APS8, a Polymeric Alkylpyridinium Salt Blocks α7 nAChR and Induces Apoptosis in Non-Small Cell Lung Carcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
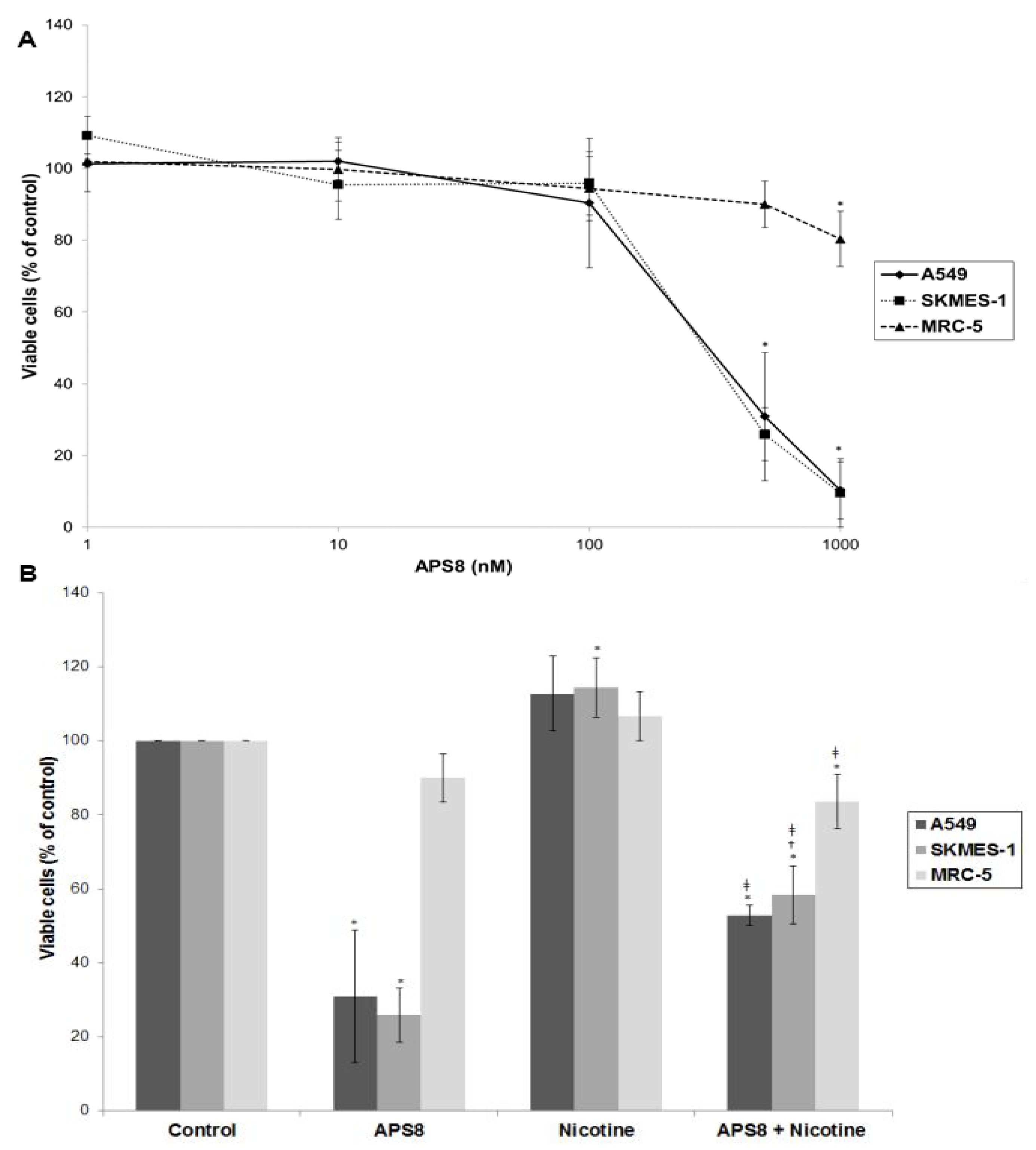
2.1. APS8 Induces Cytotoxicity in Lung Cancer (LC) Cells
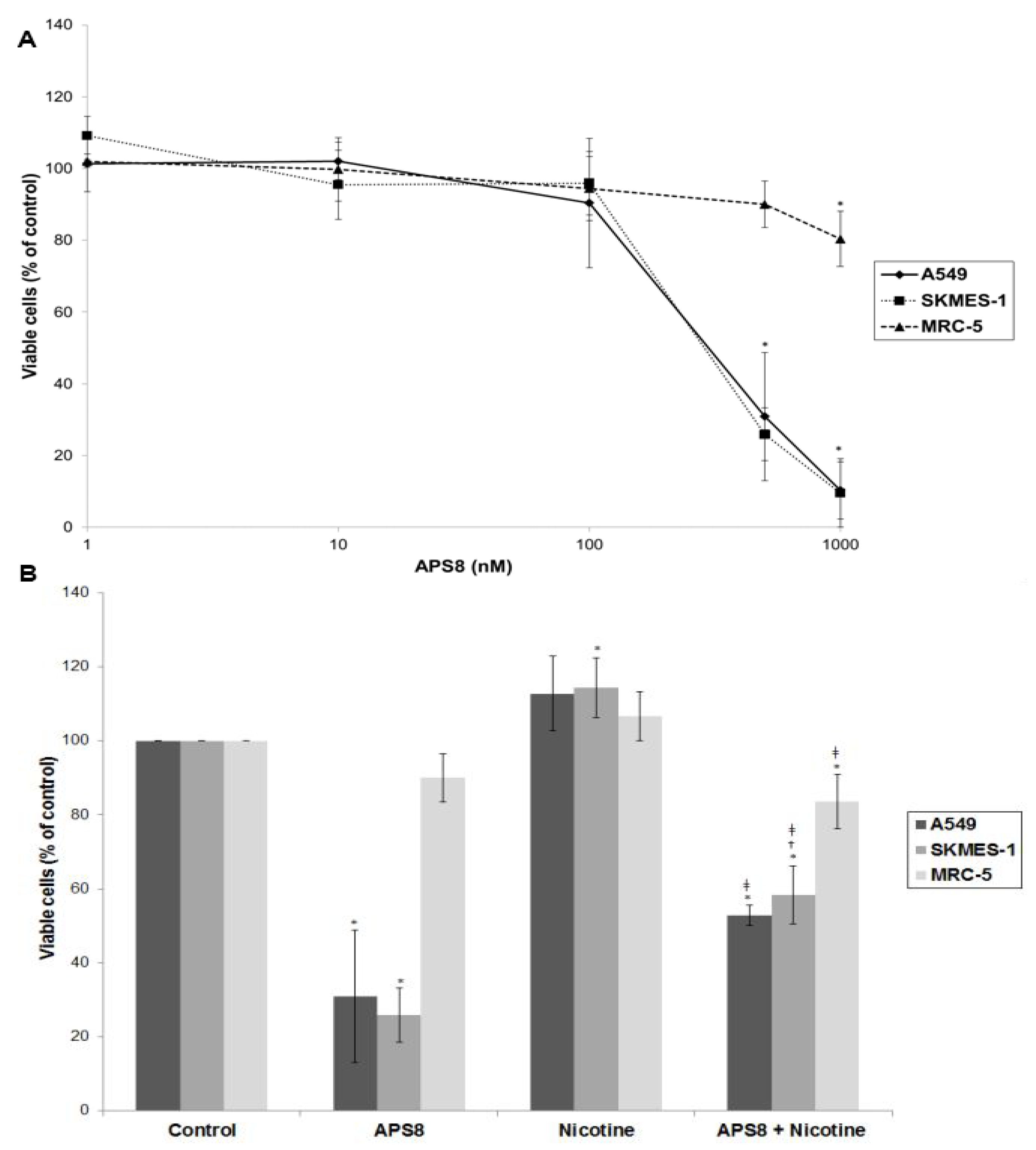
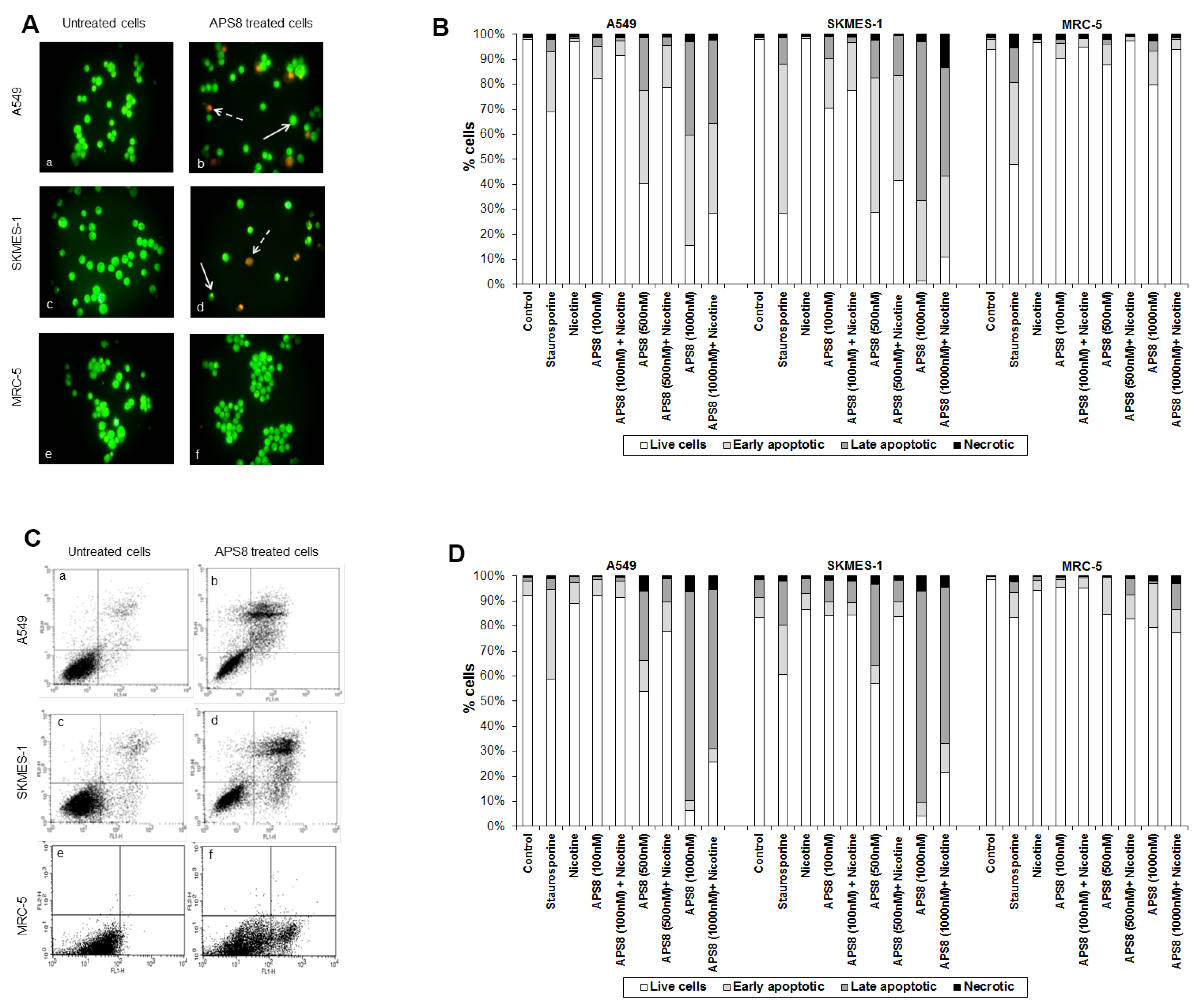
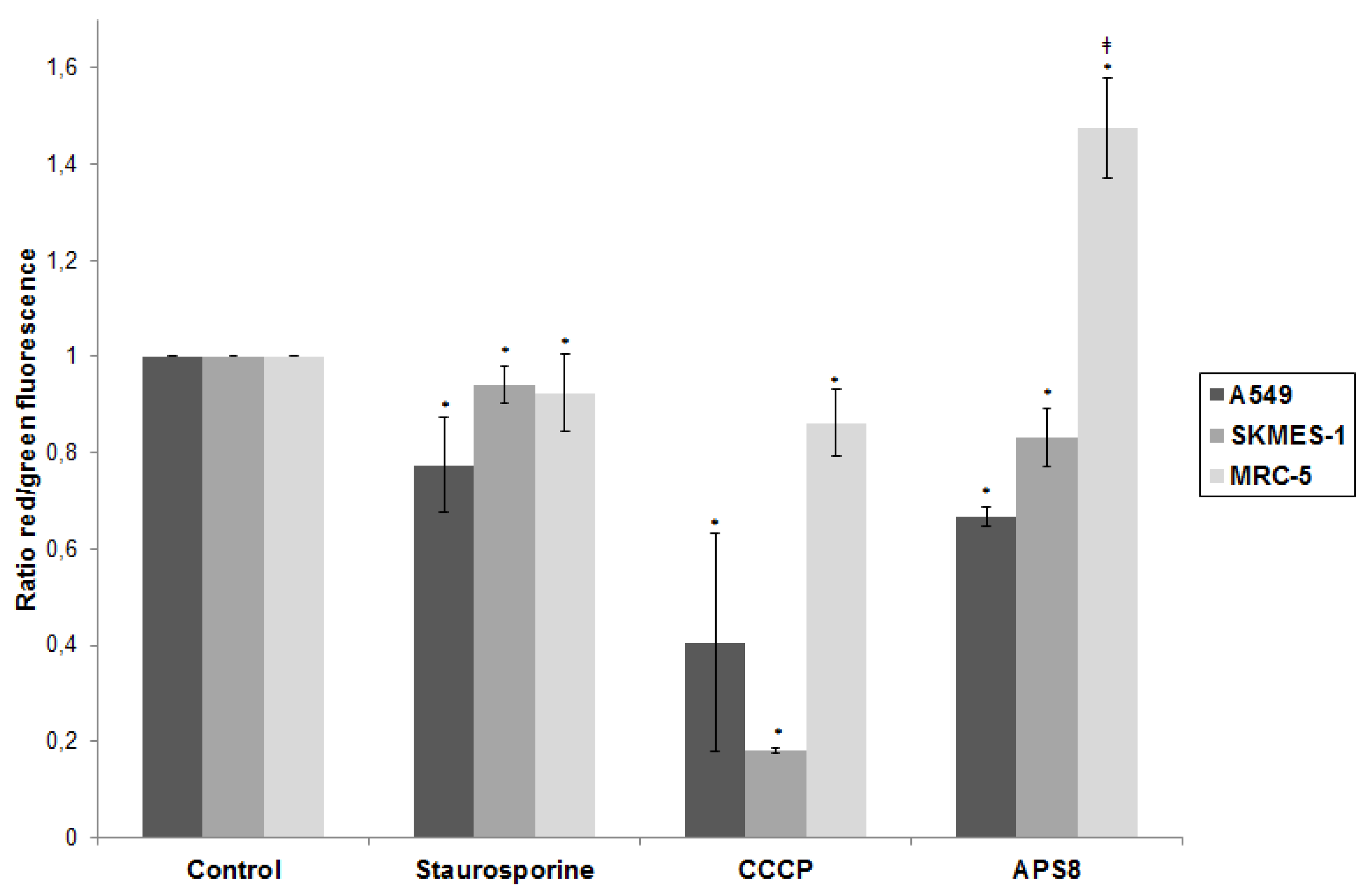
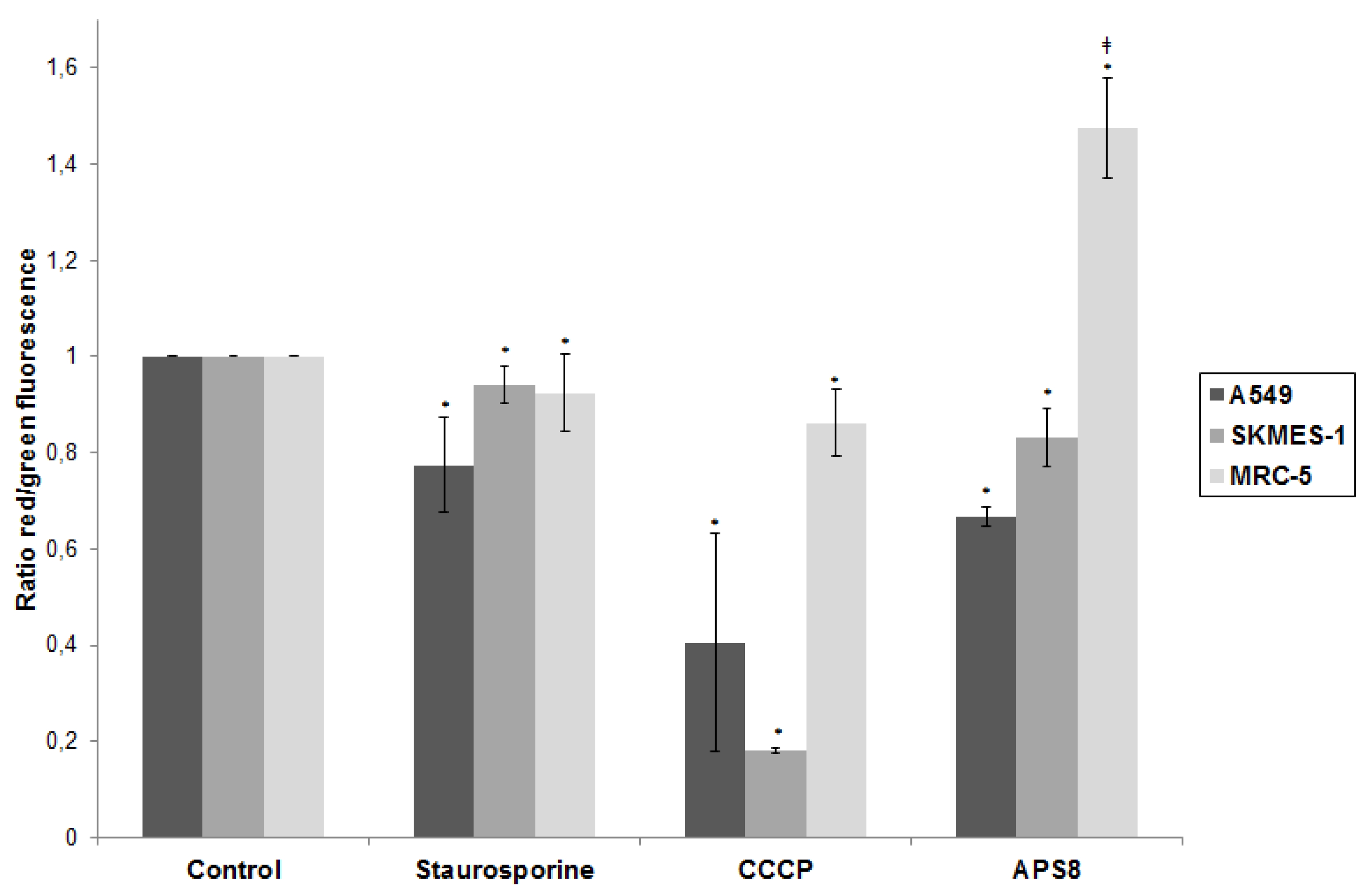
2.2. APS8 Induces Mitochondrial Depolarization in LC Cells
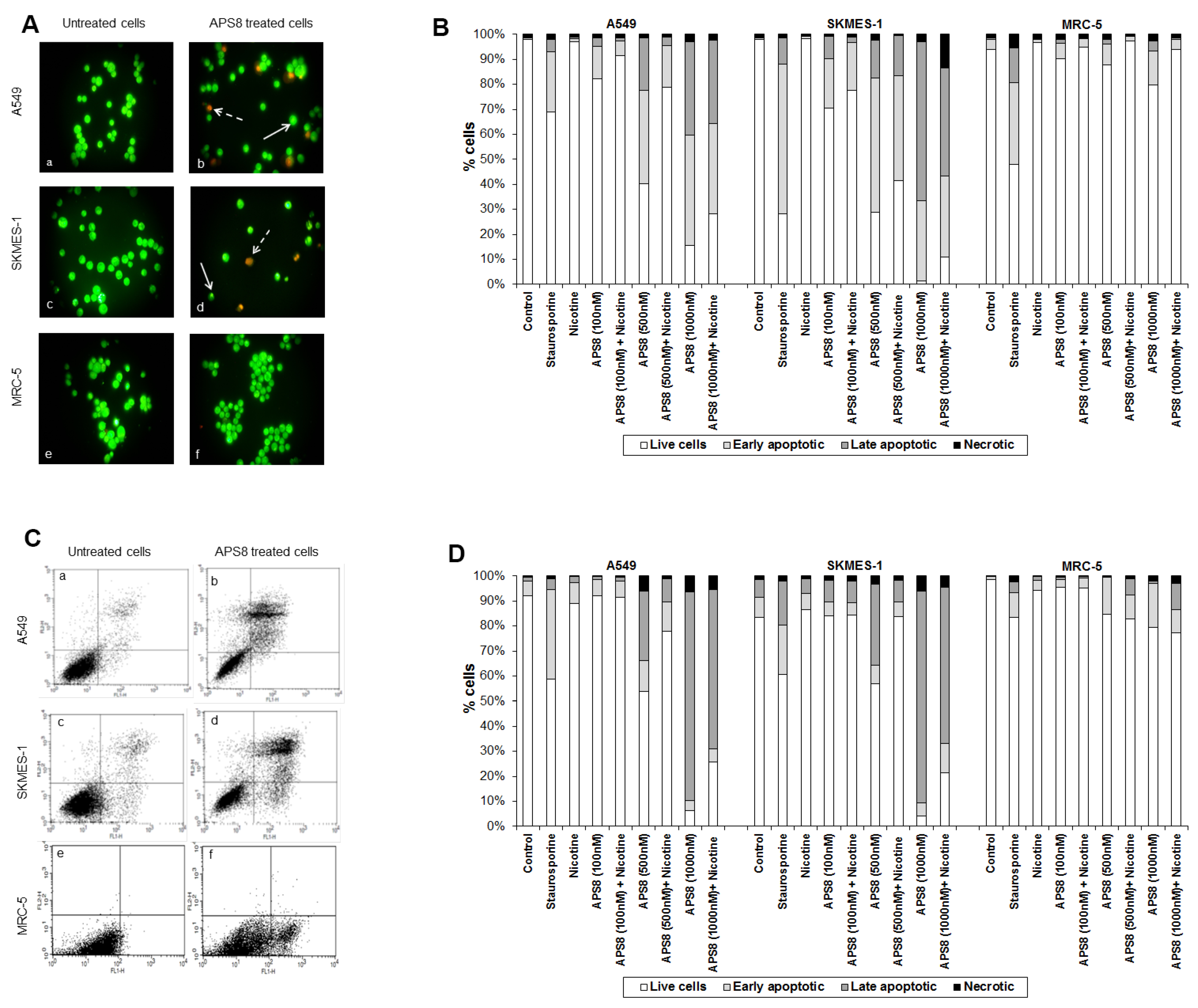
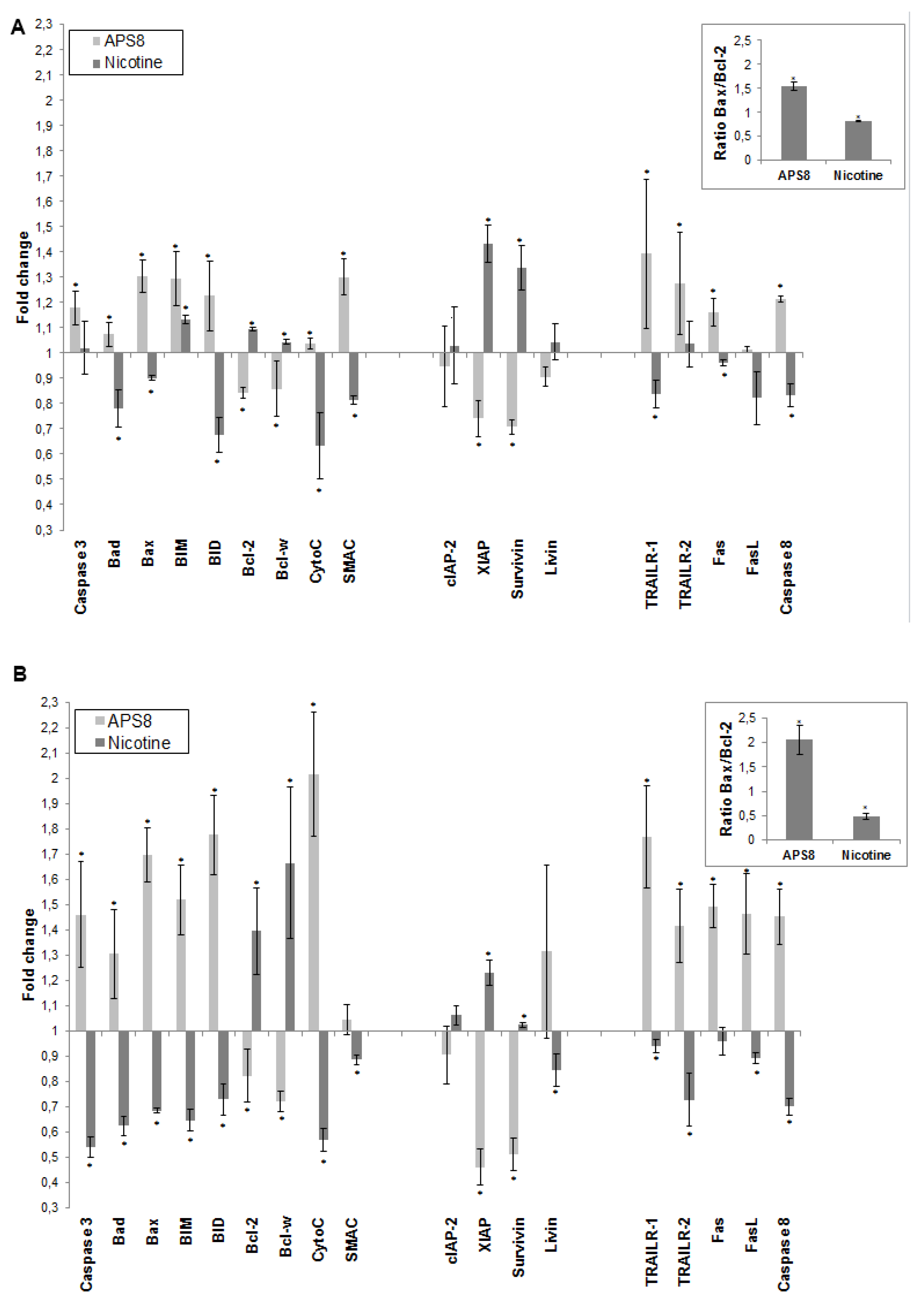
2.3. APS8 Treatment Results in Increased Expression of Pro-Apoptotic Proteins and Down-Regulation of Anti-Apoptotic Proteins in LC Cells
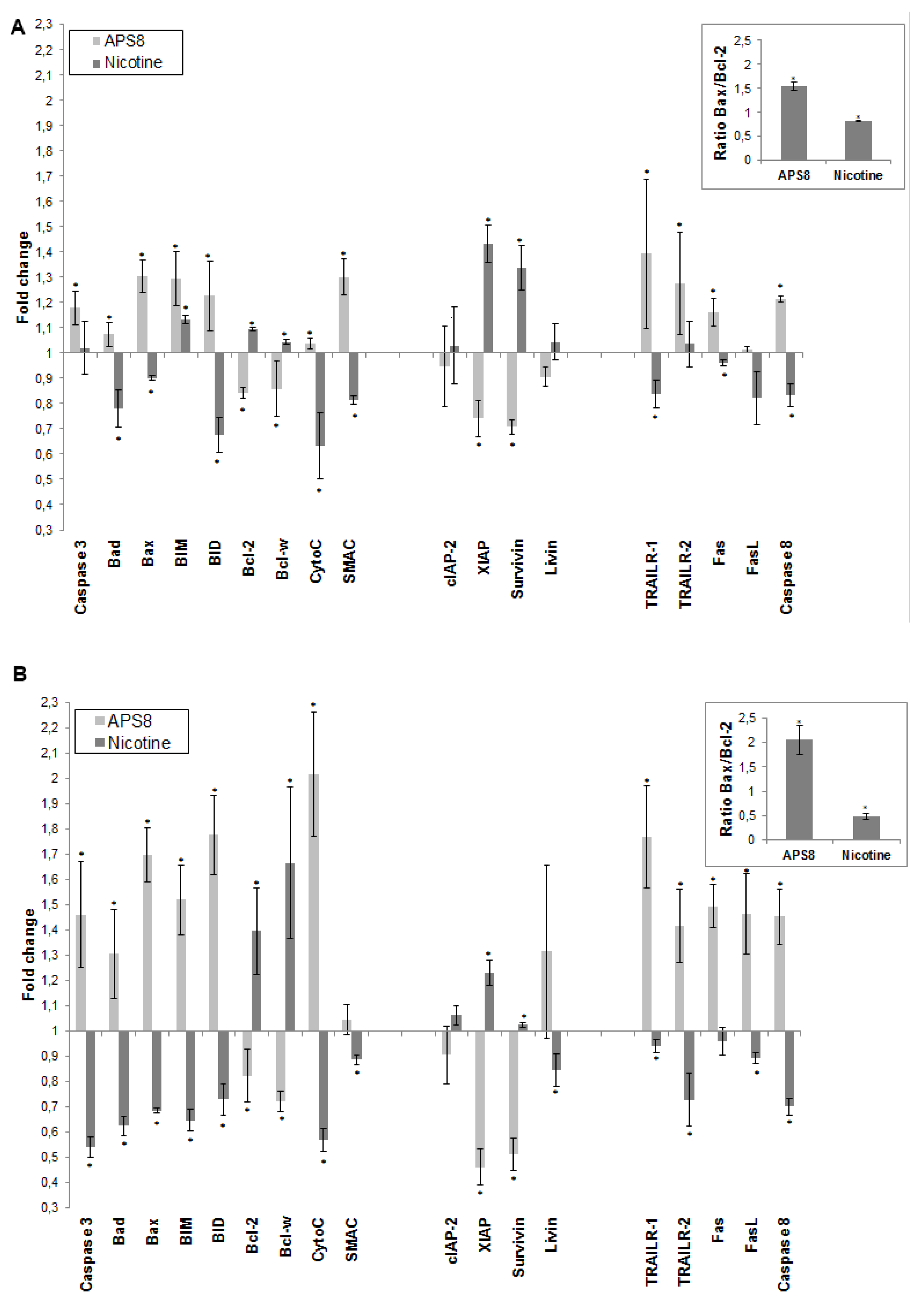
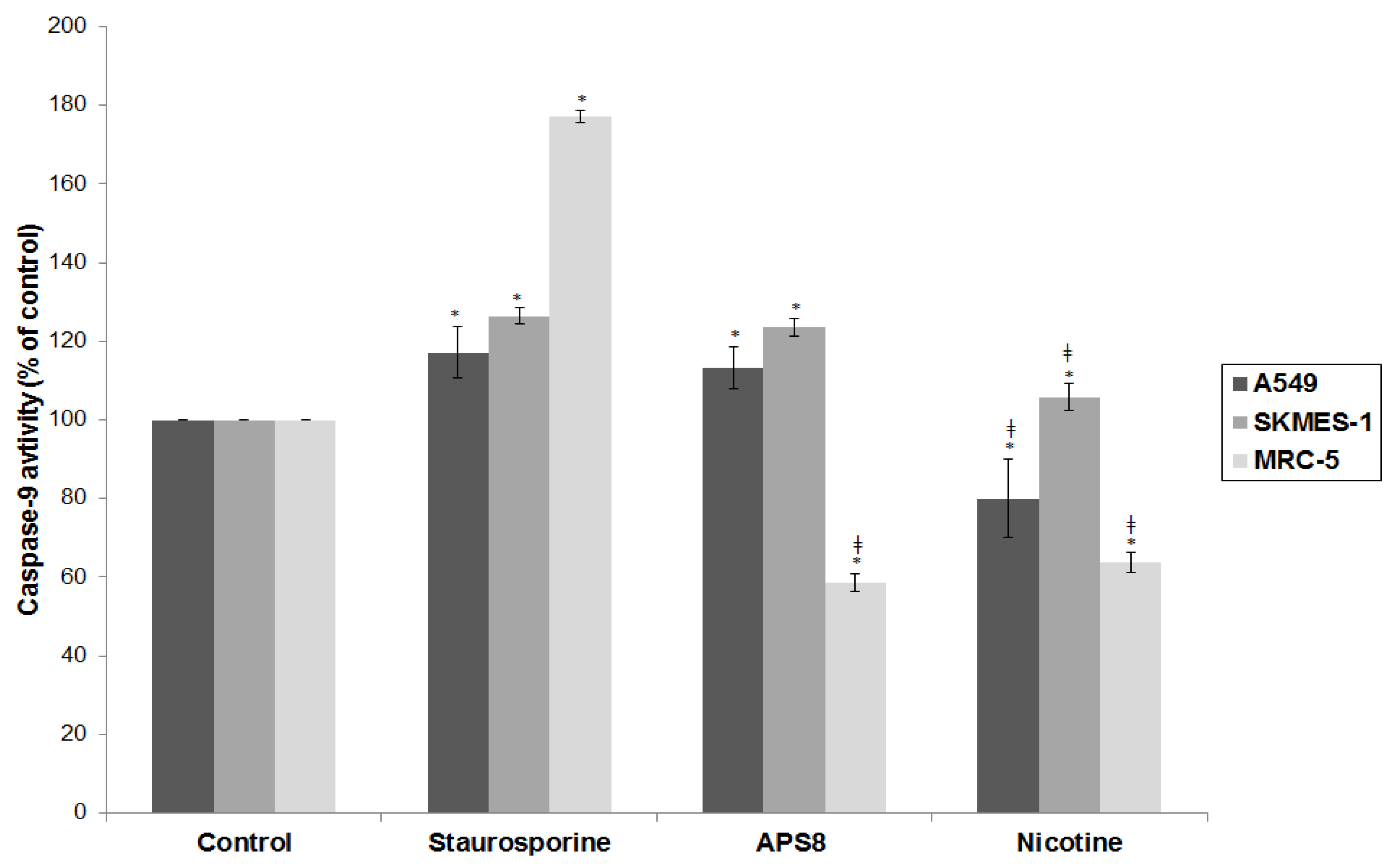
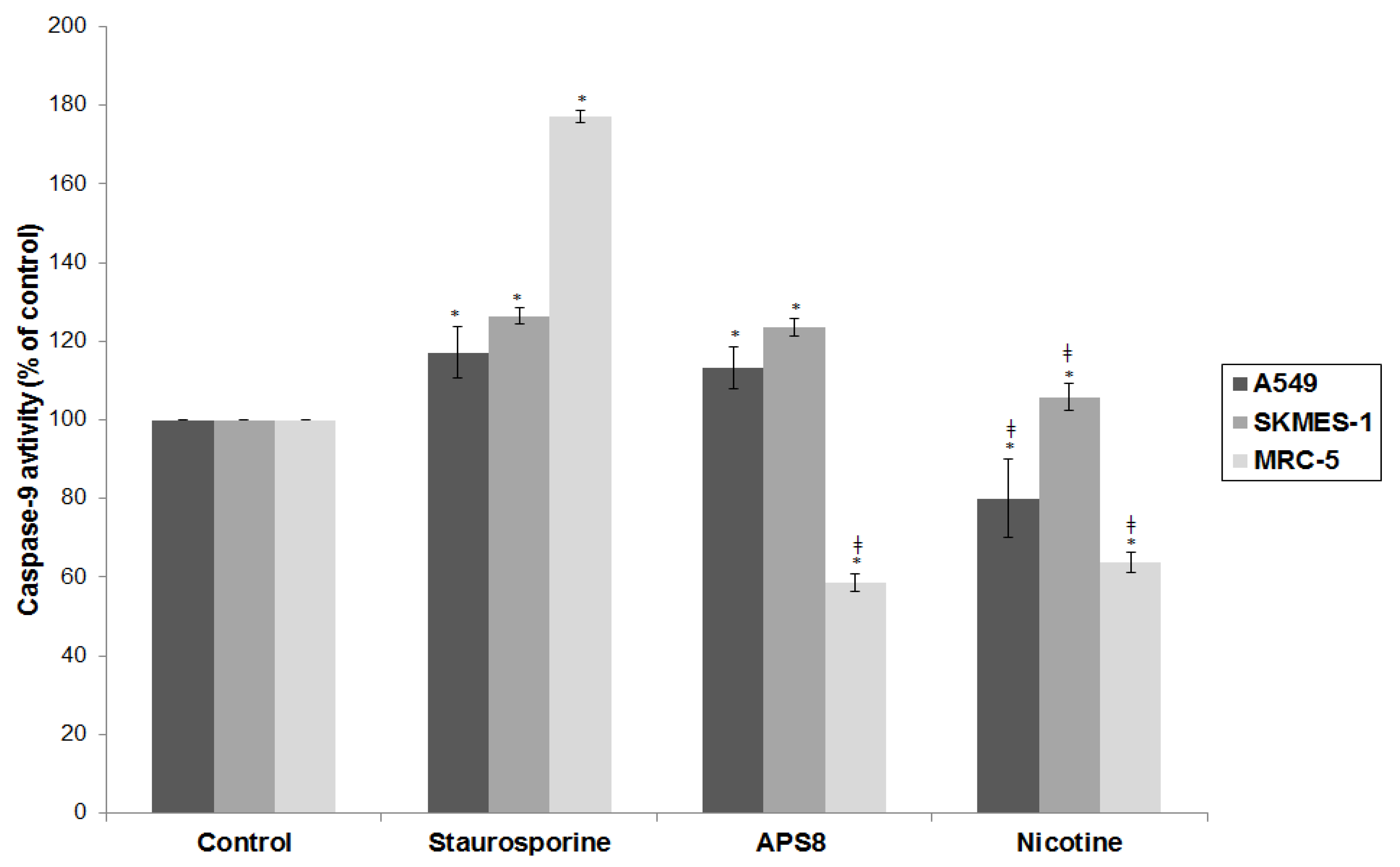
2.4. APS8 Treatment of Cancer Cell Lines Results in Activation of Caspase-9
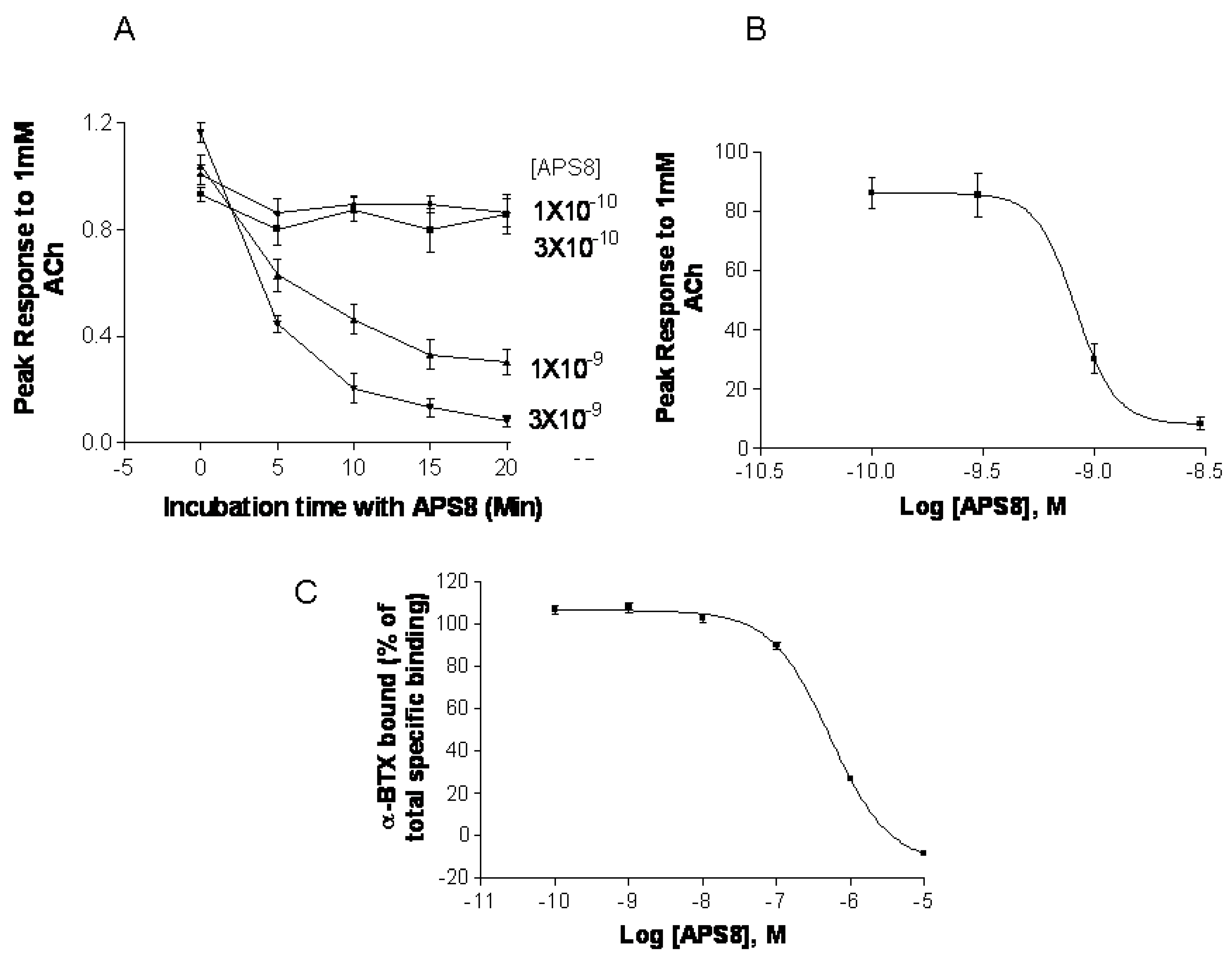
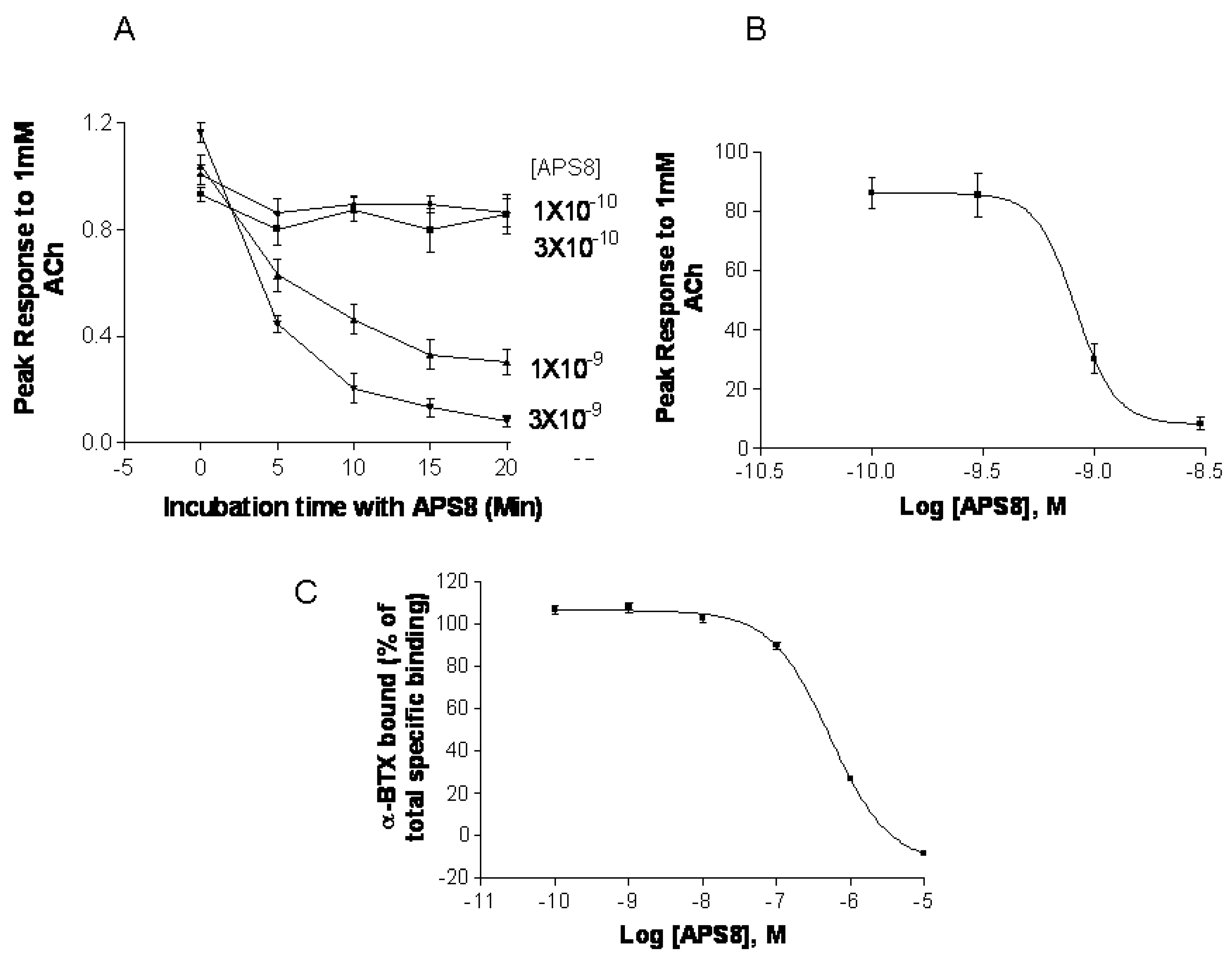
2.5. APS8 is a Negative Regulator of Human α7 nAChRs
3. Experimental Section
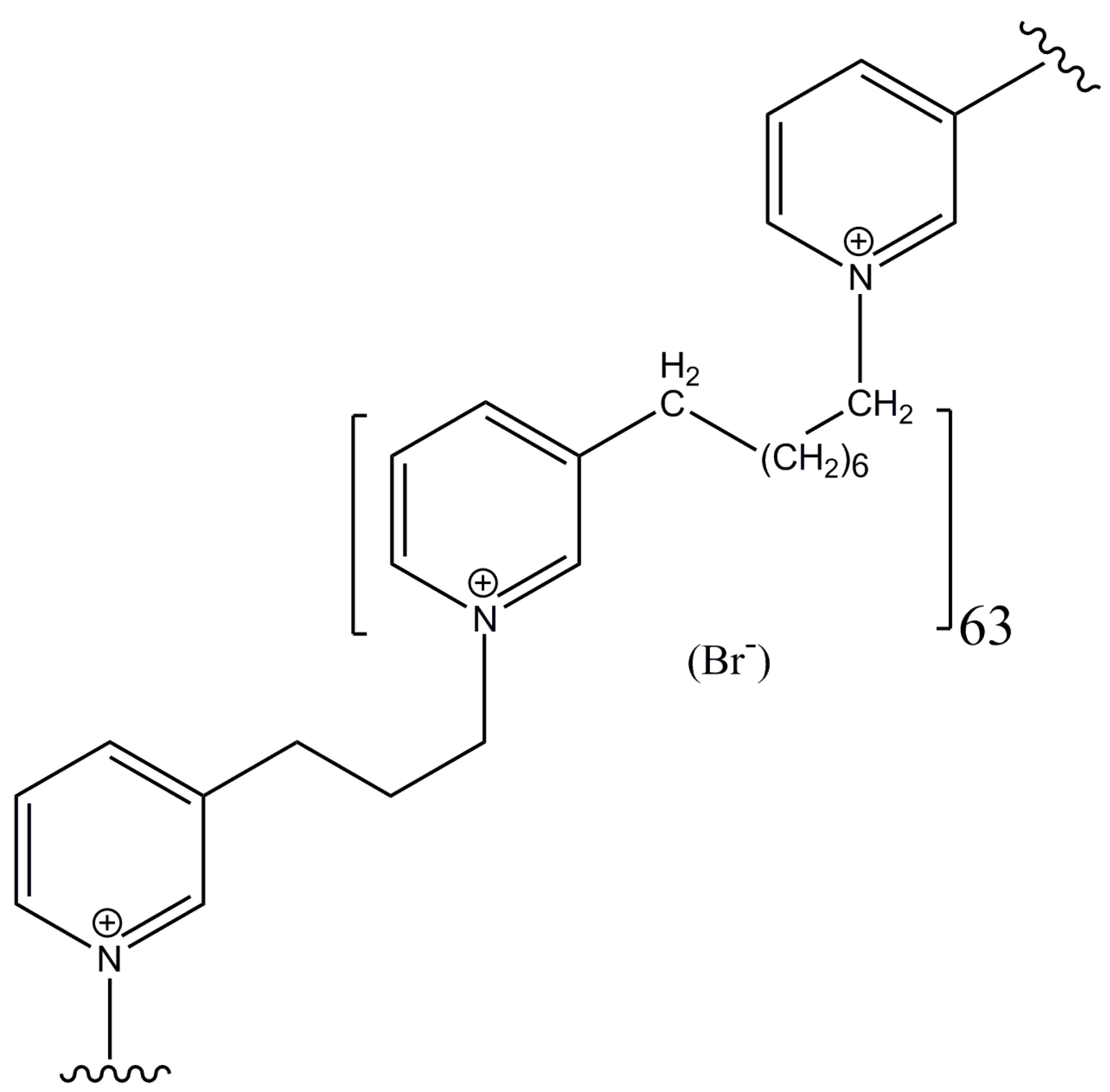
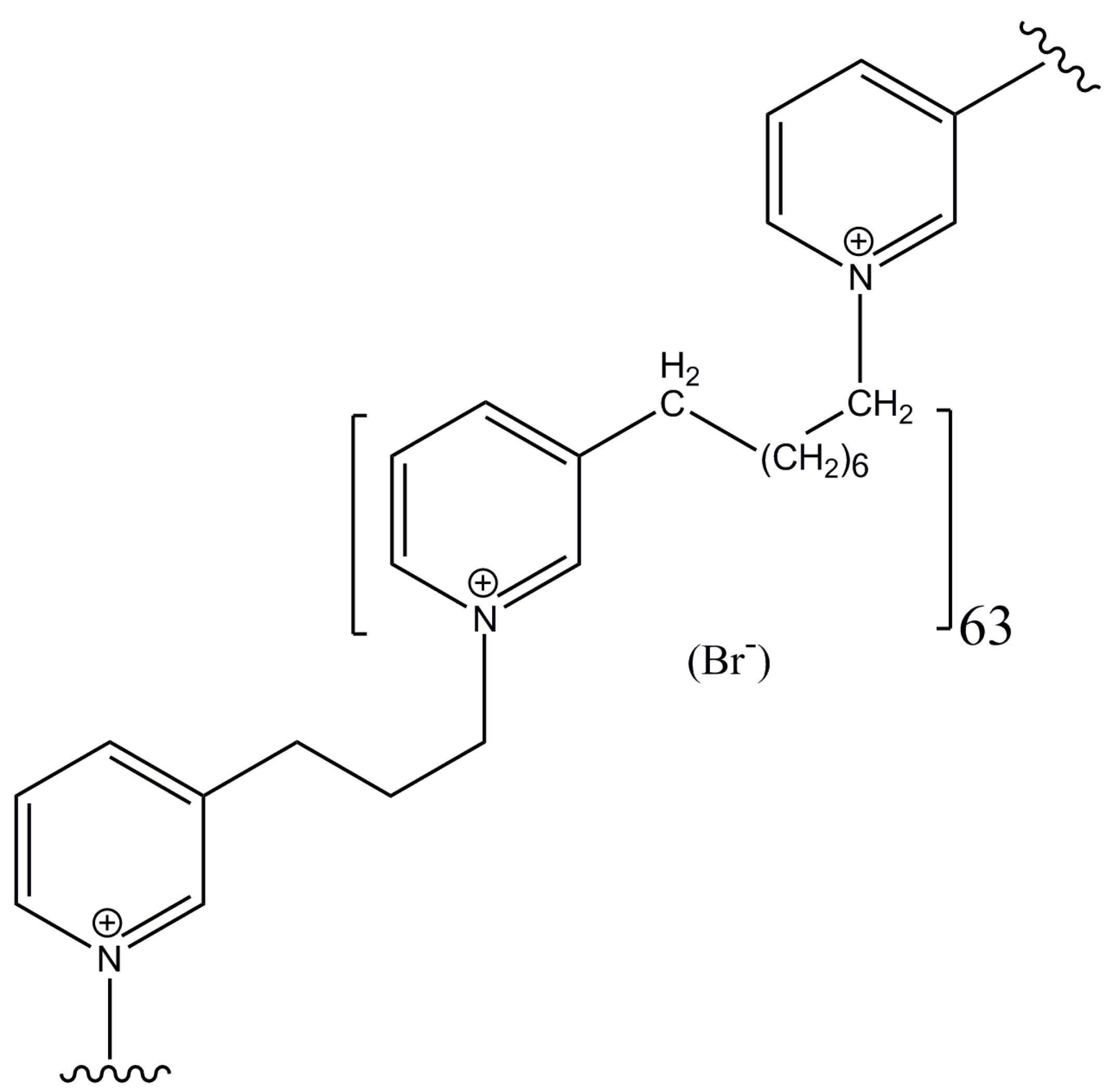
3.1. Synthetic Poly-APS Analog and Cell Cultures
3.2. Cell Viability Assay
3.3. Apoptotic Morphology Assessment
3.4. Analysis of Apoptosis by Annexin V and Propidium Iodide Staining
3.5. Mitochondrial Permeability
3.6. Expression of Pro-Apoptotic and Anti-Apoptotic Proteins
3.7. Caspase-9 Activity
3.8. Effect of APS8 on α7 nAChRs
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Singhal, S.; Vachani, A.; Antin-Ozerkis, D.; Kaiser, L.; Albelda, S.M. Prognostic implications of cell cycle, apoptosis, and angiogenesis biomarkers in non-small cell lung cancer: A review. Clin. Cancer Res. 2005, 11, 3974–3986. [Google Scholar] [CrossRef]
