2. Results and Discussion
Live colonies of Xenia elongata were grown in the coral laboratory at the Marine Biotechnology Center, Institute of Marine and Coastal Sciences, Rutgers University. For extraction, 200 g of the colonies were isolated and frozen. A methanol (or dichloromethane) soluble fraction was extracted from frozen coral tissue, lyophilized and dissolved in DMSO. The extract that induced apoptosis was subsequently fractionated and purified by analytical RPHPLC. Using this strategy, compounds with proapoptotic activity were isolated from the whole tissue extracts. Chemical structures of the two compounds (2 and 3) were ascertained by standard spectroscopic techniques as described below.
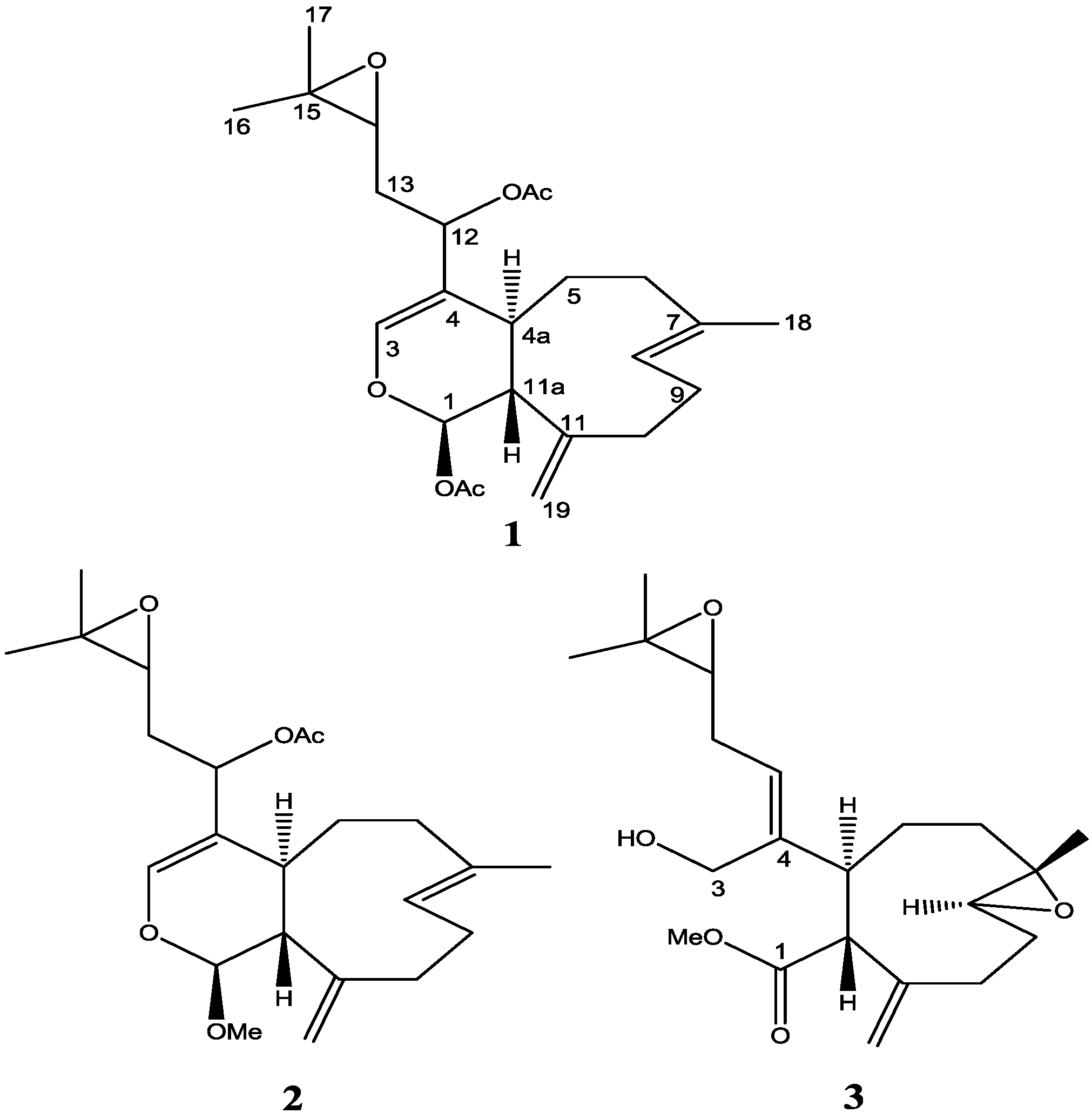
The molecular formula of
2 was established as C
23H
34O
5 on the basis of HRESIMS (
m/
z 413.2299 [M + Na]
+ calcd. for C
23H
34O
5Na, 413.2298). This indicated a difference of 28 mass units and probably a carbonyl group, compared to compound
1 [
3]. The
1H NMR (
Supplementary Figure S1) of
2 indicated clearly the existence of the following functional groups: an 1-methoxydihydropyran moiety [δ 5.35 (d,
J = 1.4 Hz), H-1 and 6.51 (s), H-3], a terminal methylene [δ 5.05 (s) and 5.15 (s), H-19, H-19′], two methyls α to oxygen [δ 1.33 (s), H-16, H-17], an epoxy signal at [δ 2.75 (dd,
J = 5.9, 6.2 Hz), H-14] and a vinyl methyl group [1.60 (s), H-18]. The NMR spectra of compounds
1 and
2 were compared in CDCl
3 and were found to be similar, with the exception of the carbon at C-1 [δ 99.3] (
Supplementary Figure S2) for
2 instead of C-1 [δ 91.7] for
1 in addition to that
2 has one less carbonyl signal, suggesting that a methoxy group is attached to C-1 in
2 instead of acetate group in
1. The deshielding effect of the methoxy group to the carbon at C-1 position is more pronounced compared to the deshielding effect of an acetate group to the carbon at the same position.
