 7a (19 mg, 74%) as a light yellow solid. mp 190.5–192.0 °C; 1H NMR (400 MHz, DMSO-d6) δ 6.10 (s, 1H), 7.19 (d, J = 8.0 Hz, 2H), 7.29–7.33 (m, 4H) ppm; 13C NMR (DMSO-d6, 100 MHz) δ 109.69, 110.24, 116.80, 117.21, 118.60, 118.66, 120.03, 122.32, 122.57, 124.96, 129.10, 129.26, 129.80, 129.90, 129.92, 134.57, 135.43, 156.09, 156.47, 167.58, 167.58, 172.66, 181.88, 183.14 ppm; HRMS (M + Na+) calcd. for C24H12Cl4N2NaO8 618.9245, found 618.9258; IR (KBr): 3075, 2956, 2919, 2851, 1707, 1631, 1599, 1446, 1394, 1294, 1228, 1023, 995, 885, 760 cm−1. HPLC purity, 98.6% (flow rate: 1 mL/min; column: Waters C18, 5 μm, 150 × 4.6 mm; wavelength: UV 254 nm; temperature: 25 °C; mobile phase: MeOH:H2O = 65:35; tR = 5.1 min).
3-Hydroxy-4-(2,3,7-trichloro-13-oxo-10-(diethylphosphonyl)-1,13-dihydrobenzo[g]dipyrrolo[2,1-b:3′,2′-d][1,3]oxazocine-5-carbonyl)diethyl phosphonate (5) and tetraethyl((4,4′,5,5′-tetrachloro-1′H-[1,3′-bipyrrole]-2,2′-dicarbonyl)bis(3-hydroxy-4,1-phenylene))bis(phosphonate) (5a). Under N2, 2 (50 mg, 0.06 mmol), diethyl phosphonate (52 mg, 0.36 mmol), Pd(PPh3)4 (7.6 mg, 0.006 mmol) and i-Pr2NEt (48 mg, 0.36 mmol) were dissolved in anhydrous MeCN (5 mL). The reaction was heated to reflux and stirred for 10 h. The reaction mixture was quenched with water (10 mL) and extracted with EtOAc (15 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by flash column chromatography (50% EtOAc/petroleum ether, Rf = 0.2) to give 5 (25 mg, 54%) and 5a (20 mg, 43%) as a yellow solid.
5: mp 122.8–124.3 °C; 1H NMR (400 MHz, CDCl3) δ 1.36 (t, J = 6.8 Hz, 12H), 4.12–4.24 (m, 8H), 5.71 (s, 1H), 7.36 (t, J = 8.4 Hz, 1H), 7.47 (d, J = 15.2 Hz, 1H), 7.87 (m, 2H), 8.08 (dd, J = 7.6, 5.2 Hz, 1H), 8.17 (d, J = 13.6 Hz, 1H), 9.85 (br s, 1H), 11.27 (s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 16.02, 16.02, 16.27, 16.27, 62.64, 62.70, 62.83, 62.83, 101.22, 106.12, 121.00, 121.59, 121.74, 123.06, 124.32, 125.10, 126.46, 130.33, 132.61, 132.77, 132.89, 135.07, 135.93, 136.94, 137.75, 145.48, 156.50, 161.50, 175.82, 187.12 ppm; HRMS (M + H+) calcd. for C30H30Cl3N2O10P2 745.0441, found 745.0454; IR (KBr): 3421, 3338, 3123, 3078, 2983, 2925, 2855, 1614, 1579, 1461, 1258, 1232, 1050, 1021, 796 cm−1. HPLC purity, 97.2% (flow rate: 1 mL/min; column: Agilent ZORBAX 300SB-C8, 5 μm, 150 × 4.6 mm; wavelength: UV 254 nm; temperature: 25 °C; mobile phase: MeOH:H2O = 80:20; tR = 6.8 min).
