Trypanocidal Activity of Marine Natural Products



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
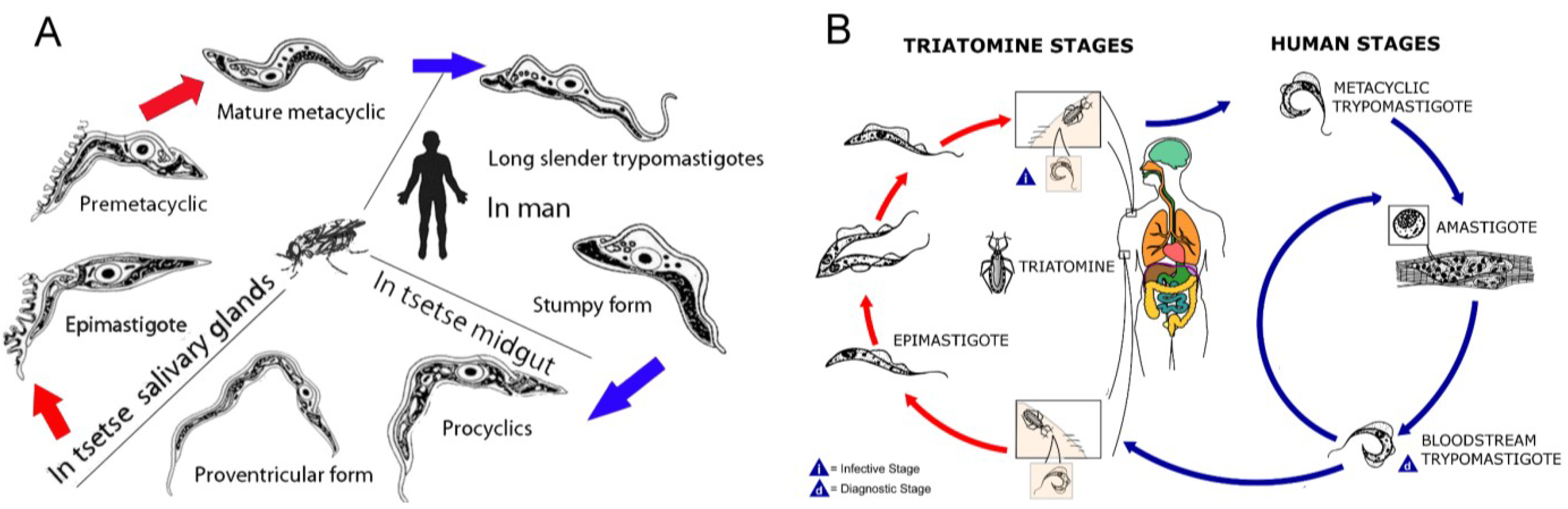
:1. Introduction
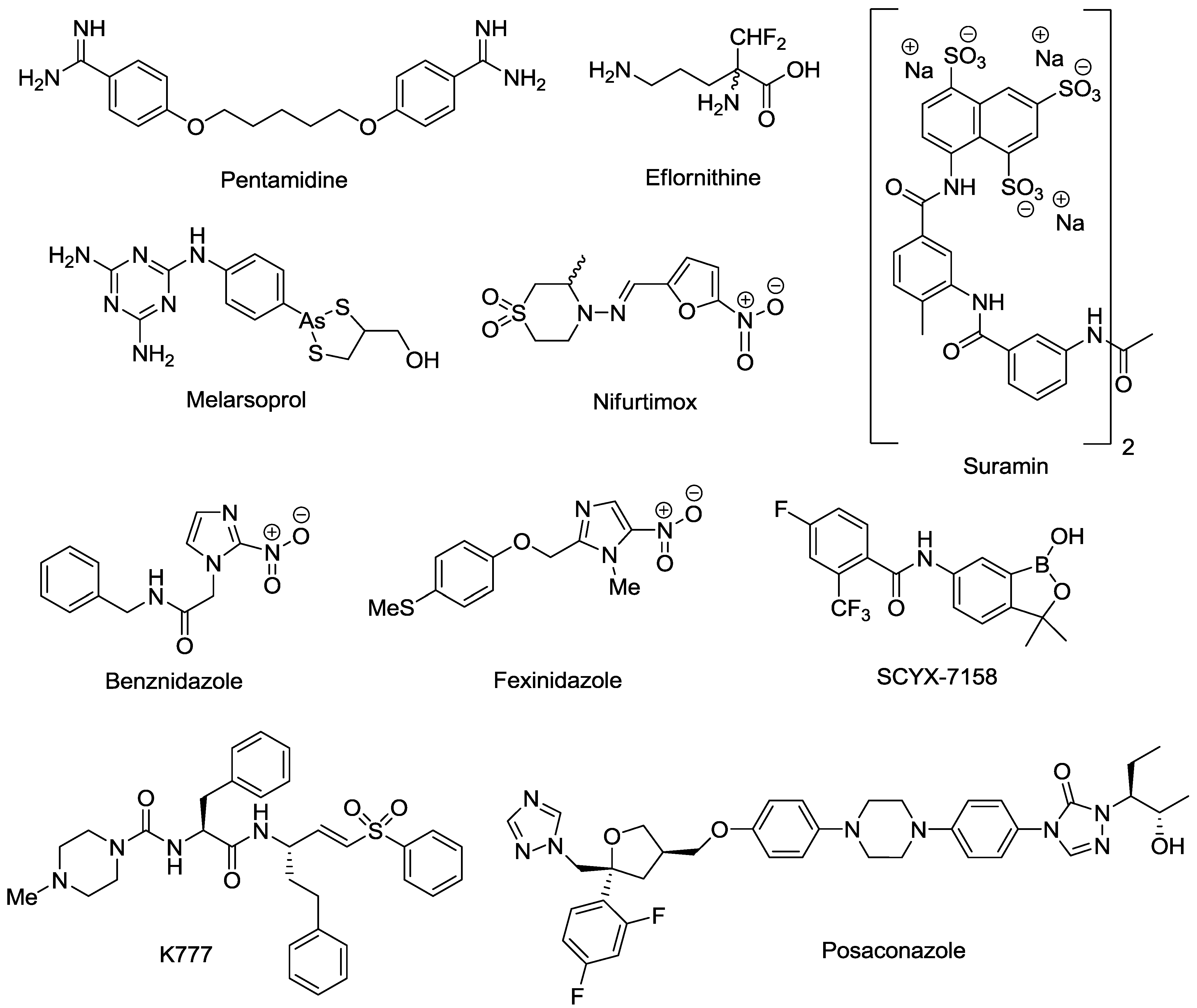
