Cyclodextrin Formulation of the Marine Natural Product Pseudopterosin A Uncovers Optimal Pharmacodynamics in Proliferation Studies of Human Umbilical Vein Endothelial Cells


Abstract
:1. Introduction
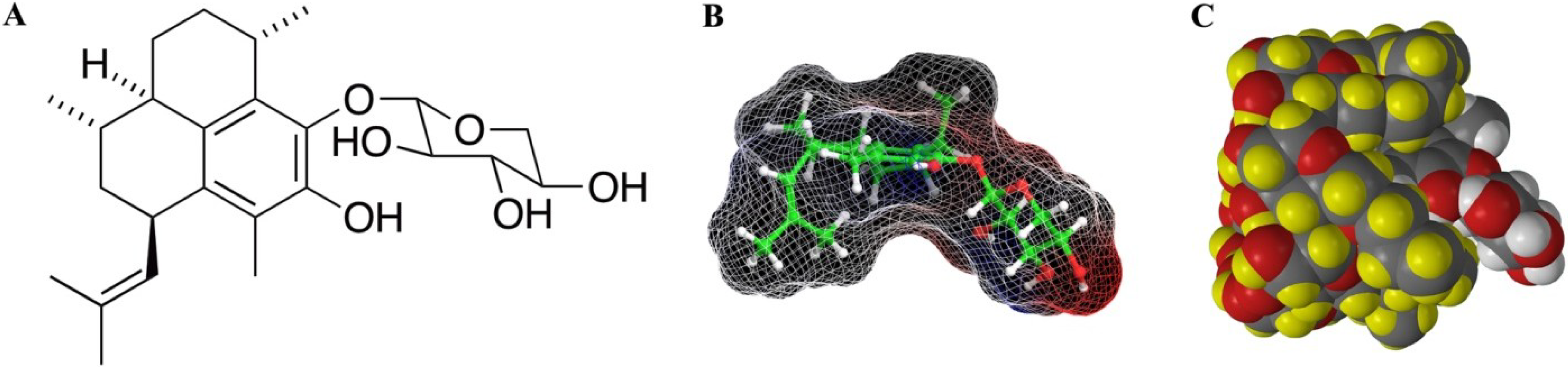
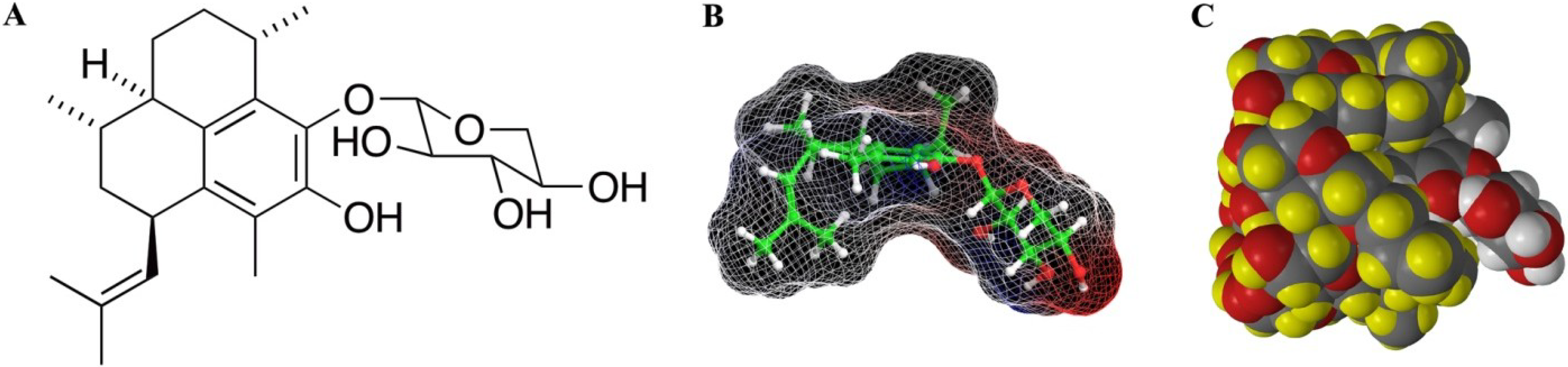
2. Results/Discussion
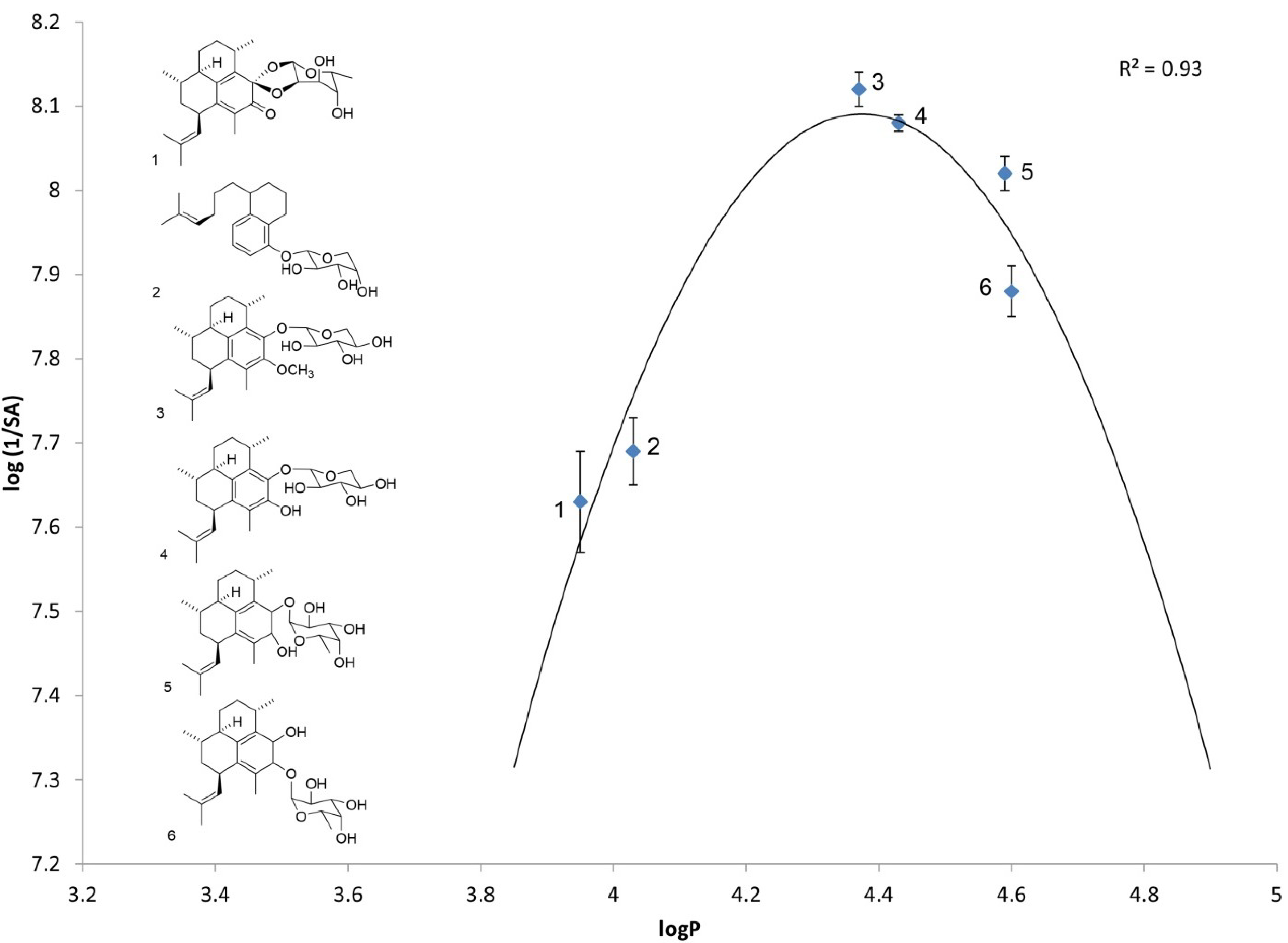
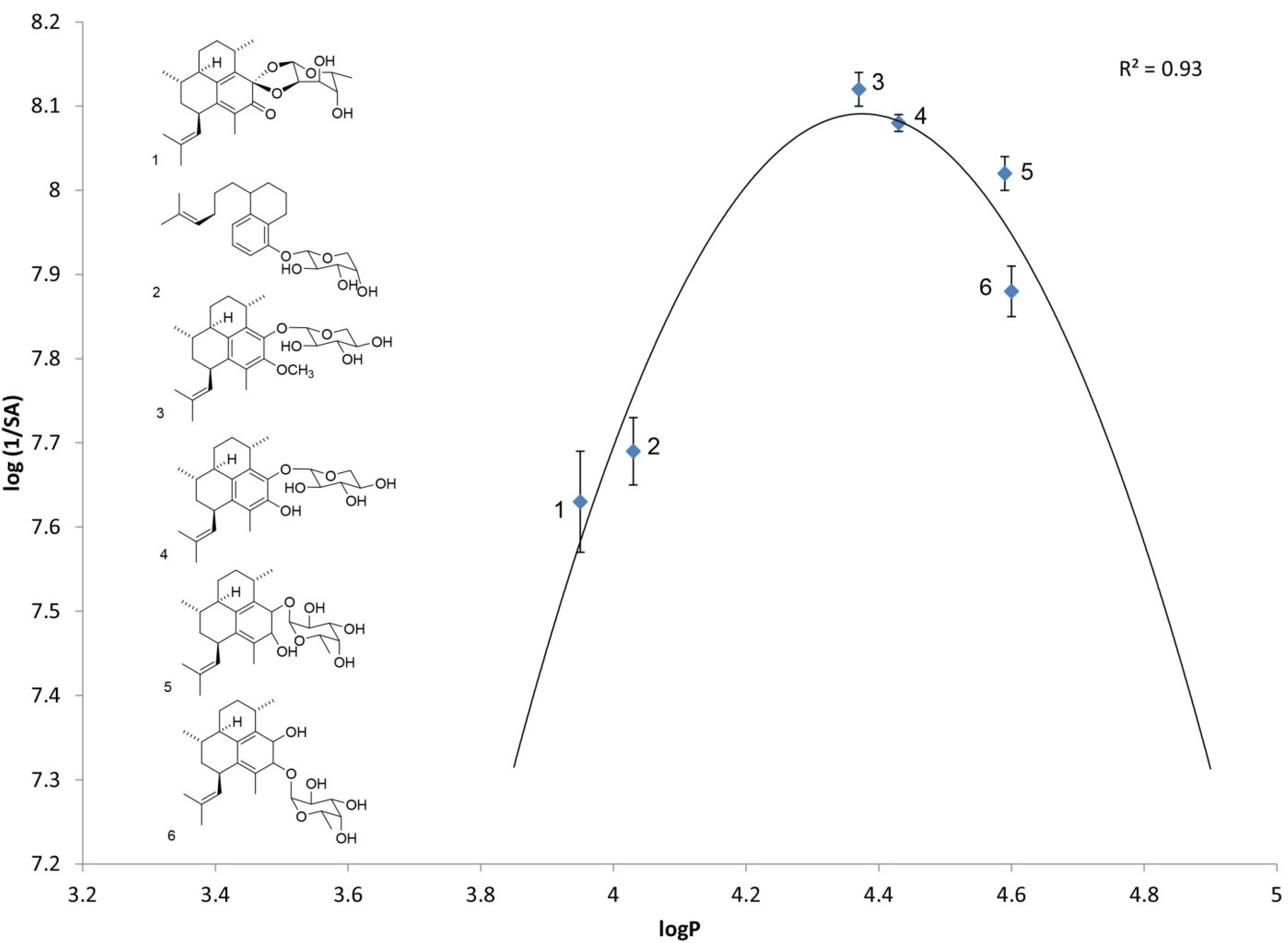
2.1. Log P as a Link to Specific Activity