5a: mp 100.7–101.5 °C; 1H NMR (400 MHz, CDCl3) δ1.24–1.36 (m, 12H), 3.98–4.22 (m, 8H), 6.15 (s, 1H), 6.91 (dd, J = 11.6, 8.0 Hz, 1H), 7.24–7.27 (m, 1H), 7.30 (d, J = 14.4 Hz, 1H), 7.40 (d, J = 14.8 Hz, 1H), 7.52 (t, J = 14.4 Hz, 1H), 7.56 (dd, J = 7.6, 2.8 Hz, 1H), 8.00 (br s, 1H), 11.12 (s, 1H), 11.44 (br s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 16.22, 16.22, 16.28, 16.28, 62.69, 62.69, 62.74, 62.74, 108.90, 111.95, 117.68, 120.73, 121.03, 121.38, 121.47, 121.59, 122.07, 122.65, 122.84, 124.79, 130.95, 133.35, 135.84, 137.67, 160.29, 160.48, 161.32, 161.52, 185.51, 187.50 ppm; HRMS (M + H+) calcd. for C30H31Cl4N2O10P2 781.0208, found 781.0220; IR (KBr): 3416, 3214, 2964, 2926, 2867, 1631, 1449, 1406, 1259, 1222, 1022, 938, 800, 671 cm−1. HPLC purity, 97.0% (flow rate, 1 mL/min; column: Phenomenex C6-phenyl, 5 μm, 150 × 4.6 mm; wavelength: UV 254 nm; temperature: 25 °C; mobile phase: MeOH:H2O = 80:20; tR= 4.0 min).
3-Hydroxy-4-(2,3,7-trichloro-13-oxo-10-phosphoryl-1,13-dihydrobenzo[g]dipyrrolo[2,1-b:3′,2′-d][1,3]oxazocine-5-carbonyl) phosphonic acid (8). To a solution of 5 (40 mg, 0.054 mmol) in MeCN (3 mL) was added Me3SiBr (230 mg, 1.50 mmol) via a syringe at room temperature under N2. The reaction was heated to reflux and stirred for 24 h. The reaction mixture was concentrated in vacuum. The residue was purified by reverse-phase flash column chromatography (6% AcOH, 47% H2O, 47% MeOH, Rf = 0.2) to give 8 (27 mg, 79%) as a yellow solid. mp 314.7–316.0 °C; 1H NMR (400 MHz, CD3OD) δ 5.95 (s, 1H), 7.26 (dd, J = 12.8, 8.4 Hz, 1H), 7.32 (d, J = 14.8 Hz, 1H), 7.53 (dd, J = 8.0, 4.4 Hz, 1H), 7.85 (dd, J = 12.8, 8.0 Hz, 1H), 8.08 (dd, J = 8.0, 4.4 Hz, 1H), 8.15 (d, J = 13.6 Hz, 1H) ppm; 13C NMR (CD3OD, 100 MHz) δ 101.57, 101.69, 106.80, 107.56, 120.56, 121.47, 122.50, 124.67, 125.16, 125.52, 125.87, 126.60, 127.10, 130.67, 132.50, 133.46, 136.20, 141.71, 147.04, 162.26, 177.27, 186.48 ppm; HRMS (M + H+) calcd. for C22H14Cl3N2O10P2 632.9189, found 632.9193; IR (KBr): 3,790, 3,407, 2,955, 2,920, 2,850, 1,727, 1,596, 1,458, 1,401, 877 cm−1. HPLC purity, 99.7% (flow rate: 1 mL/min; column: Agilent ZORBAX 300SB-C8, 5 μm, 150 × 4.6 mm; wavelength: UV 254 nm; temperature: 25 °C; mobile phase: MeOH:H2O = 55:45; tR = 4.1 min).