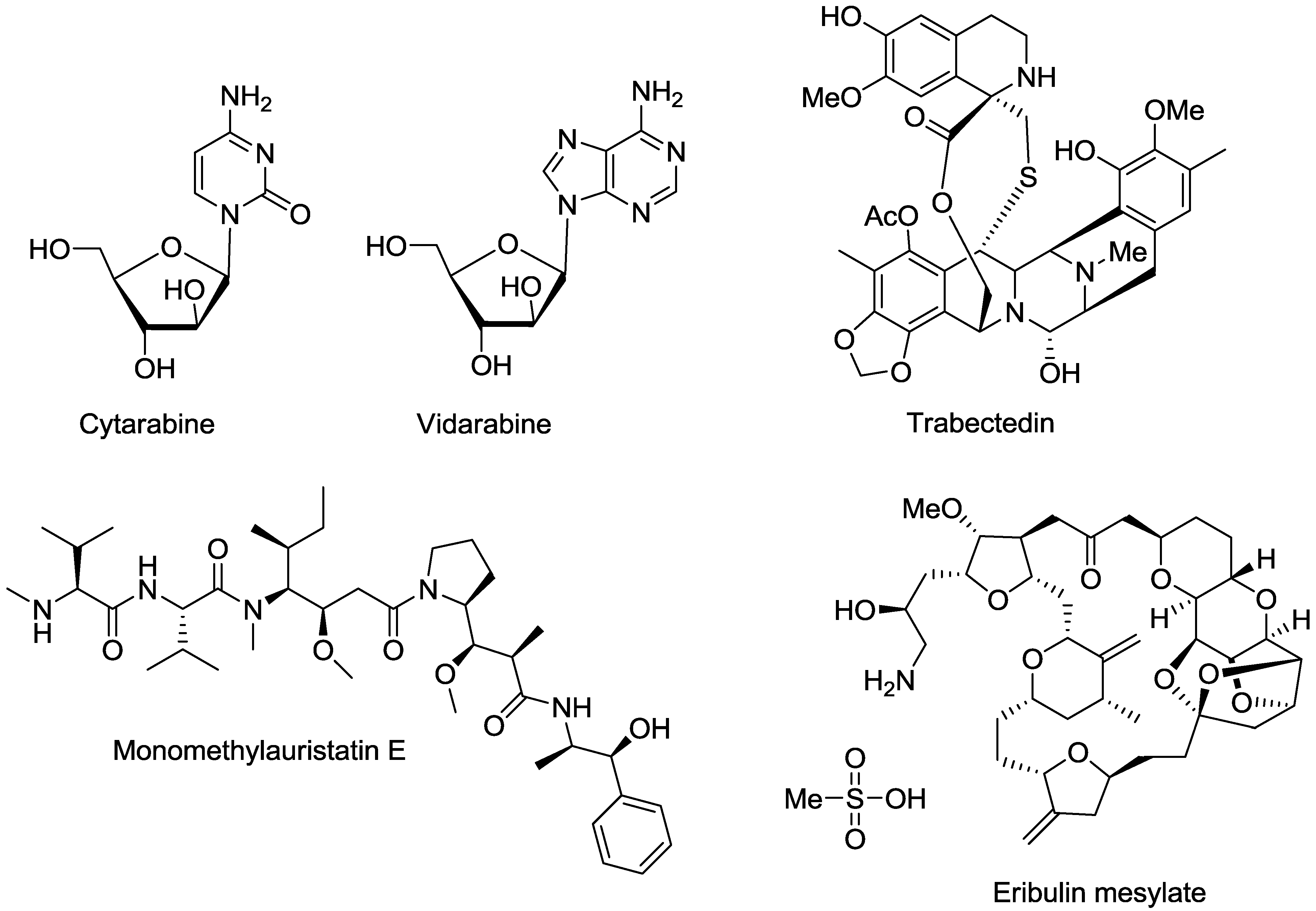
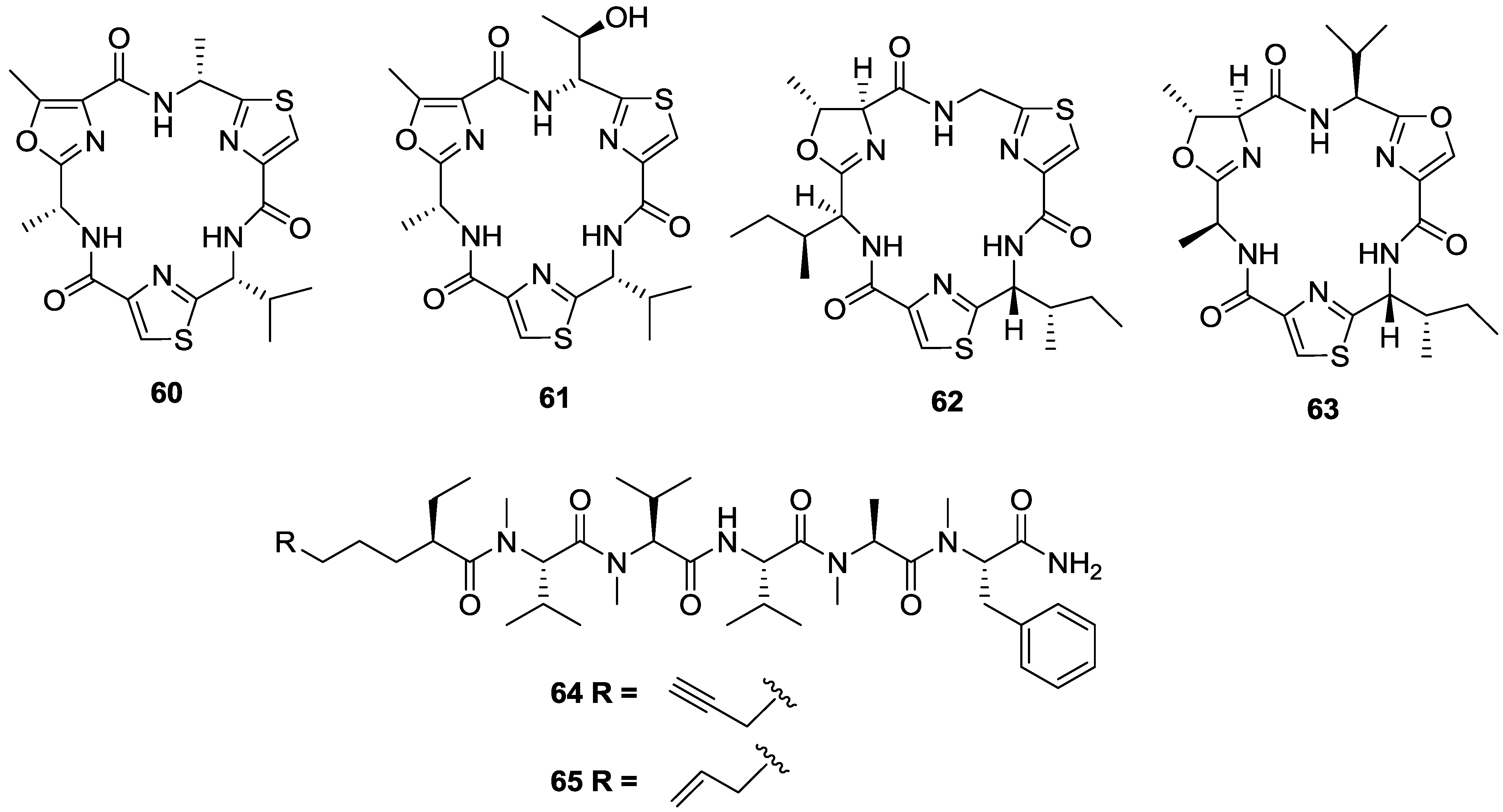