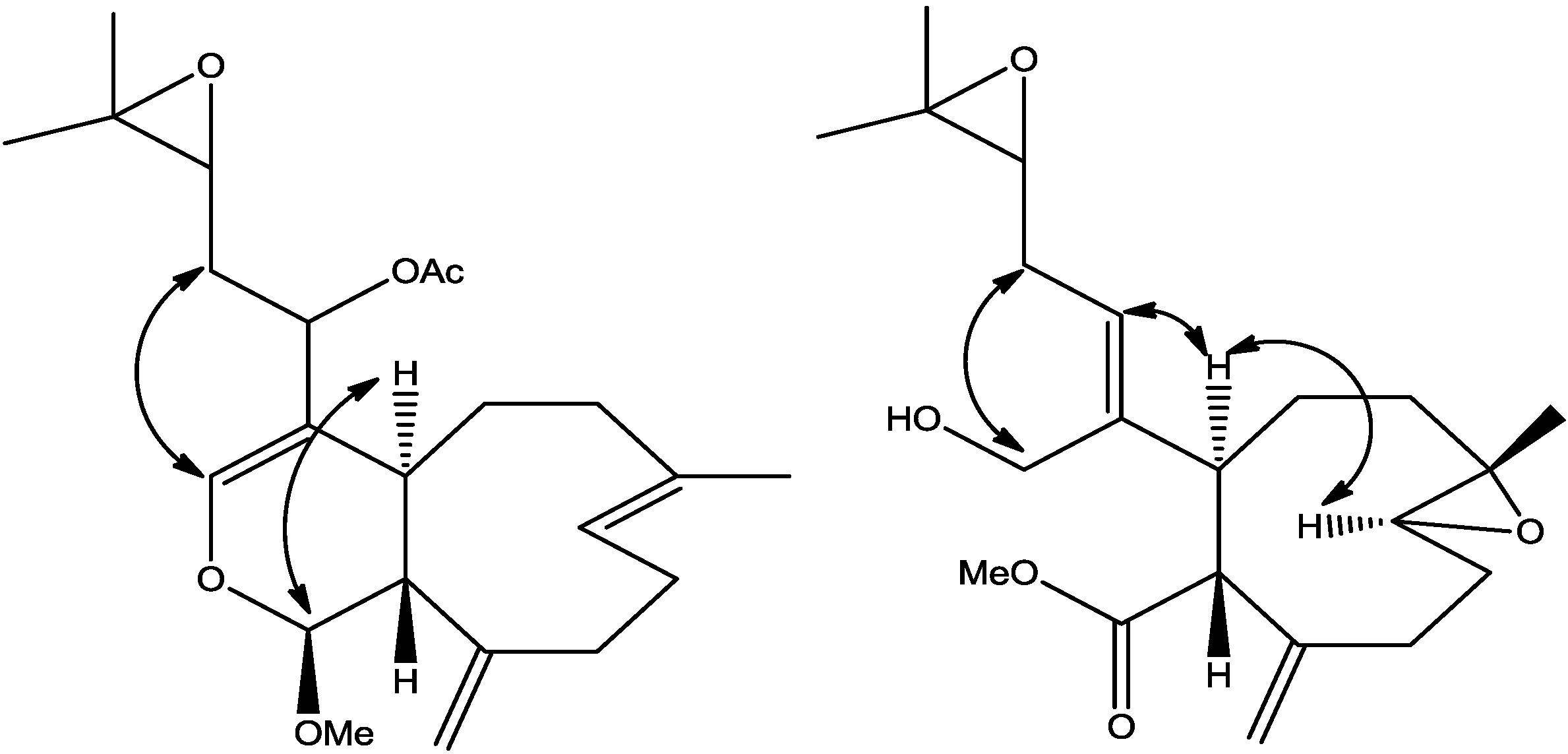
The relative stereochemistry of
2 was established with the same method as described in our previous work [
3] which is based on ROESY data (
Supplementary Figure S3), coupling constant analyses and chemical shifts comparison to
1 and xeninculin [
7], tsitsixenicin A [
8,
9] and related compounds [
7,
9]. The coupling constant (
J = 12 Hz) between H-4a and H-11a suggests a trans ring junction. The small coupling constant (
J = 1.4 Hz) between H-1 and H-11a would favor the α
-position for H-1 if the six-member ring was in a quasi-boat conformation. A closer look of the crystal structure of xenicin and essentially the six-member ring, which is also present in
2, suggests that H-1, if it is in the α
-position, should display ROESY cross peak correlation to H-4a. In order to verify this suggestion, a high scan ROESY experiment was performed. Careful analysis of the ROESY data, particularly in the cross region of H-1 and H-4a, revealed unambiguously that these two protons display a ROESY cross peak correlation (