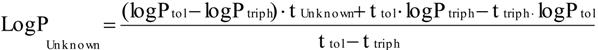

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analog number | Analog name | Direct log P | log(1/SA) |
|---|---|---|---|
| (n = 4) | (n = 10) | ||
| 1 | Ps keto ketal [4] | 3.95 ± 0.02 | 7.63 ± 0.06 |
| 2 | arabinopyranoside of seco-PsA [13] | 4.03 ± 0.05 | 7.69 ± 0.04 |
| 3 | PsA methyl ether | 4.37 ± 0.02 | 8.12 ± 0.02 |
| 4 | PsA | 4.42 ± 0.06 | 8.08 ± 0.01 |
| 5 | iso-PsE | 4.59 ± 0.05 | 8.02 ± 0.02 |
| 6 | PsE | 4.60 ± 0.06 | 7.88 ± 0.03 |
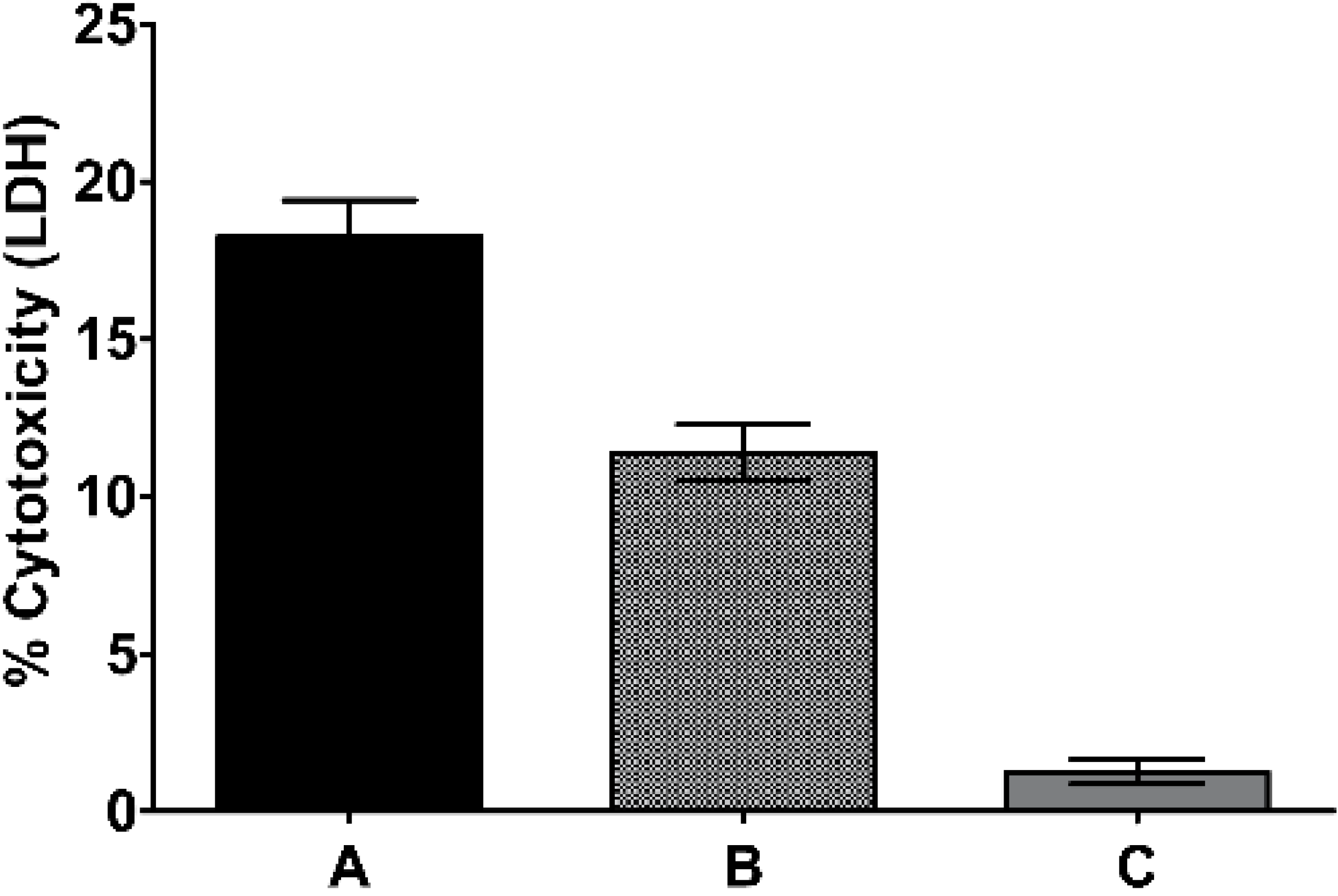
2.2. Traditional Formulation to Assess Cytotoxicity
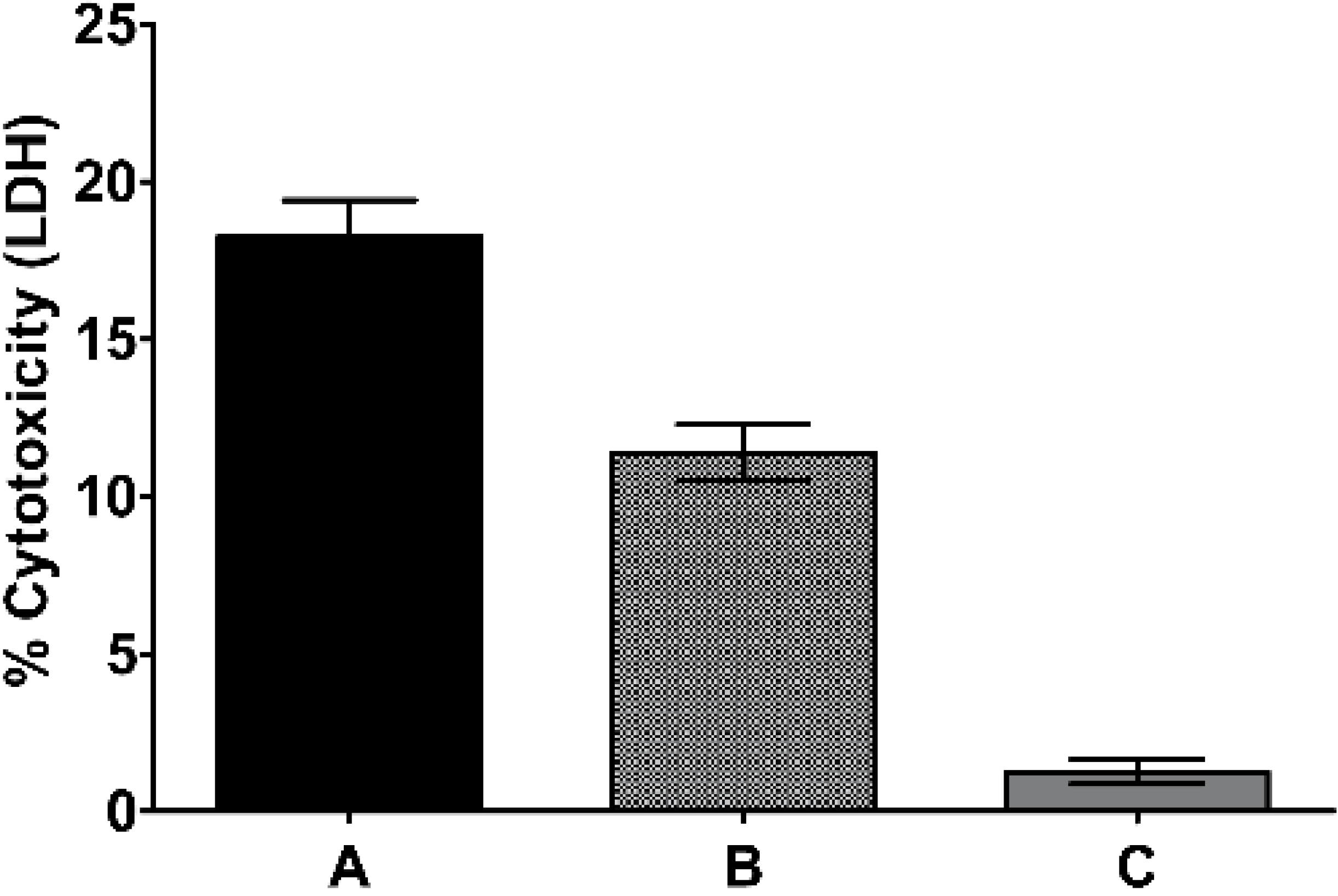
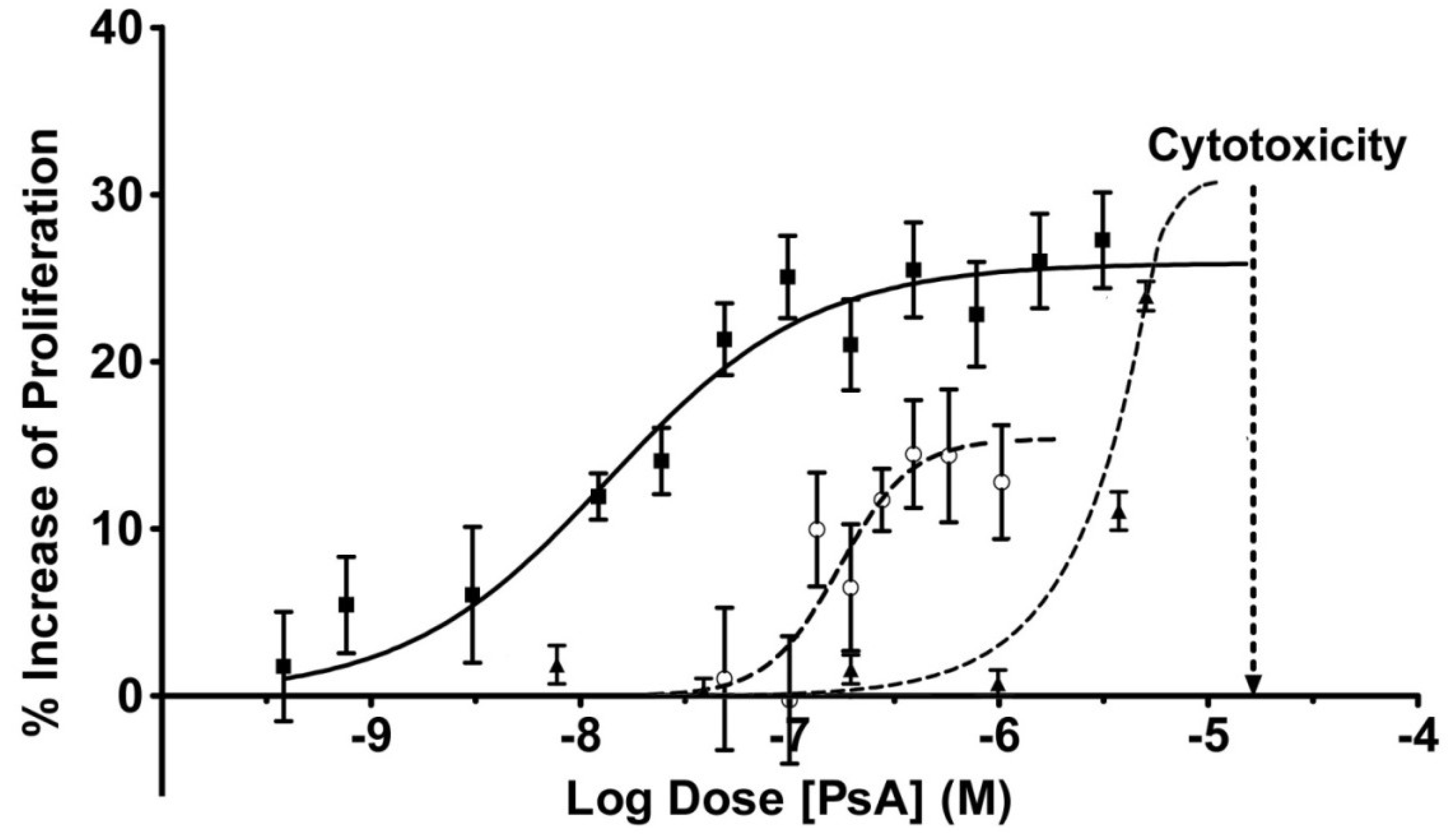
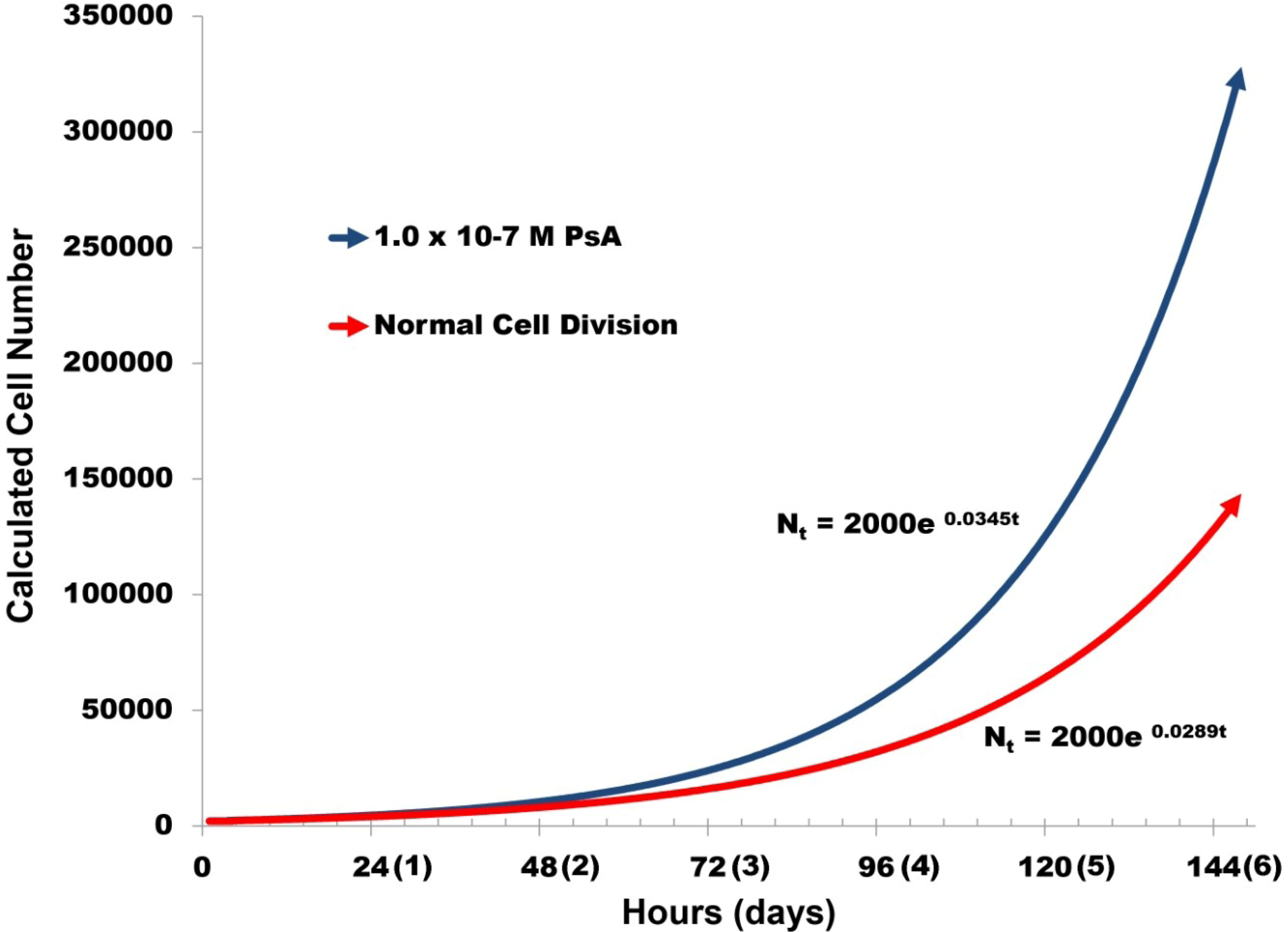
2.3. Pseudopterosin Induces HUVEC Proliferation
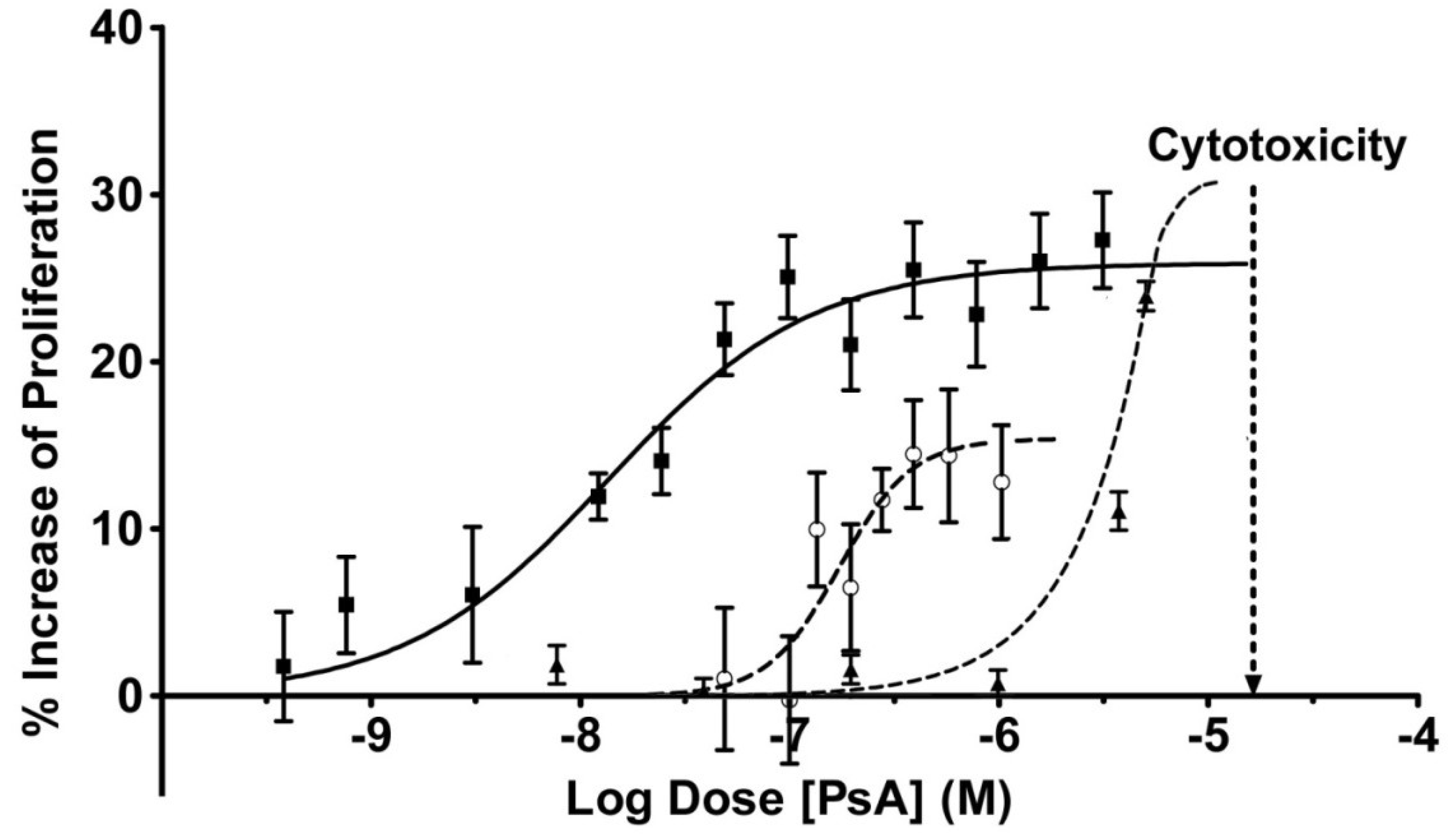
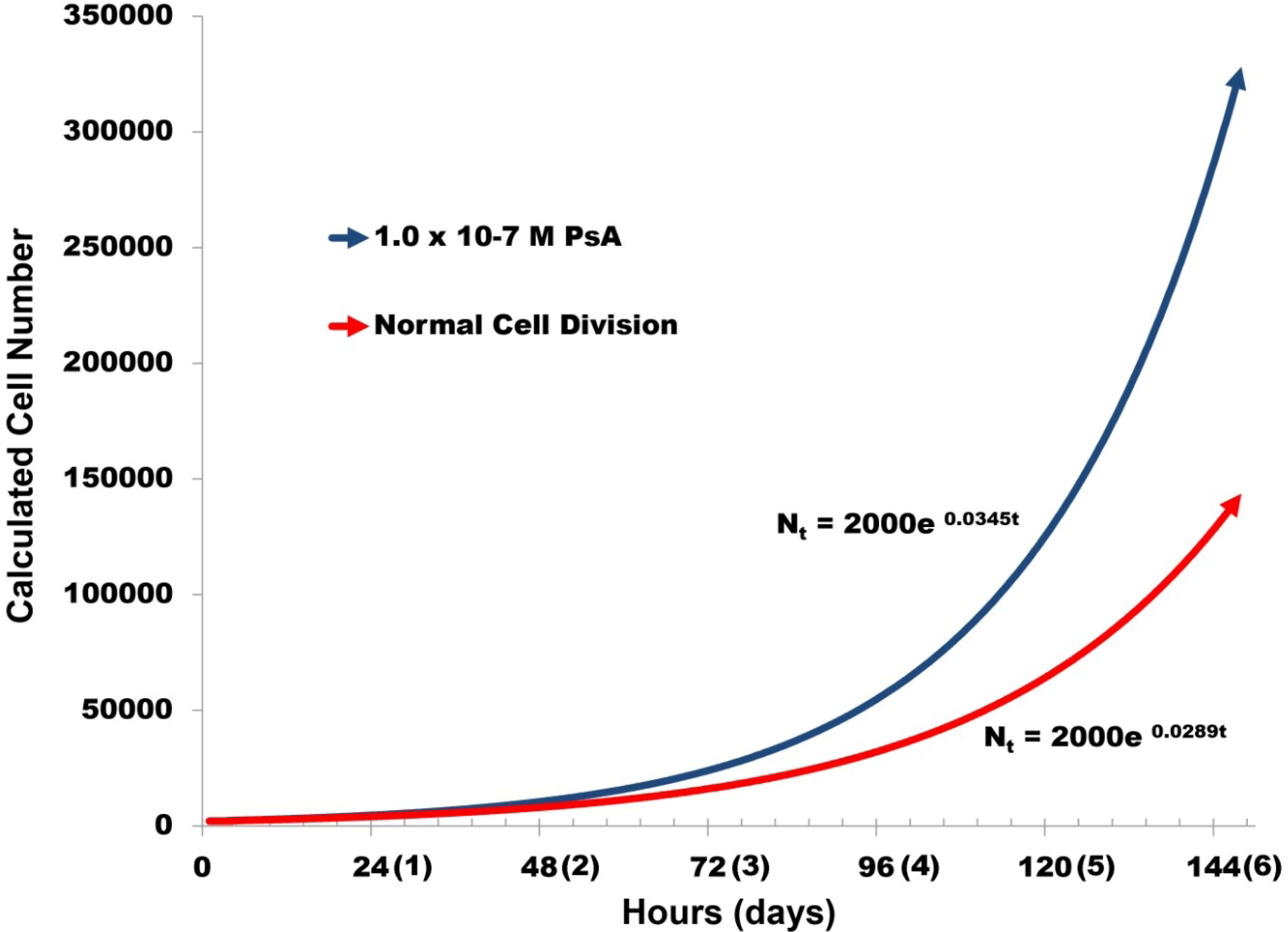