2. Marine Natural Products with Reported in Vitro Activity against the Trypanosome Species T. cruzi, T. brucei or T.b. rhodesiense
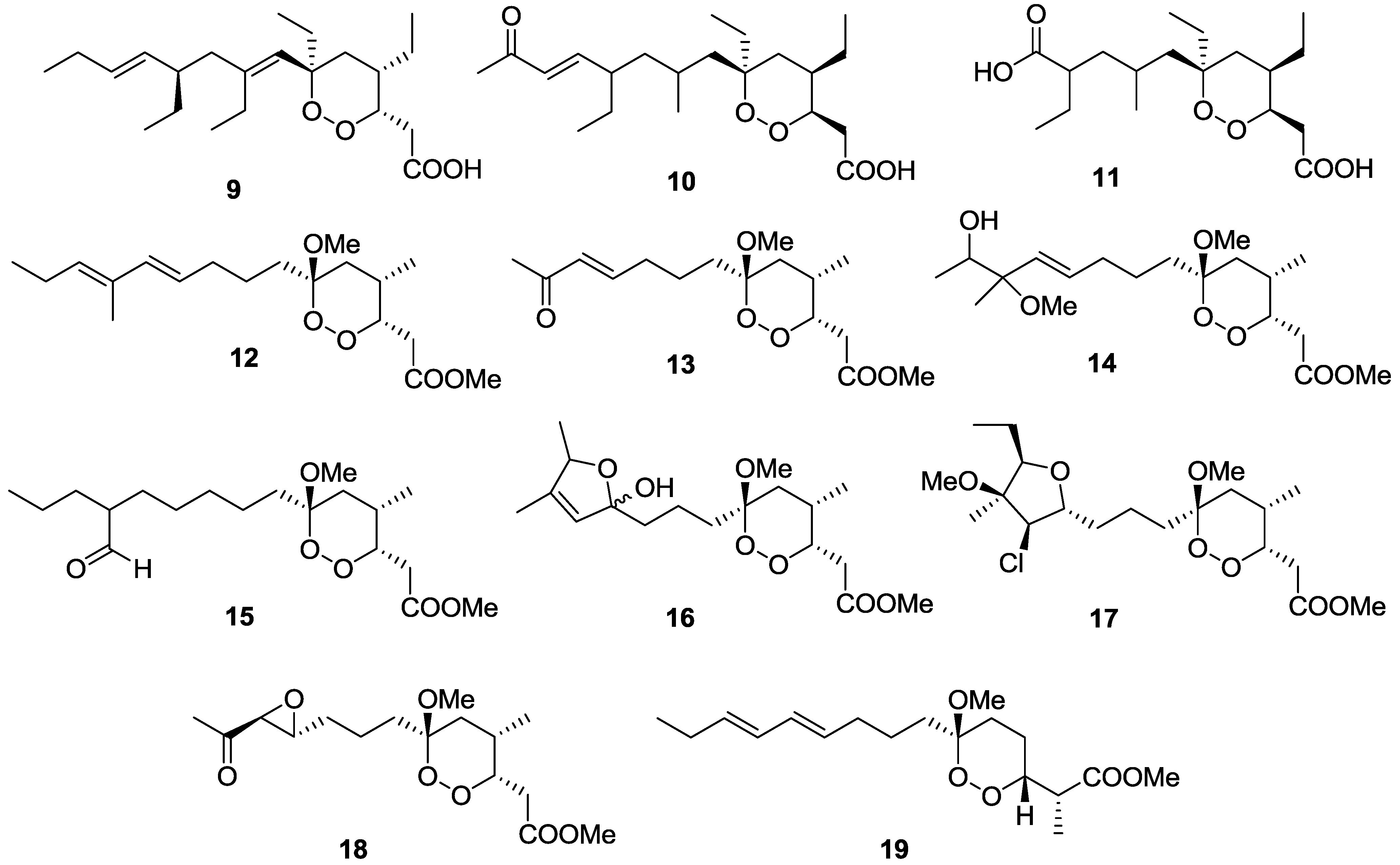
2.1. Terpenes