Figure 2). Based on this result, it appears that the configurations at C-1, C-4a, and C-11a in compound
2 are essentially the same as those in
1 and xeninculin, in which the protons H-1 and H-4a are on the α face of the ring and the proton H-11a is oriented on the β
-side. This observation confirmed that the stereochemistry of this proton at H-1 position is well conserved and common to any molecule with dihydropyran moiety in the xenicane diterpene class. Previous attempts to establish the stereochemistry of an acetate group at C-12 in xenicane diterpenes by chemical transformations and spectroscopy have failed [
9,
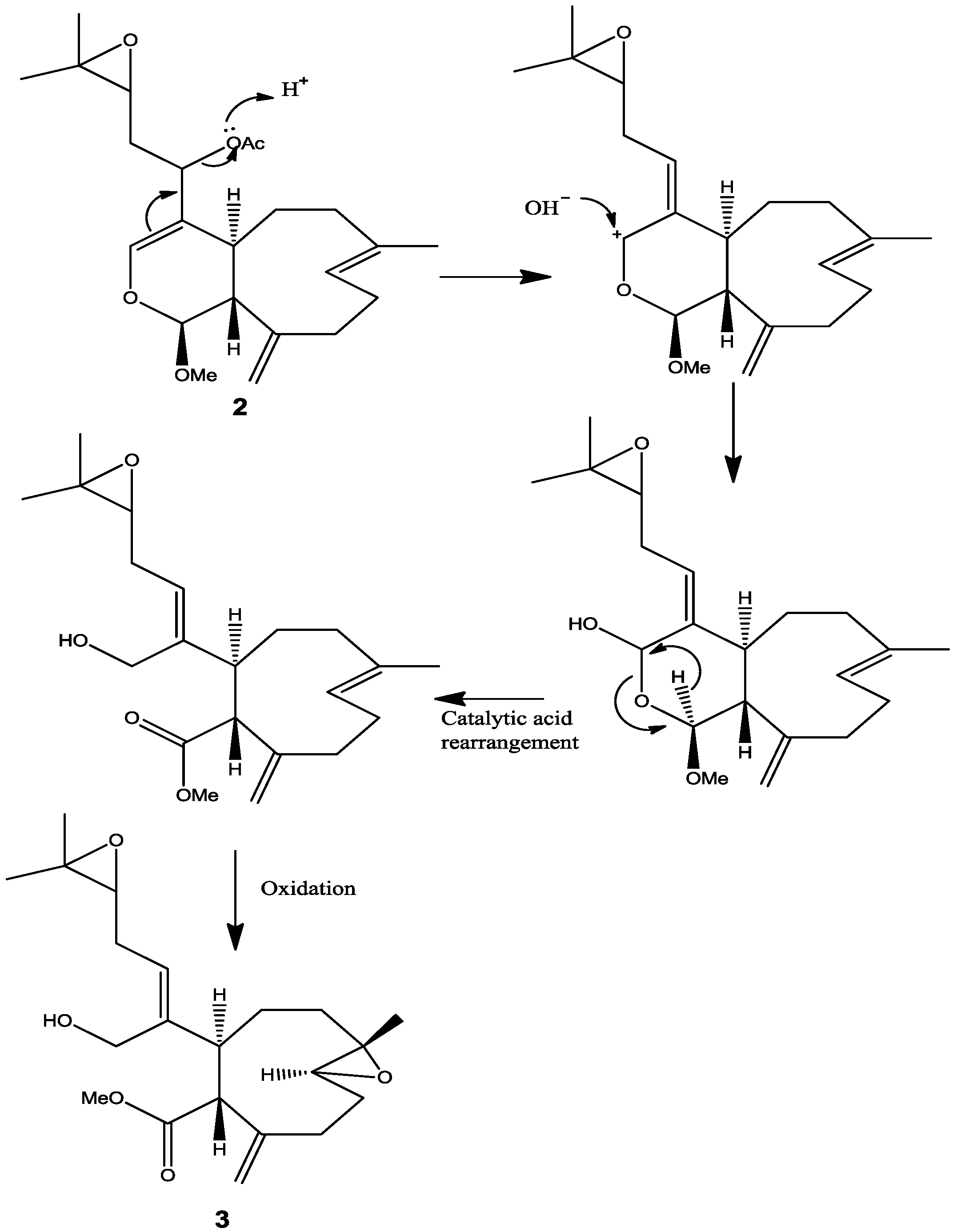
10], and therefore the stereochemistry at C-12 remains unassigned. This information suggested that the acetate group attached to C-12 is very reactive and probably leads to further transformation of the molecule as described by the proposed rearrangement in
Scheme 1. From the above results, the structure of
2 was formulated as shown.
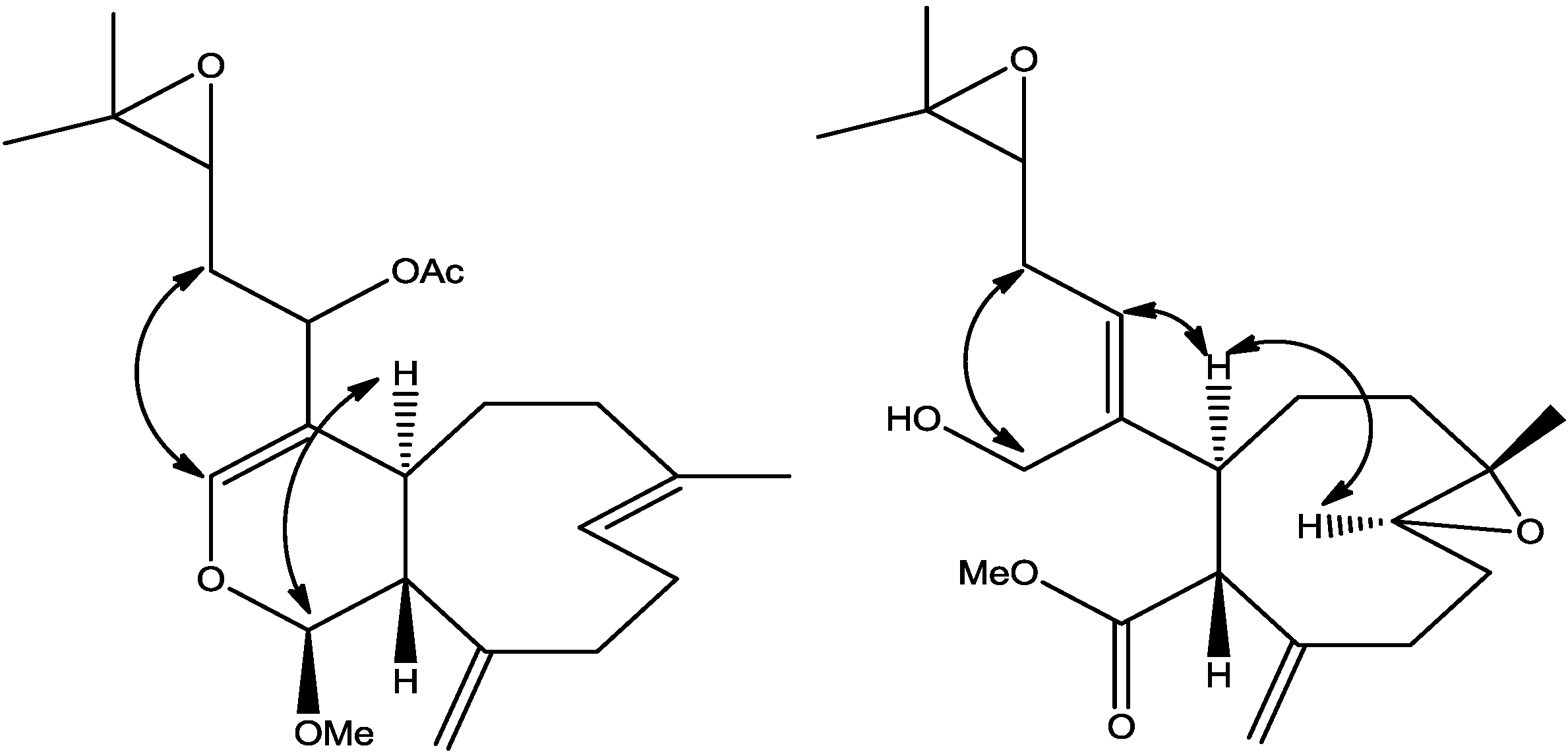