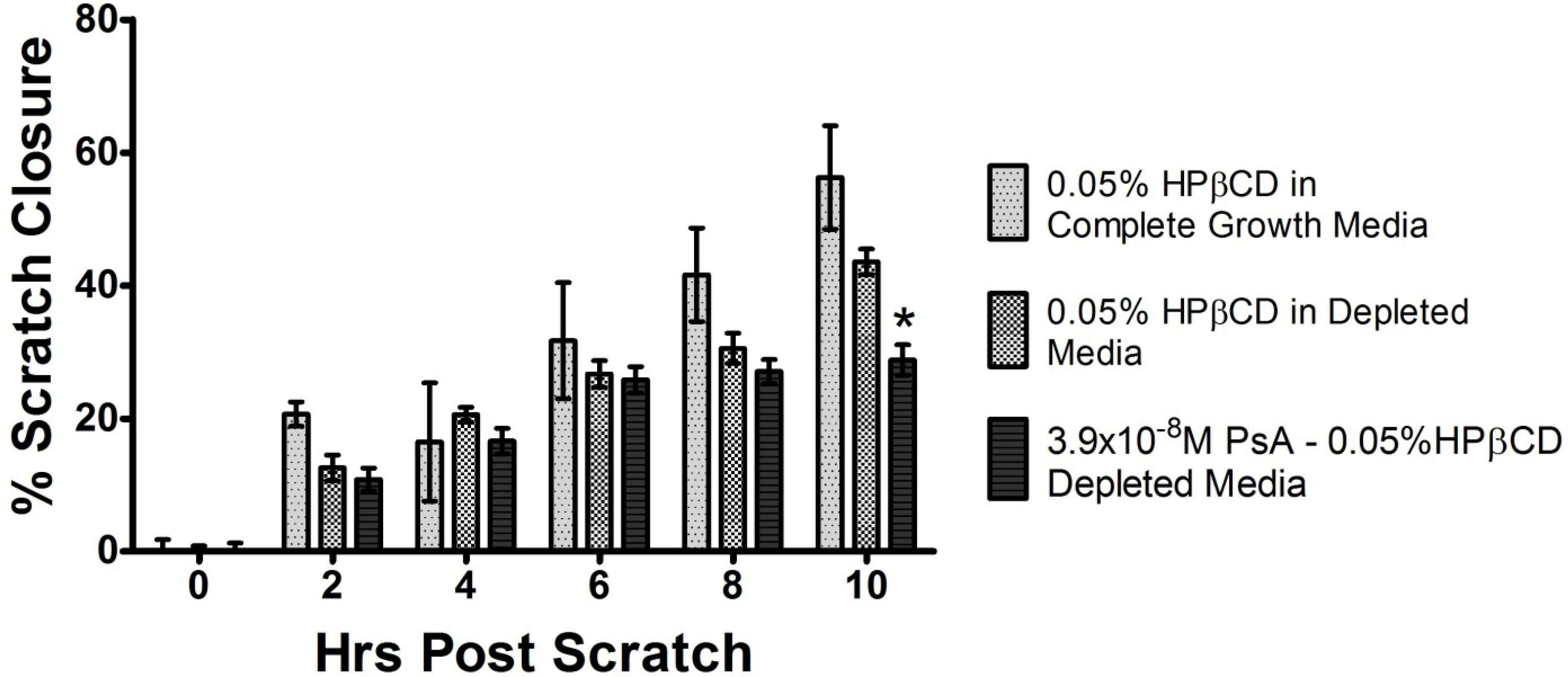
2.4. PsA Inhibits Normal HUVEC Migration
3. Materials and Methods
3.1. Cell Culture and Reagents
3.2. Equipment
3.3. Extraction and Purification from P. elisabethae
3.4. Biological Activity-Inhibition of Inflammation
3.5. Formation of Cyclodextrin Inclusion Complex
3.6. Drug Solubilization Verification
3.7. HUVEC Cell Culture
3.8. WST-1 Cell Proliferation Assay
3.9. Scratch Migration Assay