((4,4′,5,5′-Tetrachloro-1′H-[1,3′-bipyrrole]-2,2′-dicarbonyl)bis(3-hydroxy-4,1-phenylene))diphosphonic acid (8a). To a solution of 5a (18 mg, 0.023 mmol) in MeCN (3 mL) was added Me3SiBr (99 mg, 0.65 mmol) via a syringe at room temperature under N2. The reaction was heated to reflux and stirred for 24 h. The reaction mixture was concentrated in vacuum. The residue was purified by reverse-phase flash column chromatography (6% AcOH, 30% H2O, 64% MeOH, Rf = 0.2) to give 8a (13 mg, 84%) as a yellow solid. mp 317.6–318.7 °C; 1H NMR (400 MHz, CD3OD) δ 6.29 (s, 1H), 7.05 (s, 1H), 7.26–7.37 (m, 5H) ppm; 13C NMR (CD3OD, 100 MHz) δ 110.32, 110.38, 112.63, 114.07, 118.96, 120.50, 121.60, 122.41, 123.23, 123.83, 125.59, 125.94, 126.34, 127.43, 129.35, 130.79, 132.80, 136.61, 159.27, 159.82, 185.99, 187.14 ppm; HRMS (M + H+) calcd. for C22H15Cl4N2O10P2 668.8956, found 668.8958; IR (KBr): 2,955, 2,919, 2,850, 1,626, 1,464, 1,020, 799 cm−1. HPLC purity, 99.5% (flow rate: 1 mL/min; column: Agilent ZORBAX 300SB-C8, 5 μm, 150 × 4.6 mm; wavelength: UV 254 nm; temperature: 25 °C; mobile phase: MeOH:H2O = 55:45; tR = 4.0 min).
3.2. Enzyme-Linked Immunosorbent Assay (ELISA) and Western Blotting Following Treatment of Intact Human Breast Cancer Cells
ELISAs were performed using a similar procedure as previously described [12]. Briefly, 40 nM of biotinylated Bim BH3 peptide (Biomatik, Wilmington, DE, USA) in SuperBlock blocking buffer (Thermo Scientific Pierce, Rockford, IL, USA) was incubated in high-binding capacity streptavidin-coated plates (Thermo Scientific Pierce, Rockford, IL, USA) for 2 h. Compounds were diluted in 120 µL of PBS containing 10 nM of GST-Mcl-1 or GST-Bcl-xL in 1.5-mL tubes for 15 min. Wells were washed with wash buffer (PBS containing 0.05% Tween-20) and then 100 µL of the compound/GST-protein mixture was transferred to the wells. The plates were incubated for 2 h, and then, the wells were washed with wash buffer. HRP-conjugated anti-GST antibody (Bethyl Laboratories, Montgomery, TX, USA) was diluted 1:2000 in SuperBlock, and 100 µL were transferred to each well. The plate was incubated for 1 h, and then, the wells were washed with wash buffer followed by PBS. One hundred microliters of SureBlue TMB Microwell Peroxidase Substrate (VWR International, Radnor, PA, USA) was added to each well, and the plates were developed for 5–10 min. One hundred microliters of 1 N HCl was added to each well to stop the reaction, and the absorbance was read at 450 nm using a µQuant plate reader (Bio-Tek Instruments, Winooski, VT, USA). Treatment of the human breast cancer (MDA-MB-468) cells and Western blotting were performed using the methods described by us previously [22].
4. Conclusions
This article describes general synthetic routes to access novel symmetrical and cyclic marinopyrrole derivatives and evaluation of their in vitro activity against the binding of the pro-survival proteins, Mcl-1 and Bcl-xL, to the pro-apoptotic protein, Bim. The efforts were focused on improving anti-Mcl-1/Bim and Bcl-xL/Bim potency. The synthetic methods paved the way toward diverse sets of both symmetrical and cyclic marinopyrroles. SAR studies of marinopyrrole derivatives have clearly demonstrated that: (i) replacing the chlorines with bromines within the bispyrrole core improved the potency by two-fold (1 vs. 9); (ii) symmetrical marinopyrroles with substituents in the para-position to the carbonyl group are more potent against Mcl-1/Bim than Bcl-xL/Bim (Figure 2); (iii) the same trend was observed for cyclic marinopyrroles (Figure 3); (iv) cyclic marinopyrrole 3 is six- and seven-fold more potent than 1 against Mcl-1/Bim and Bcl-xL/Bim, respectively (Figure 3); (v) the cyclic marinopyrroles with certain substituents (OSO2SF3 and CO2Me) in the para-position to the carbonyl group are excellent “leads” for further optimization. In summary, we have designed and synthesized a series of novel symmetrical and cyclic marinopyrroles with improved potency against both Mcl-1 and Bcl-xL. Further optimization is actively ongoing, and the activity, selectivity and absorption, distribution, metabolism and excretion (ADME)/tox data of these compounds will be published in due course.