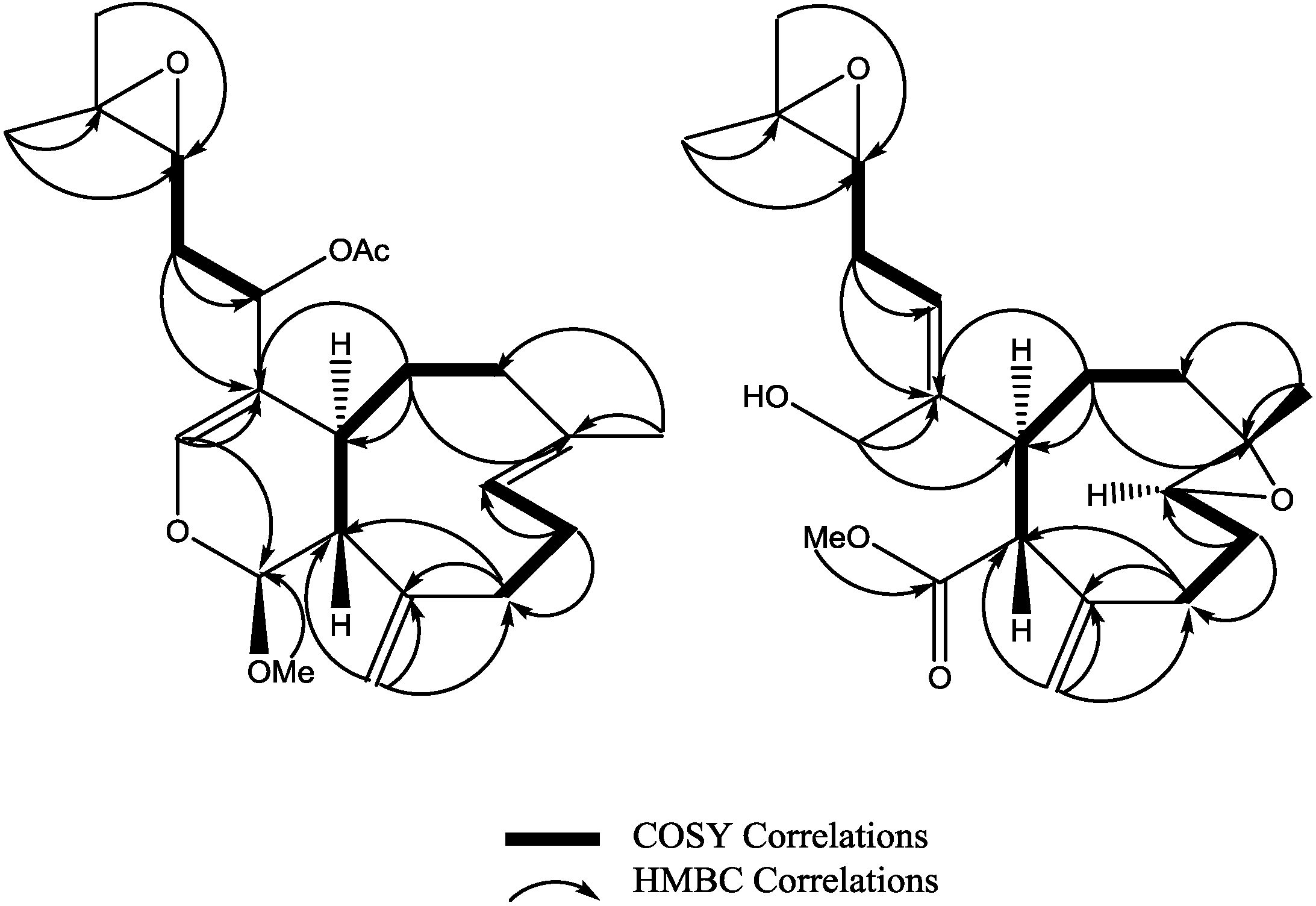
Figure 2.
Selected ROESY cross peaks for compounds (2) and (3).
Figure 2.
Selected ROESY cross peaks for compounds (2) and (3).
A molecular formula of C
21H
32O
5 for
3 was determined from HRESIMS data (
Supplementary Figure S4 Low resolution).
1H NMR (
Supplementary Figure S5) of
3 indicated the presence of two epoxy signals at [δ 2.80 (dd,
J = 11.5, 3.2 Hz), H-8] and [δ 2.76 (dd,
J = 7.2, 6.1 Hz), H-14], one methyl α to oxygen [δ 1.16 (s), H-18] and a terminal methylene [δ 5.00 (s) and 5.20 (s), H-19, H-19′]. The presence of two equivalent protons at δ [3.72, d(6)], H-3 for an isolated methylene attached to oxygen indicated that
3 has a primary alcohol functional group at C-3 which also revealed that the dihydropyran moiety presents in
1 and
2 is absent in
3. The NMR data of
3 were analogous to those of diterpene xenitacin isolated from the Formosan soft coral
Xenia umbellata [
4] except that xenitacin had an acetate group attached to C-3 instead of a hydroxy group for
3 at C-3 (
Supplementary Figure S6).