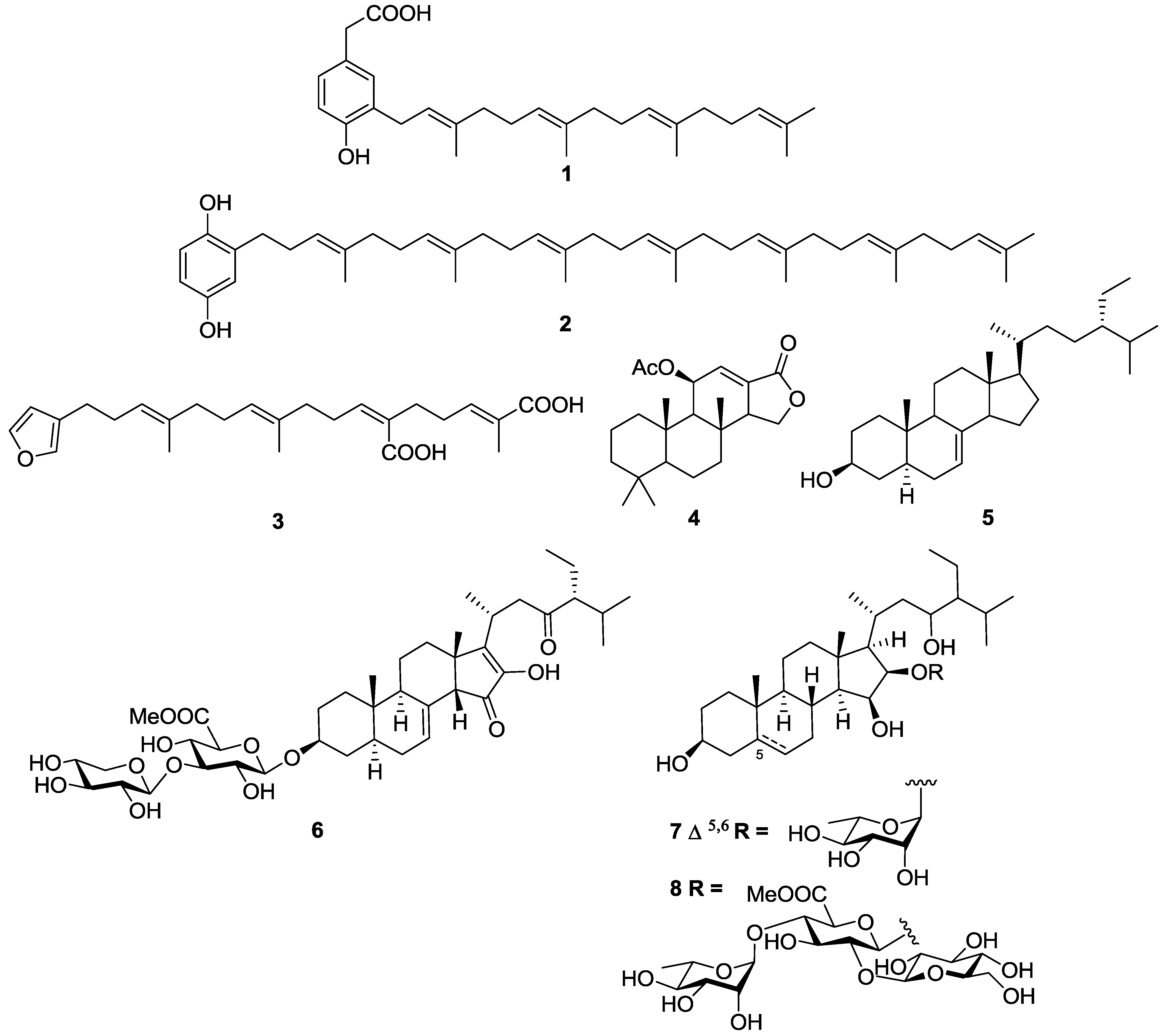
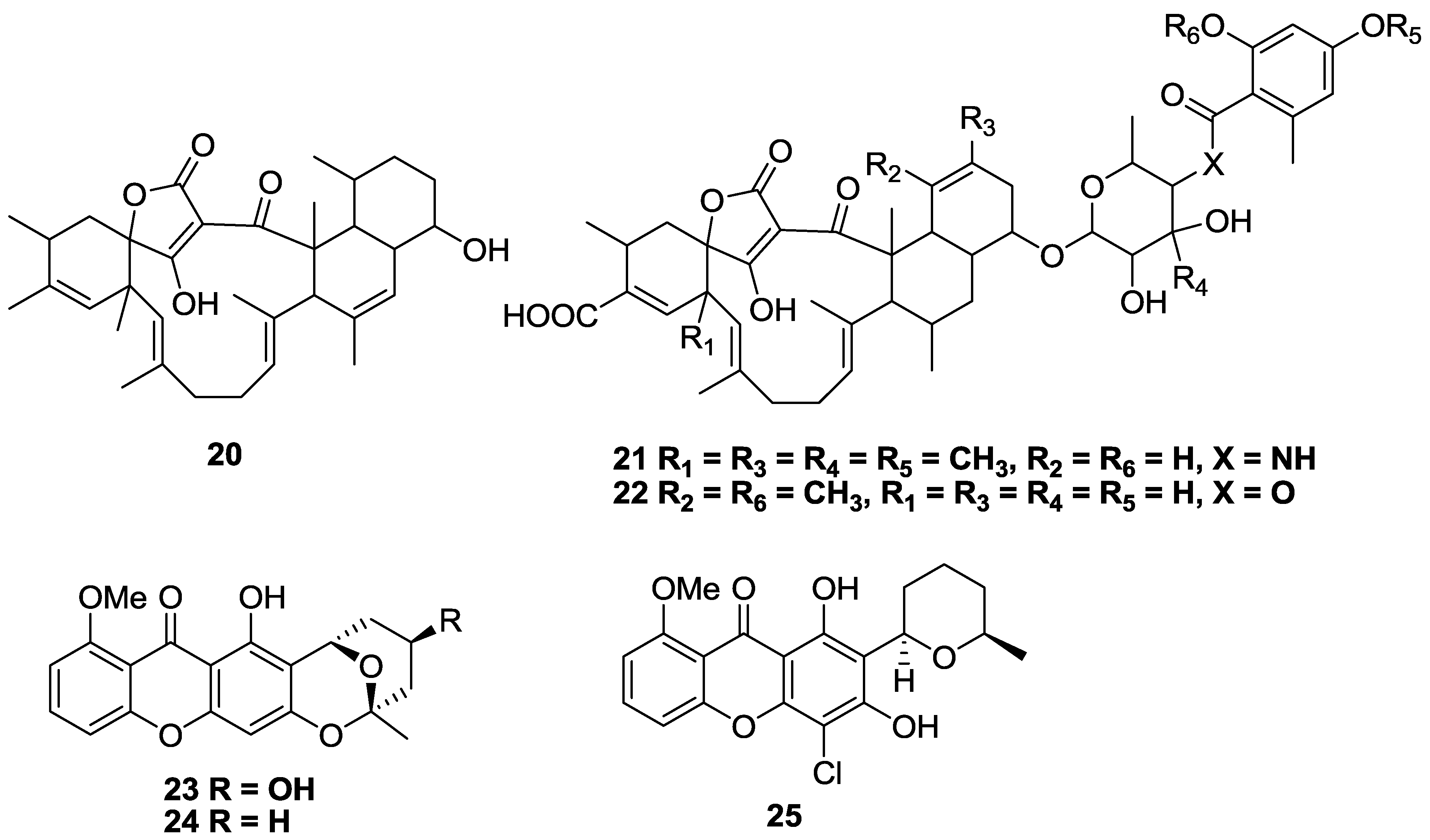
2.2. Polyketides and Xanthones