- Petersen, S.; Petersen, S. Towards a genetic-based classification of human lung cancer. Anal. Cell. Pathol. 2001, 22, 111–121. [Google Scholar]
- Wang, Y.; Pereira, E.F.; Maus, A.D.; Ostlie, N.S.; Navaneetham, D.; Lei, S.; Albuquerque, E.X.; Conti-Fine, B.M. Human bronchial epithelial and endothelial cells express a7 nicotinic acetylcholine receptors. Mol. Pharmacol. 2001, 60, 1201–1209. [Google Scholar]
- Plummer, H.K.; Dhar, M.; Schuller, H.M. Expression of the alpha7 nicotinic acetylcholine receptor in human lung cells. Respir. Res. 2005, 6, 29. [Google Scholar] [CrossRef]
- Russo, P.; Catassi, A.; Cesario, A.; Servent, D. Development of novel therapeutic strategies for lung cancer: Targeting the cholinergic system. Curr. Med. Chem. 2006, 13, 3493–3512. [Google Scholar] [CrossRef]
- Lam, D.C.; Girard, L.; Ramirez, R.; Chau, W.S.; Suen, W.S.; Sheridan, S.; Tin, V.P.; Chung, L.P.; Wong, M.P.; Shay, J.W.; et al. Expression of nicotinic acetylcholine receptor subunit genes in non-small-cell lung cancer reveals differences between smokers and nonsmokers. Cancer Res. 2007, 67, 4638–4647. [Google Scholar] [CrossRef]
- Schuller, H.M. Is cancer triggered by altered signaling of nicotinic acetylcholine receptors? Nat. Rev. Cancer 2009, 9, 195–205. [Google Scholar] [CrossRef]
- Egleton, R.D.; Brown, K.C.; Dasgupta, P. Nicotinic acetylcholine receptors in cancer: Multiple roles in proliferation and inhibition of apoptosis. Trends Pharmacol. Sci. 2008, 29, 151–158. [Google Scholar] [CrossRef]