The relative stereochemistry of
3 was investigated with the aid of a ROESY spectrum. The
Z-configuration was assigned to Δ
4(12) double bond based on the observed ROESY cross peak between H-12 [δ 5.45] and H-4a [δ 3.29]. The configuration of the two chiral centers at C-4a and C-11a was deduced by ROESY data and comparison with those of xenitacin [
4]. The large coupling constant (
J = 12 Hz) between H-4a [δ 3.29] and H-11a [δ 3.45] suggests that they have a configuration opposite to each other [
4]. The observed ROESY cross peak between H-4a [δ 3.29] and H-8 [δ 2.80] (
Figure 2,
Supplementary Figure S7) suggests that they have similar configuration to xenitacin [
4]. Therefore the structure of
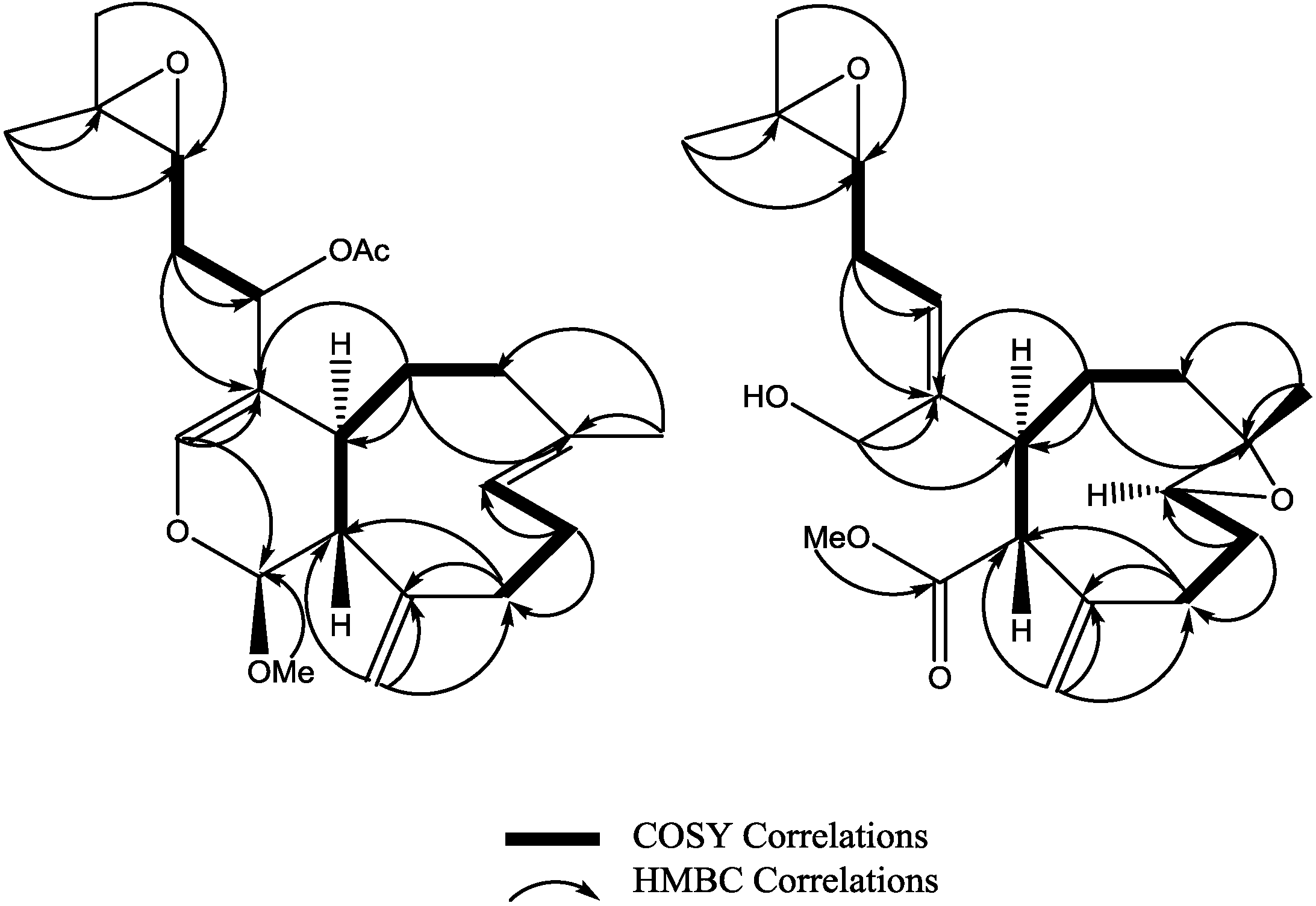
3 was established as shown. Key HMBC and selected COSY correlations were established for
2 and
3 in order to assign all carbons framework in their structures (
Figure 3 and
Supplementary Figures S8–S13).
Figure 3.
Key HMBC and selected COSY correlations for compounds (2) and (3).
Figure 3.
Key HMBC and selected COSY correlations for compounds (2) and (3).
In order to assess the pro-apoptotic activities of these compounds as a marker for their potential anticancer efficacy, methanol soluble fractions of
Xenia extracts were tested for apoptosis induction in an MTT assay using the apoptosis competent W2 and apoptosis resistant D3 cells, as described in our previous work [
3]. Compounds
2 and
3 induce measurable cell death in W2 but not in D3 cells (
Supplementary Figures S14 and S15). To quantify apoptosis induction, viability was determined using an MTT assay [
11]. Measurements were taken at time 0 and 48 h after addition of compounds in different concentrations. Viability was calculated as the difference between time 0 (addition of compound) and 48 h. Apoptosis induction was defined as at least 20% death of W2 cells and a 10% or higher growth of D3 cells by MTT assay. The optimal concentration of the compounds for apoptosis induction was determined based on the dose response of the two cells lines by MTT assay. The results reveal a significant induction of apoptosis by the whole tissue extract and by individual compounds (11.5 µM for
2 and 1.2 µM for
3), with the most remarkable induction shown by compound
3 (1.2 µM) which is ten times more active than compound
1 previously characterized [
3]. Treatment of W2 and D3 cells with 0.1 µM staurosporine, a protein kinase inhibitor and potent inducer of apoptosis, was included as a positive control for apoptosis induction and for comparison. Thus, the diterpenes we have isolated and characterized effectively and specifically activate apoptosis in immortalized mammalian epithelial cells.
There are efforts underway to synthesize optically active xenicane diterpene [
12] and work is in progress for the synthesis of diterpene from
Xenia elongata. One synthetic compound (
Supplementary Figure S16) from this work which has the same structure of
3 but does not have the side chain was also tested for comparison of activity. The result revealed that this synthetic compound did not induce apoptosis, which suggested that the side chain is critical for the activity. It is also reported before that a similar structure, xenitacin, isolated from the Formosan soft coral
Xenia umbellata [
4], is the most active compound in its group in a cytotoxicity assay against P-388, HT-29 and A549 cells [
4]. These bioactivity results may suggest that a conversion or rearrangement toward the most active structure may undergo within the organism itself or even in the cell in order to increase the efficacy of affecting or killing cancer cells. A proposed conversion or rearrangement of
2 to
3 is displayed (
Scheme 1). It is also possible that
1 will convert to
2 by enzymatic reaction of deacetylation followed by methylation and then
2 will be converted to
3.
Scheme 1.
Proposed rearrangement of 2 under acidic conditions, to give 3.
Scheme 1.
Proposed rearrangement of 2 under acidic conditions, to give 3.