3.10. Statistical Analysis
4. Conclusion
Acknowledgements
Conflicts of Interest
References and Notes
- Look, S.A.; Fenical, W.; Jacobs, R.S.; Clardy, J. The pseudopterosins—Anti-inflammatory and analgesic natural-products from the sea whip Pseudopterogorgia-elisabethae. Proc. Natl. Acad. Sci. USA 1986, 83, 6238–6240. [Google Scholar] [CrossRef]
- Mayer, A.M.; Jacobson, P.B.; Fenical, W.; Jacobs, R.S.; Glaser, K.B. Pharmacological characterization of the pseudopterosins: Novel anti-inflammatory natural products isolated from the Caribbean soft coral, Pseudopterogorgia elisabethae. Life Sci. 1998, 62, PL401–PL407. [Google Scholar] [CrossRef]
- Luedke, E.S. The Identification and Characterization of Pseudopterosins: Anti-Inflammatory Agents Isolated from the Gorgonian Coral Pseudopterogorgia elisabethae. Ph.D. Thesis, University of California, Santa Barbara, CA, USA, 21 June 1990. [Google Scholar]
- Zhong, W.; Little, R.D. Exploration and determination of the redox properties of the pseudopterosin class of marine natural products. Tetrahedron 2009, 65, 10784–10790. [Google Scholar] [CrossRef]
- Mayer, A.M.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- Vimrx Pharmaceuticals, New Drug Application VM 301 for Use in Phase I/IIA Clinical Trials. IND 60,606. 12 July 2000.