- Lee, W.; Jiang, Z.; Liu, J.; Haverty, P.M.; Guan, Y.; Stinson, Y.; Yue, P.; Zhang, Y.; Pant, K.P.; Bhatt, D.; et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature 2011, 465, 473–477. [Google Scholar]
- Richman, D.P.; Arnason, B.G. Nicotinic acetylcholine receptor: Evidence for a functionally distinct receptor in human lymphocytes. Proc. Natl. Acad. Sci. USA 1979, 76, 4632–4635. [Google Scholar] [CrossRef]
- Grando, S.A.; Horton, R.M.; Pereira, E.F.; Diethelm-Okita, B.M.; George, P.M.; Albuquerque, E.X.; Conti-Fine, B.M. A nicotinic acetylcholine receptor regulating cell adhesion and motility is expressed in human keratinocytes. J. Investig. Dermatol. 1995, 105, 774–781. [Google Scholar]
- Macklin, K.D.; Maus, A.D.; Pereira, E.F.; Albuquerque, E.X.; Conti-Fine, B.M. Human vascular endothelial cells express functional nicotinic acetylcholine receptors. J. Pharmacol. Exp. Ther. 1998, 287, 435–439. [Google Scholar]
- Heeschen, C.; Jang, J.J.; Weis, M.; Pathak, A.; Kaji, S.; Hu, R.S.; Tsao, P.S.; Johnson, F.L.; Cooke, J.P. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat. Med. 2001, 7, 833–839. [Google Scholar] [CrossRef]
- Wessler, I.; Kirkpatrick, C.J. Acetylcholine beyond neurons: The nonneuronal cholinergic system in humans. Br. J. Pharmacol. 2008, 154, 1558–1571. [Google Scholar] [CrossRef]
- Carlisle, D.L.; Hopkins, T.M.; Gaither-Davis, A.; Silhanek, M.J.; Luketich, J.D.; Christie, N.A.; Siegfried, J.M. Nicotine signals through muscle-type and neuronal nicotinic acetylcholine receptors in both human bronchial epithelial cells and airway fibroblasts. Respir. Res. 2004, 5, 1–16. [Google Scholar] [CrossRef]
- Zeidler, R.; Albermann, K.; Lang, S. Nicotine and apoptosis. Apoptosis 2007, 12, 1927–1943. [Google Scholar] [CrossRef]