In order to determine the mode of action of diterpenes from
Xenia elongata, a connectivity analysis for MCF-7 breast cancer cells treated with compound
1 was performed. The objective is to analyze changes in gene expression in a breast cancer cell line exposed to a soft coral diterpene, which is compound
1 in this experiment, and to compare the resulting signature to perturbagen signatures in the Connectivity Map [
13].
To prepare a 0.12 M stock solution in DMSO, 1.6 mg of 1 was used. Proliferating MCF-7 breast cancer cells were treated in triplicate with either a 120 µM final concentration of 1 (final DMSO concentration 0.1%), or were left untreated for 8 h. Cells were harvested before the RNA was extracted then purified, reverse transcribed, and hybridized to an Affymetrix array by the CINJ Transcriptional Profiling Core. The resulting gene expression data were analyzed.
Expression levels of 22,277 genes in three untreated and three treated samples were obtained using an Affymetrix array. For this initial analysis, the 11,400 genes were used for which all six samples provided clearly measurable signals. (Genes for which any sample was determined to be “A” or “M” were excluded, and only those for which all six were called “P” by the Affymetrix software were kept).
The package “siggenes” [
14] was used in the bioconductor set of gene expression packages for the R statistical system. Two different methods were considered for obtaining the most differentially expressed genes. The first method is known as “significance analysis of arrays” or “SAM” and was first proposed by Tusher
et al. [
15]. This method computes a modified t-statistic comparing treated to control samples for each gene. It then identifies differentially expressed genes so that the false discovery rate (FDR) is a pre-specified value. The second method, known as “Empirical Bayes Analysis of Microarrays” or “EBAM” [
16] also computes the modified t-statistic. Unlike SAM, however, it computes the posterior probability that each gene is differentially expressed. Each of the two methods can be used to create a list of genes that are differentially expressed. The two lists will generally be different, although there is usually some overlap.
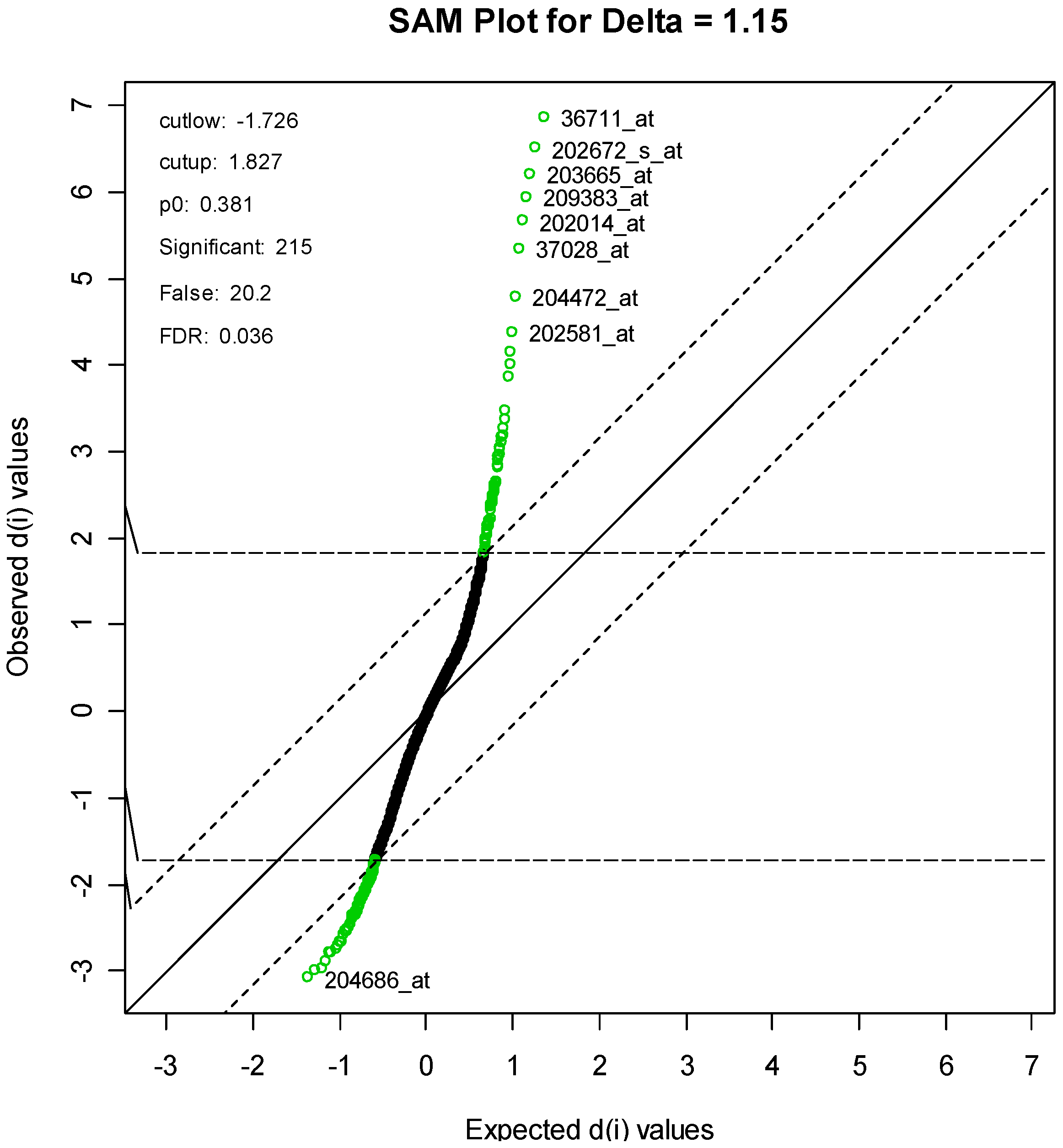
For the SAM method, the parameter delta was set to 1.15. This produced an FDR of 3.58%, and resulted in a list of 215 genes, of which an estimated 20 genes are falsely declared to be differentiated. A SAM plot is given in
Figure 4, with some of the genes labeled. A complete list is given in
Supplementary Table S1.