- Kell, D.B.; Dobson, P.D.; Oliver, S.G. Pharmaceutical drug transport: The issues and the implications that it is essentially carrier-mediated only. Drug Discov. Today 2011, 16, 704–714. [Google Scholar] [CrossRef]
- Leeson, P.D.; Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef]
- Hansch, C.; Anderson, S.M. The structure-activity relationship in barbiturates and its similarity to that in other narcotics. J. Med. Chem. 1967, 10, 745–753. [Google Scholar] [CrossRef]
- Zheng, B.; West, L.M. Estimating the lipophilicity of natural products using a polymeric reversed phase HPLC method. J. Liq. Chromatogr. Relat. Technol. 2009, 33, 118–132. [Google Scholar] [CrossRef]
- Donovan, S.F.; Pescatore, M.C. Method for measuring the logarithm of the octanol–water partition coefficient by using short octadecyl–poly(vinyl alcohol) high-performance liquid chromatography columns. J. Chromatogr. A 2002, 952, 47–61. [Google Scholar] [CrossRef]
- Tanis, V.M.; Moya, C.E.; Jacobs, R.S.; Little, R.D. ynthesis and evaluation of the bioactivity of simplified analogs of the seco-pseudopterosins; progress toward determining a pharmacophore. Tetrahedron 2008, 64, 10649–10663. [Google Scholar] [CrossRef]
- Andrews, P.R.; Craik, D.J.; Martin, J.L. Functional group contributions to drug-receptor interactions. J. Med. Chem. 1984, 27, 1648–1657. [Google Scholar] [CrossRef]
- Trengove, N.J.; Stacey, M.C.; Macauley, S.; Bennett, N.; Gibson, J.; Burslem, F.; Murphy, G.; Schultz, G. Analysis of the acute and chronic wound environments: The role of proteases and their inhibitors. Wound Repair Regen. 1999, 7, 442–452. [Google Scholar] [CrossRef]
- Lauer, G.; Sollberg, S.; Cole, M.; Flamme, I.; Sturzebecher, J.; Mann, K.; Krieg, T.; Eming, S.A. Expression and proteolysis of vascular endothelial growth factor is increased in chronic wounds. J. Investig. Dermatol. 2000, 115, 12–18. [Google Scholar] [CrossRef]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in wound repair: Molecular and cellular mechanisms. J. Investig. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef]
- Lauder, H.; Frost, E.E.; Hiley, C.R.; Fan, T.P. Quantification of the repair process involved in the repair of a cell monolayer using an in vitro model of mechanical injury. Angiogenesis 1998, 2, 67–80. [Google Scholar] [CrossRef]
- Lee, P.C.; Salyapongse, A.N.; Bragdon, G.A.; Shears, L.L.; Watkins, S.C.; Edington, H.D.; Billiar, T.R. Impaired wound healing and angiogenesis in eNOS-deficient mice. Am. J. Physiol. 1999, 277, H1600–H1608. [Google Scholar]
- Pitha, J.; Milecki, J.; Fales, H.; Pannell, L.; Uekama, K. Hydroxypropyl-beta-cyclodextrin: Preparation and characterization; Effects on solubility of drugs. Int. J. Pharm. 1986, 29, 73–82. [Google Scholar] [CrossRef]
- Uekama, K.; Hirayama, F.; Irie, T. Cyclodextrin drug carrier systems. Chem. Rev. 1998, 98, 2045–2076. [Google Scholar] [CrossRef]
- Ettouati, W.S.; Jacobs, R.S. Effect of pseudopterosin A on cell division, cell cycle progression, DNA, and protein synthesis in cultured sea urchin embryos. Mol. Pharm. 1987, 31, 500–505. [Google Scholar]
- Correa, H.; Aristizabal, F.; Duque, C.; Kerr, R. Cytotoxic and antimicrobial activity of pseudopterosins and seco-pseudopterosins isolated from the octocoral Pseudopterogorgia elisabethae of San Andres and Providencia Islands (Southwest Caribbean Sea). Mar. Drugs 2011, 9, 334–343. [Google Scholar] [CrossRef]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef]
- Mydlarz, L.D.; Jacobs, R.S.; Boehnlein, J.; Kerr, R.G. Pseudopterosin biosynthesis in Symbiodinium sp., the dinoflagellate symbiont of Pseudopterogorgia elisabethae. Chem. Biol. 2003, 10, 1051–1056. [Google Scholar] [CrossRef]
- PMA and the test compounds were dissolved in acetone so that they could be administered simultaneously. The acetone evaporates rapidly and the control shows that the initial presence of acetone does not affect the results. A reviewer has expressed concern that PMA and the various pseudopterosins might react/interact during the mixing time and influence the results, and that they should have been administered separately. Since the total time of incubation is >3 h, any reaction/interaction would have to occur during the initial mixing in order to influence the results. Given the structures of PMA and the pseudopterosins as well as the known chemistries of each, we consider it unlikely, but acknowledge the reviewers’ viewpoint.
- Miller-Keane; O’Toole, M.T. Miller-Keane Encyclopedia and Dictionary of Medicine, Nursing, and Allied Health, 7th ed.; Saunders: Philadelphia, PA, USA, 2005. [Google Scholar]
- Tonnesen, M.G.; Feng, X.; Clark, R.A. Angiogenesis in wound healing. J. Investig. Dermatol. Symp. Proc. 2000, 5, 40–46. [Google Scholar] [CrossRef]
- Marti, H.H.; Risau, W. Angiogenesis in ischemic disease. Thromb. Haemost. 1999, 82 (Suppl. 1), 44–52. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Day, D.R.; Jabaiah, S.; Jacobs, R.S.; Little, R.D. Cyclodextrin Formulation of the Marine Natural Product Pseudopterosin A Uncovers Optimal Pharmacodynamics in Proliferation Studies of Human Umbilical Vein Endothelial Cells. Mar. Drugs 2013, 11, 3258-3271. https://doi.org/10.3390/md11093258
Day DR, Jabaiah S, Jacobs RS, Little RD. Cyclodextrin Formulation of the Marine Natural Product Pseudopterosin A Uncovers Optimal Pharmacodynamics in Proliferation Studies of Human Umbilical Vein Endothelial Cells. Marine Drugs. 2013; 11(9):3258-3271. https://doi.org/10.3390/md11093258
Chicago/Turabian StyleDay, Daniel R., Suraya Jabaiah, Robert S. Jacobs, and R. Daniel Little. 2013. "Cyclodextrin Formulation of the Marine Natural Product Pseudopterosin A Uncovers Optimal Pharmacodynamics in Proliferation Studies of Human Umbilical Vein Endothelial Cells" Marine Drugs 11, no. 9: 3258-3271. https://doi.org/10.3390/md11093258
APA StyleDay, D. R., Jabaiah, S., Jacobs, R. S., & Little, R. D. (2013). Cyclodextrin Formulation of the Marine Natural Product Pseudopterosin A Uncovers Optimal Pharmacodynamics in Proliferation Studies of Human Umbilical Vein Endothelial Cells. Marine Drugs, 11(9), 3258-3271. https://doi.org/10.3390/md11093258