- Resende, R.R.; Adhikari, A. Cholinergic receptor pathways involved in apoptosis, cell proliferation and neuronal differentiation. Cell Commun. Signal. 2009, 7, 20. [Google Scholar] [CrossRef]
- Grozio, A.; Paleari, L.; Catassi, A.; Servent, D.; Cilli, M.; Piccardi, F.; Paganuzzi, M.; Cesario, A.; Granone, P.; Mourier, G.; et al. Natural agents targeting the a7-nicotinic–receptor in NSCLC: A promising prospective in anti-cancer drug development. Int. J. Cancer 2008, 122, 1911–1915. [Google Scholar]
- Al-Wadei, H.A.; Al-Wadei, M.H.; Schuller, H.M. Cooperative regulation of non-small cell lung carcinoma by nicotinic and beta-adrenergic receptors: A novel target for intervention. PLos One 2012, 7, e29915. [Google Scholar]
- Paleari, L.; Negri, E.; Catassi, A.; Cilli, M.; Servent, D.; D’Angelillo, R.; Cesario, A.; Russo, P.; Fini, M. Inhibition of nonneuronal a7-nicotinic receptor for lung treatment. Am. J. Respir. Crit. Care Med. 2009, 179, 1141–1150. [Google Scholar] [CrossRef]
- Schwartsmann, G. Marine organisms and other novel natural sources of new cancer drugs. Ann. Oncol. 2000, 11, 235–243. [Google Scholar]
- Sipkema, D.; Franssen, M.C.; Osinga, R.; Tramper, J.; Wijffels, R.H. Marine sponges as pharmacy. Mar. Biotechnol. 2005, 7, 142–162. [Google Scholar] [CrossRef]
- Mayer, A.M.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.R.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef]
- Sepčić, K.; Batista, U.; Vacelet, J.; Maček, P.; Turk, T. Biological activities of aqueous extracts from marine sponges and cytotoxic effects of 3-alkylpyridinium polymers from Reniera sarai. Comp. Biochem. Physiol. 1997, 117C, 47–53. [Google Scholar]