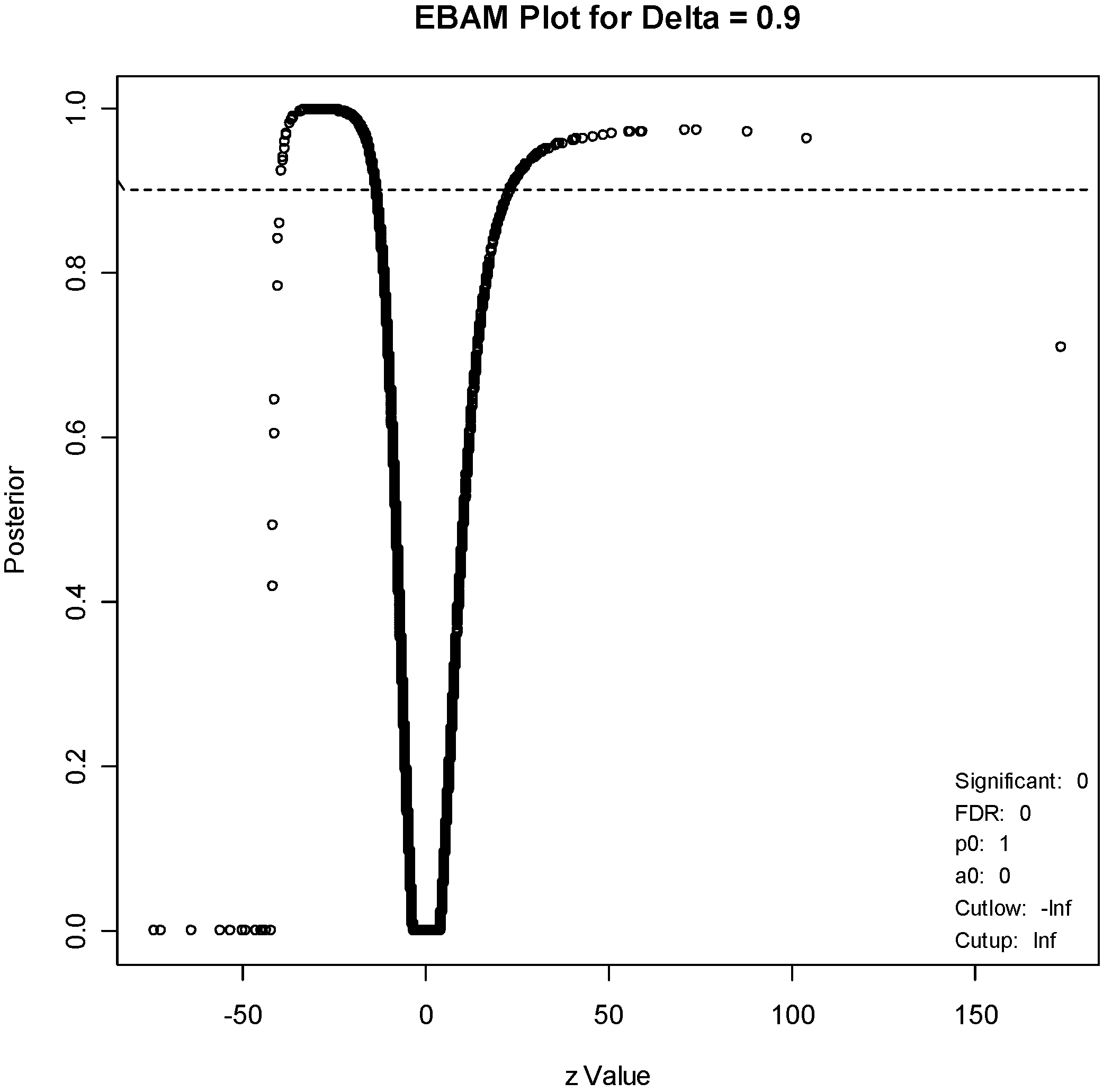
For the EBAM method, the required posterior probability was set to 0.7, which resulted in an FDR of 15.4%, to generate a list of about the same number of genes (211). An EBAM plot of posterior probabilities is given in
Figure 5, and a complete list of differentially expressed genes is given in
Supplementary Table S2. A list of the 61 genes that are present on both lists is given in
Supplementary Table S3. One gene, with code “36711_at,” is clearly at the top of both lists. This protein is a novel MAFF (v-maf musculoaponeurotic fibrosarcoma (avian) oncogene family, protein F)-LIKE protein.
The diterpene connectivity scores for the 453 “instances” (representing treatment of various cell lines with 164 different compounds) are shown in
Supplementary Tables S4 and S5. Common drug targets among the mostly highly connected instances include the proteasome, calmodulin, and histone deacetylases (HDACs). The presence of structurally distinct compounds that yield high connectivity scores suggests that the diterpene
1 acts as a proteasome and calmodulin inhibitor. Conversely, instances associated with the most highly ranked negative scores (corresponding to a gene expression profile similar to a reversal of the diterpene signature) include compounds that target the PI3 kinase/mTOR pathway. Additional inferences can be made from “permuted results” provided by the connectivity analysis (
Table 1). In particular, analysis of permuted results supports the idea that the diterpene
1 is affecting cells in a manner similar to that of HSP90 and HDAC inhibitors and in a manner opposite to that of PI3 kinase/mTOR inhibitors. Note that the proteasome inhibitors are not included in the permuted results because they are represented by single instances.
Figure 4.
SAM Plot, with some genes labeled.
Figure 4.
SAM Plot, with some genes labeled.
Figure 5.
EBAM plot. The points above the dotted line correspond to genes with a posterior probability of 0.9 of being differentially expressed.
Figure 5.
EBAM plot. The points above the dotted line correspond to genes with a posterior probability of 0.9 of being differentially expressed.
Table 1.
Permuted results.
Table 1.
Permuted results.
| Rank | CMAP name | Score | N | Enrichment | P value | Target |
|---|
| 1 | sirolimus | −0.254 | 10 | −0.770 | 0.0000 | mTOR |
| 2 | 17-allylamino-geldanamycin | 0.496 | 18 | 0.611 | 0.0000 | HSP90 |
| 3 | trichostatin A | 0.565 | 12 | 0.714 | 0.0000 | HDACs |
| 4 | LY-294002 | −0.038 | 17 | −0.506 | 0.0001 | PI3 kinase |
| 5 | geldanamycin | 0.544 | 6 | 0.759 | 0.0004 | HSP90 |
| 6 | 5253409 | 0.748 | 2 | 0.971 | 0.0009 | ? |
| 7 | 5224221 | 0.758 | 2 | 0.967 | 0.0015 | ? |
| 8 | vorinostat | 0.684 | 2 | 0.956 | 0.0032 | HDAC |
| 9 | tetraethylene-pentamine | −0.084 | 6 | −0.662 | 0.0055 | proteases |
| 10 | wortmannin | −0.248 | 8 | −0.565 | 0.0071 | PI3 kinase |
Each row in the table summarizes the performance of all instances produced with the same perturbagen. The table displays the cmap name for those perturbagens, the arithmetic mean of the connectivity scores for each of those instances (labeled “score”), the number of those instances (labeled “n”), a measure of the enrichment of those instances in the list of all instances ordered by descending order of connectivity score and up score (labeled “enrichment”) and a permutation p-value for that enrichment score (labeled “p”). Enrichment scores and p-values are not provided for perturbagens represented by only one instance or where the mean of the connectivity scores for their instances is zero.