Abbreviations
| ADME | absorption, distribution, metabolism and excretion |
| calcd. | calculated |
| DCM | dichloromethane |
| dd | double doublet |
| br | broad |
| DPPP | bis(diphenylphosphino)propane |
| DIEA | diisopropylethylamine |
| DMF | dimethylformamide |
| DMSO | dimethyl sulfoxide |
| EtOAc | ethyl acetate |
| ESI | electrospray ionization |
| HPLC | high performance liquid chromatography |
| HPO(OEt)2 | diethyl phosphanate |
| HRMS | high resolution mass spectrometry |
| HRP | horseradish peroxidase |
| IBX | 2-iodoxybenzoic acid |
| IR | infrared |
| KBr | potassium bromide |
| KF | potassium fluoride |
| LC | liquid chromatography |
| MeCN | acetonitrile |
| MeOH | methyl alcohol |
| MDA-MB-468 | breast cancer cell line |
| mp | melting point |
| MRSA | methicillin-resistant Staphylococcus aureus |
| NCS | N-chlorosuccinimide |
| NaI | sodium iodide |
| NMR | nuclear magnetic resonance |
| PBS | phosphate-buffered saline |
| SAR | structure activity relationship |
| s | singlet |
| TBAF | tetrabutylammonium fluoride |
| TBDMS | t-butyldimethylsilyl |
| TBDMSCl | t-butyldimethylsilyl chloride |
| Tf | trifluoromethanesulfonyl |
| THF | tetrahydrofuran |
| TMB | 3,3′,5,5′-Tetramethylbenzidine |
| tox | toxicity |
Supplementary Files
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (21321061 and 21132006), the National Science and Technology Major Project of China (2011ZX09401-304) and the National Basic Research Program of China (973 program, 2010CB833200) to Yong Qin, as well as funds from the Skaggs Institute for Chemical Biology, a grant from the National Institutes of Health (U.S.A.) to K.C. Nicolaou, a graduate fellowship from the National Science Foundation to Nicholas L. Simmons, and start-up funds (partial support) from the Moffitt Cancer Center and the University of Nebraska Medical Center to Rongshi Li. We are grateful to ChemAxon Software for pKa and log p calculations.
Author Contributions
Conceived and designed the experiments: RL, SMS, YQ. Performed the experiments: CC, HS, JL, LP, MEB, YL. Analyzed the data: RL, SMS, YQ. Contributed materials/reagents: KCN, NLS. Wrote the first draft of the manuscript: RL. Contributed to the writing of the manuscript: CC, JL, NLS, RL, SMS.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hughes, C.C.; Prieto-Davo, A.; Jensen, P.R.; Fenical, W. The marinopyrroles, antibiotics of an unprecedented structure class from a marine Streptomyces sp. Org. Lett. 2008, 10, 629–631. [Google Scholar] [CrossRef]
- Hughes, C.C.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Structures, reactivities, and antibiotic properties of the marinopyrroles A–F. J. Org. Chem. 2010, 75, 3240–3250. [Google Scholar] [CrossRef]
- Cheng, C.; Pan, L.; Chen, Y.; Song, H.; Qin, Y.; Li, R. Total synthesis of (±)-marinopyrrole A and its library as potential antibiotic and anticancer agents. J. Comb. Chem. 2010, 12, 541–547. [Google Scholar] [CrossRef]
- Kanakis, A.A.; Sarli, V. Total synthesis of (±)-marinopyrrole A via copper-mediated N-arylation. Org. Lett. 2010, 12, 4872–4875. [Google Scholar]
- Nicolaou, K.C.; Simmons, N.L.; Chen, J.S.; Haste, N.M.; Nizet, V. Total synthesis and biological evaluation of marinopyrrole A and analogues. Tetrahedron Lett. 2011, 52, 2041–2043. [Google Scholar] [CrossRef]
- Liu, Y.; Haste, N.M.; Thienphrapa, W.; Nizet, V.; Hensler, M.; Li, R. Marinopyrrole derivatives as potential antibiotic agents against methicillin-resistant Staphylococcus aureus (I). Mar. Drugs 2012, 10, 953–962. [Google Scholar] [CrossRef]