- Sepčić, K.; Guella, G.; Mancini, I.; Pietra, F.; Dalla Serra, M.; Menestrina, G.; Tubbs, K.; Maček, P.; Turk, T. Characterization of anticholinesterase-active 3-alkylpyridinium polymers from the marine sponge Reniera sarai in aqueous solutions. J. Nat. Prod. 1997, 60, 991–996. [Google Scholar] [CrossRef]
- Zovko, A.; Vaukner Gabrič, M.; Sepčić, K.; Pohleven, F.; Jaklič, D.; Gunde-Cimerman, N.; Lu, Z.; Edrada-Ebel, R.; Houssen, W.E.; Mancini, I.; et al. Antifungal and antibacterial activity of 3-alkylpyridinium polymeric analogues of marine toxins. Int. Biodeterior. Biodegrad. 2012, 68, 71–77. [Google Scholar] [CrossRef]
- Turk, T.; Frangež, R.; Sepčić, K. Mechanisms of toxicity of 3-alkylpyridinium polymers from marine sponge Reniera sarai. Mar Drugs. 2007, 5, 157–167. [Google Scholar] [CrossRef]
- Paleari, L.; Trombino, S.; Falugi, C.; Gallus, L.; Carlone, S.; Angelini, C.; Sepčić, K.; Turk, T.; Faimali, M.; Noonan, D.M.; et al. Marine sponge-derived polymeric alkylpyridinium salts as a novel tumor chemotherapeutic targeting the cholinergic system in lung tumors. Int. J. Oncol. 2006, 29, 1381–1388. [Google Scholar]
- Houssen, W.E.; Lu, Z.; Edrada-Ebel, R.A.; Chatzi, C.; Tucker, S.J.; Sepčić, K.; Turk, T.; Zovko, A.; Shen, S.; Mancini, I.; et al. Chemical synthesis and biological activities of 3-alkyl pyridinium polymeric analogues of marine toxins. J. Chem. Biol. 2010, 3, 113–125. [Google Scholar] [CrossRef]
- Grandič, M.; Sepčić, K.; Turk, T.; Juntes, P.; Frangež, R. In vivo toxic and lethal cardiovascular effects of a synthetic polymeric 1,3-dodecylpyridinium salt in rodents. Toxicol. Appl. Pharmacol. 2011, 255, 86–93. [Google Scholar] [CrossRef]