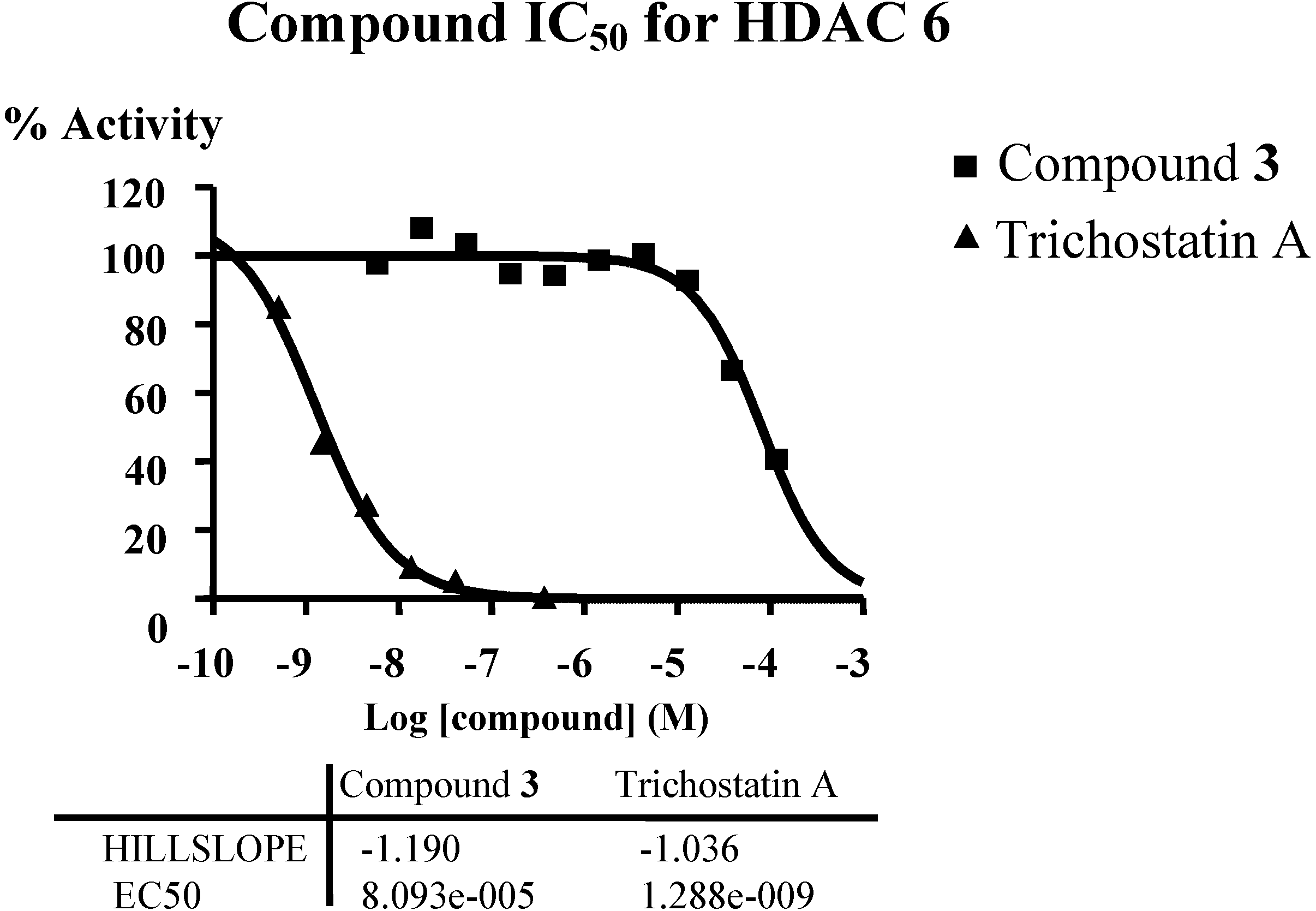
In order to verify the predicted mode of action of diterpene from connectivity analysis, HDAC inhibitory assays were performed. Given the availability of compound material and the structural similarity of all isolated diterpenes, only compound
3 was subjected to enzymatic inhibition test against the class I (HDAC1, HDAC2, HDAC3 and HDAC8), the class II A (HDAC4, HDAC5, HDAC7 and HDAC9) and the class II B (HDAC6) enzymes. HDAC inhibitors Trichostatin A (TSA) and TMP269 (TMP) were used as reference compounds. The results showed that compound
3 up to 50 or 100 µM did not significantly inhibit the class I (HDAC1,
Supplementary Table S6; HDAC2,
Supplementary Table S7; HDAC3,
Supplementary Table S8; HDAC8,
Supplementary Table S9) as well as the class II A (HDAC4,
Supplementary Table S10; HDAC5,
Supplementary Table S11; HDAC7,
Supplementary Table S12; HDAC9,
Supplementary Table S13), though compound
3 inhibited class II B HDAC6 with IC
50 around 80 µM (
Figure 6,
Supplementary Table S14). The predicted mode of action of diterpene was verified. HDAC6 appears to be the key deacetylase regulating Hsp90; treatment of breast cancer cells with Hsp90 inhibitors or HDAC inhibitor (vorinostat) induces breast cancer cell differentiation [
17]. Cell death signaling is complex and it is clear that there are multiple alternate pathways, especially in cancer cell types. The HDAC6 inhibition pathway is among several pathways that the diterpene may trigger simultaneously, thus explaining the difference of potency between the apoptosis induction assay and the HDAC6 inhibition assay.
Figure 6.
Inhibition of HDAC6 by compound 3, IC50 around 80 µM.
Figure 6.
Inhibition of HDAC6 by compound 3, IC50 around 80 µM.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}