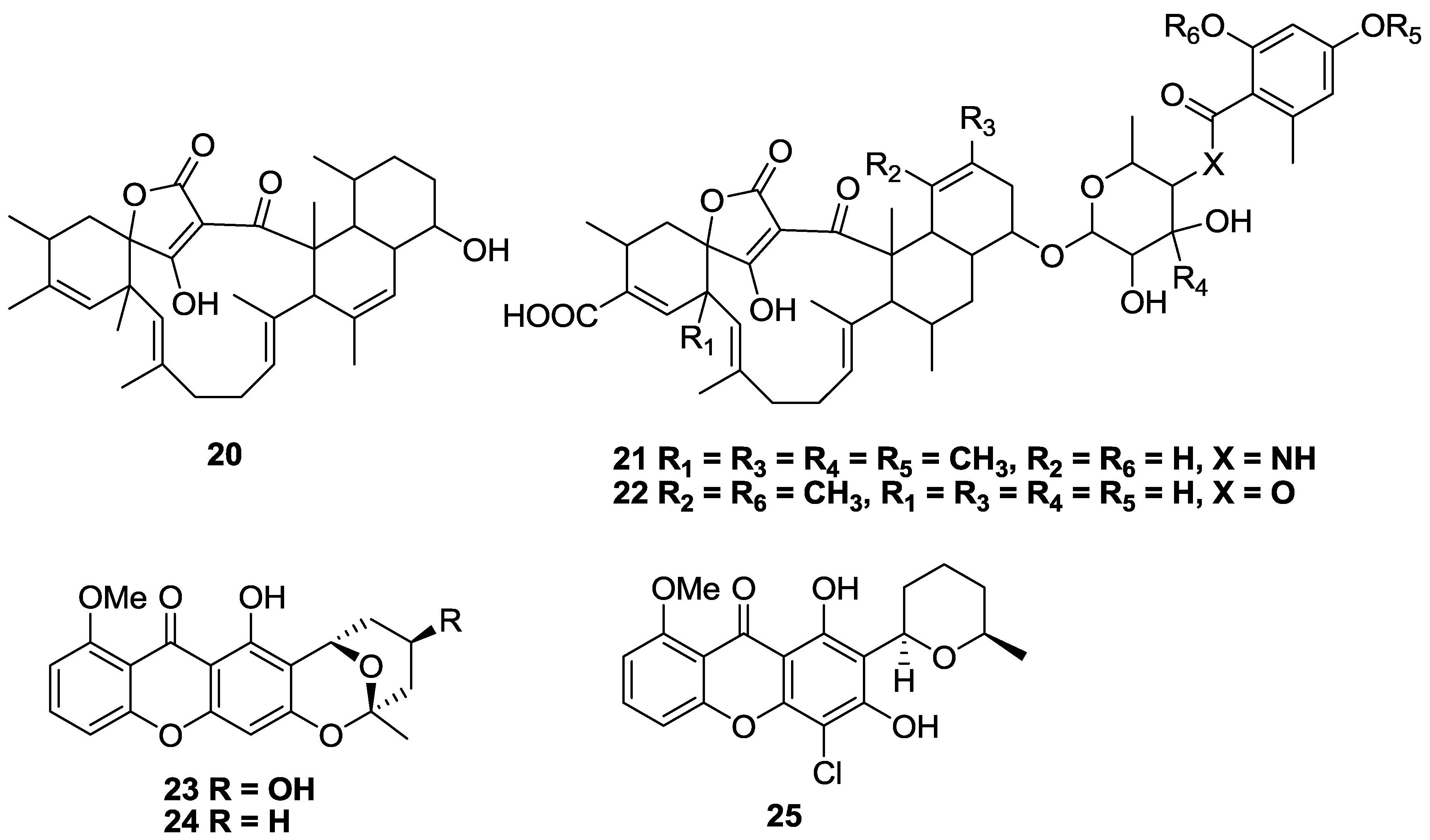
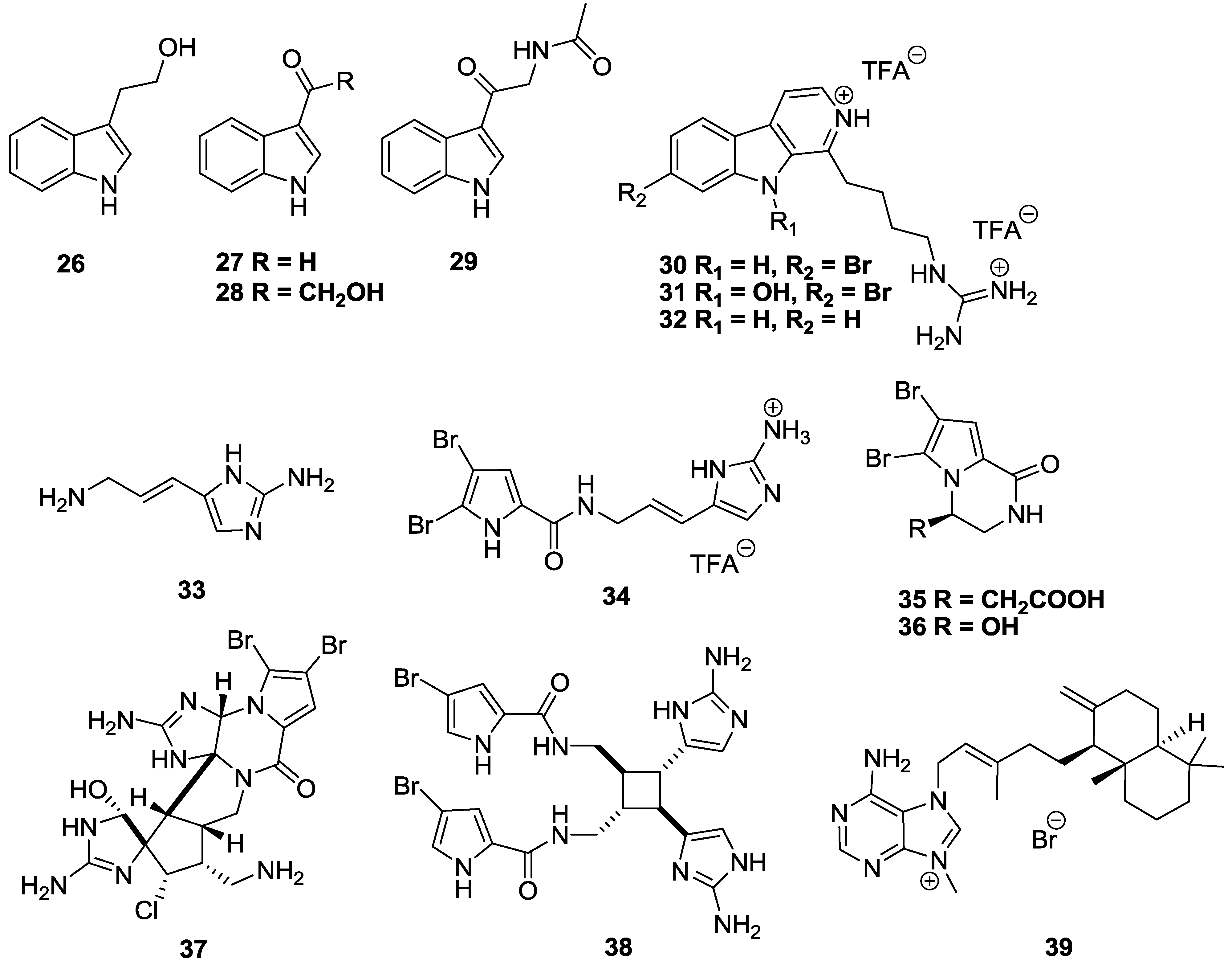