- Grandič, M.; Frangež, R. Veterinary Faculty, University of Ljubljana: Ljubljana, Slovenia, 2012; Unpublished work.
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef]
- Lau, J.K.; Brown, K.C.; Thornhill, B.A.; Crabtree, C.M.; Dom, A.M.; Witte, T.W.; Hardman, W.E.; McNees, C.A.; Stover, C.A.; Carpenter, A.B.; et al. Inhibition of cholinergic signaling causes apoptosis in human bronchioalveolar carcinoma. Cancer Res. 2013, 73, 1328–1339. [Google Scholar] [CrossRef]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef]
- Jin, Z.; Xin, M.; Deng, X. Survival function of protein kinase Cι as a novel nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-activated bad kinase. J. Biol. Chem. 2005, 280, 16045–16052. [Google Scholar] [CrossRef]
- Zhang, Q.; Tang, X.; Zhang, Z.F.; Velikina, R.; Shi, S.; Le, A.D. Nicotine induces hypoxia-inducible factor-1alpha expression in human lung cancer cells via nicotinic acetylcholine receptor-mediated signaling pathways. Clin. Cancer Res. 2007, 13, 4686–4694. [Google Scholar] [CrossRef]
- Ray, M.R.; Jablons, D.; He, B. Lung cancer therapeutics that target signaling pathways: An update. Expert Rev. Respir. Med. 2010, 4, 631–645. [Google Scholar] [CrossRef]
- Dasgupta, P.; Rastogi, S.; Pillai, S.; Ordonez-Ercan, D.; Morris, M.; Haura, E.; Chellappan, S. Nicotine induces cell proliferation by beta-arrestin-mediated activation of Src and Rb-Raf-1 pathways. J. Clin. Investig. 2006, 116, 2208–2217. [Google Scholar] [CrossRef]
- Xin, M.; Deng, X. Nicotine inactivation of the proapoptotic function of bax through phosphorilation. J. Biol. Chem. 2005, 280, 10781–10784. [Google Scholar] [CrossRef]