- Pan, L.; Cheng, C.; Song, H. Optimization of synthetic method of marinopyrrole A derivatives. Chem. J. Chin. Univ. 2012, 33, 1476–1480. [Google Scholar]
- Yamanaka, K.; Ryan, K.S.; Gulder, T.A.; Hughes, C.C.; Moore, B.S. Flavoenzyme-catalyzed atropo-selective N,C-bipyrrole homocoupling in marinopyrrole biosynthesis. J. Am. Chem. Soc. 2012, 134, 12434–12437. [Google Scholar]
- Cheng, P.; Clive, D.L.; Fernandopulle, S.; Chen, Z. Racemic marinopyrrole B by total synthesis. Chem. Commun. 2013, 49, 558–560. [Google Scholar] [CrossRef]
- Clive, D.L.J.; Cheng, P. The marinopyrroles. Tetrahedron 2013, 69, 5067–5078. [Google Scholar] [CrossRef]
- Cheng, C.; Liu, Y.; Song, H.; Pan, L.; Li, J.; Qin, Y.; Li, R. Marinopyrrole derivatives as potential antibiotic agents against methicillin-resistant Staphylococcus aureus (II). Mar. Drugs 2013, 11, 2927–2948. [Google Scholar] [CrossRef]
- Doi, K.; Li, R.; Sung, S.S.; Wu, H.; Liu, Y.; Manieri, W.; Krishnegowda, G.; Awwad, A.; Dewey, A.; Liu, X.; et al. Discovery of marinopyrrole A (Maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. J. Biol. Chem. 2012, 287, 10224–10235. [Google Scholar] [CrossRef]
- Abulwerdi, F.; Liao, C.; Liu, M.; Azmi, A.S.; Aboukameel, A.; Mady, A.S.A.; Gulappa, T.; Cierpicki, T.; Owens, S.; Zhang, T.; et al. A novel small-molecule inhibitor of Mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 2013. [Google Scholar] [CrossRef]
- Uyanik, M.; Ishihara, K.; Yamamoto, H. Catalytic diastereoselective polycyclization of homo(polyprenyl)arene analogues bearing terminal siloxyvinyl groups. Org. Lett. 2006, 8, 5649–5652. [Google Scholar] [CrossRef]
- Kim, Y.C.; Brown, S.G.; Harden, T.K.; Boyer, J.L.; Dubyak, G.; King, B.F.; Burnstock, G.; Jacobson, K.A. Structure-activity relationships of pyridoxal phosphate derivatives as potent and selective antagonists of P2X1 receptors. J. Med. Chem. 2001, 44, 340–349. [Google Scholar] [CrossRef]
- Petrakis, K.S.; Nagabhushan, T.L. Palladium-catalyzed substituions of triflates derivaed from tyrosine-containing peptides and simpler hydroxyarenens forming 4-(diethoxyphosphinyl)phenylalanines and diethyl aryphosphonates. J. Am. Chem. Soc. 1987, 109, 2831–2833. [Google Scholar] [CrossRef]
- Dixon, S.L.; Jurs, P.C. Estimation of pKa for organic oxyacids using calculated atomic charge. J. Comp. Chem. 1993, 14, 1460–1467. [Google Scholar] [CrossRef]
- Csizmadia, F.; Tsantili-Kakoulidou, A.; Panderi, I.; Darvas, F. Prediction of distribution coefficient from structure. 1. Estimation method. J. Pharm. Sci. 1997, 86, 865–871. [Google Scholar] [CrossRef]
- Liptak, M.D.; Gross, K.C.; Seybold, P.G.; Feldgus, S.; Shields, G.C. Absolute pK(a) determinations for substituted phenols. J. Am. Chem. Soc. 2002, 124, 6421–6427. [Google Scholar] [CrossRef]
- Kuhn, B.; Mohr, P.; Stahl, M. Intramolecular hydrogen bonding in medicinal chemistry. J. Med. Chem. 2010, 53, 2601–2611. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Del. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Balasis, M.E.; Forinash, K.D.; Chen, Y.A.; Fulp, W.J.; Coppola, D.; Hamilton, A.D.; Cheng, J.Q.; Sebti, S.M. Combination of farnesyltransferase and Akt inhibitors is synergistic in breast cancer cells and causes significant breast tumor regression in ErbB2 transgenic mice. Clin. Cancer Res. 2011, 17, 2852–2862. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).