- Le Novere, N.; Changeux, J.P. Molecular evolution of the nicotinic acetylcholine receptor: An example of multigene family in excitable cells. J. Mol. Evol. 1995, 40, 155–172. [Google Scholar] [CrossRef]
- Reed, J.C. Proapoptotic multidomain bcl-2/bax-family proteins: Mechanisms, physiological roles and therapeutic opportunities. Cell Death Differ. 2006, 13, 1378–1386. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- LaCasse, E.C.; Mahoney, D.J.; Cheung, H.H.; Plenchette, S.; Baird, S.; Korneluk, R.G. IAP-targeted therapies for cancer. Oncogene 2008, 27, 6252–6275. [Google Scholar] [CrossRef]
- Dasgupta, P.; Kinkade, R.; Joshi, B.; DeCook, C.; Haura, E.; Chellappan, S. Nicotine inhibits apoptosis induced by chemotherapeutic drugs by up-regulating XIAP and survivin. Proc. Natl. Acad. Sci. USA 2006, 103, 6332–6337. [Google Scholar]
- Wallach, D.; Varfolomeev, E.E.; Malinin, N.L.; Goltsev, Y.V.; Kovalenko, A.V.; Boldin, M.P. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu. Rev. Immunol. 1999, 17, 331–367. [Google Scholar] [CrossRef]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of bid by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Giard, D.J.; Aaronson, S.-A.; Todaro, G.J.; Arnstein, P.; Kersey, J.H.; Dosik, H.; Parks, W.P. In vitro cultivation of human tumors: Establishment of cell lines derived from a series of solid tumors. J. Natl. Cancer Inst. 1973, 51, 1417–1423. [Google Scholar]
- Jacobs, J.P.; Jones, C.M.; Baille, J.P. Characteristics of a human diploid cell designated MRC-5. Nature 1970, 227, 168–170. [Google Scholar] [CrossRef]
- Fogh, J. Human Tumor Cells in Vitro; Plenum Press: New York, NY, USA, 1975; pp. 115–141. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Kem, W.R.; Mahnir, V.M.; Prokai, L.; Papke, R.M.; Cao, X.F.; LeFrancois, S.; Wildeboer, K.; Porter-Papke, J.; Prokai-Tatrai, K.; Soti, F. Hydroxy metabolites of the Alzheimer’s drug candidate DMXBA (GTS-21): Their interactions with brain nicotinic receptors.; and brain penetration. Mol. Pharmacol. 2004, 65, 56–67. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zovko, A.; Viktorsson, K.; Lewensohn, R.; Kološa, K.; Filipič, M.; Xing, H.; Kem, W.R.; Paleari, L.; Turk, T. APS8, a Polymeric Alkylpyridinium Salt Blocks α7 nAChR and Induces Apoptosis in Non-Small Cell Lung Carcinoma. Mar. Drugs 2013, 11, 2574-2594. https://doi.org/10.3390/md11072574
Zovko A, Viktorsson K, Lewensohn R, Kološa K, Filipič M, Xing H, Kem WR, Paleari L, Turk T. APS8, a Polymeric Alkylpyridinium Salt Blocks α7 nAChR and Induces Apoptosis in Non-Small Cell Lung Carcinoma. Marine Drugs. 2013; 11(7):2574-2594. https://doi.org/10.3390/md11072574
Chicago/Turabian StyleZovko, Ana, Kristina Viktorsson, Rolf Lewensohn, Katja Kološa, Metka Filipič, Hong Xing, William R. Kem, Laura Paleari, and Tom Turk. 2013. "APS8, a Polymeric Alkylpyridinium Salt Blocks α7 nAChR and Induces Apoptosis in Non-Small Cell Lung Carcinoma" Marine Drugs 11, no. 7: 2574-2594. https://doi.org/10.3390/md11072574
APA StyleZovko, A., Viktorsson, K., Lewensohn, R., Kološa, K., Filipič, M., Xing, H., Kem, W. R., Paleari, L., & Turk, T. (2013). APS8, a Polymeric Alkylpyridinium Salt Blocks α7 nAChR and Induces Apoptosis in Non-Small Cell Lung Carcinoma. Marine Drugs, 11(7), 2574-2594. https://doi.org/10.3390/md11072574