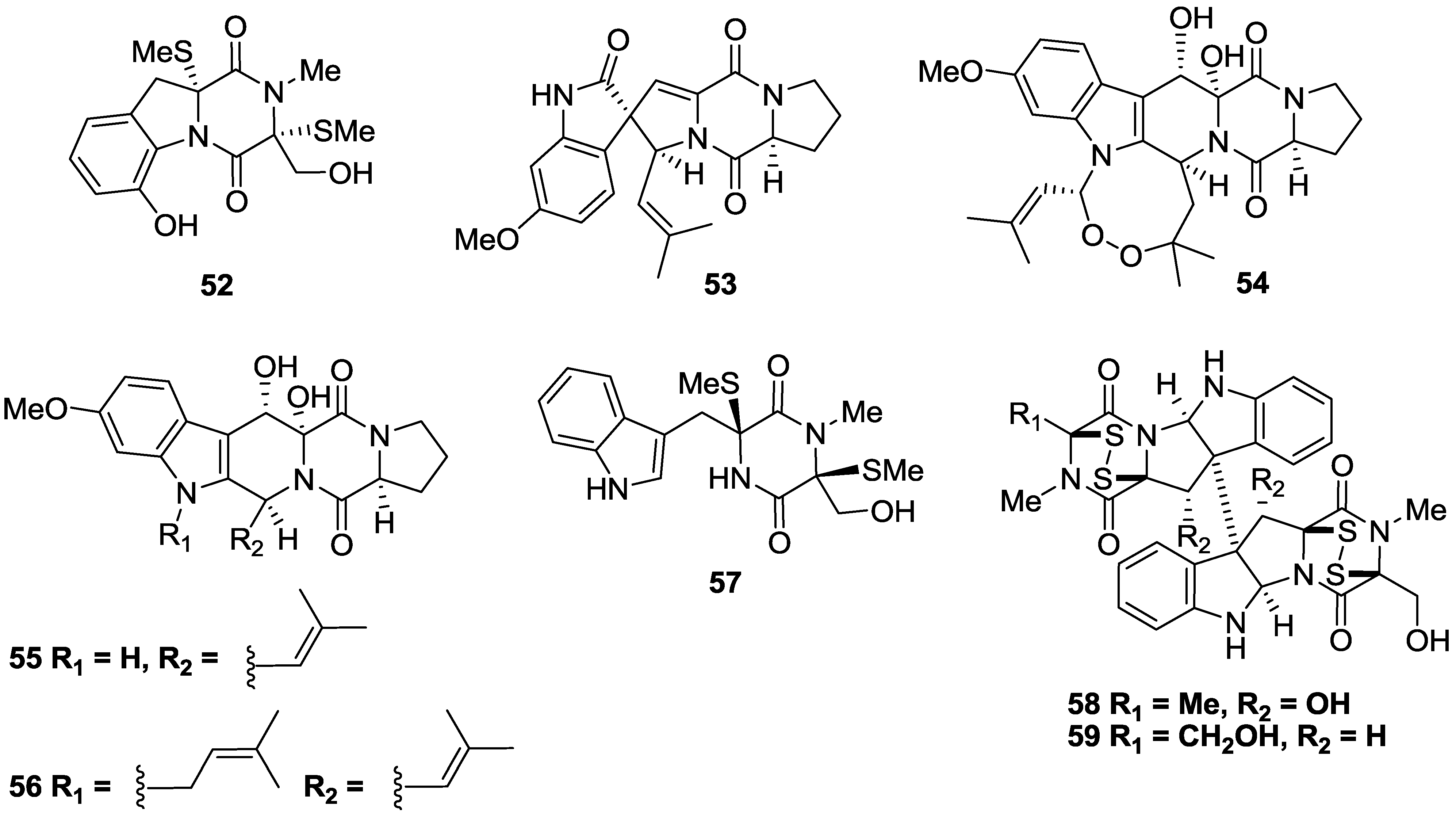
2.3. Alkaloids
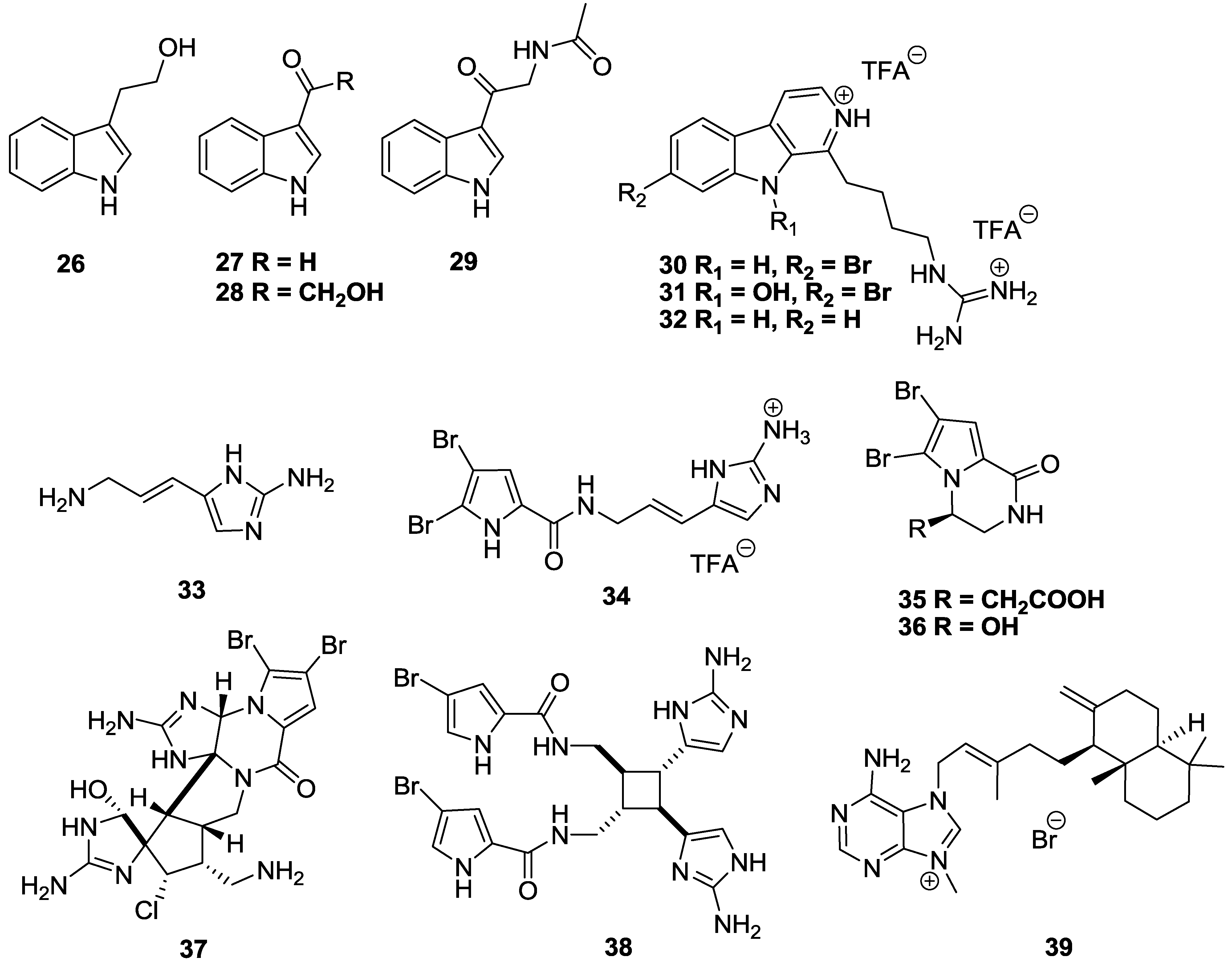
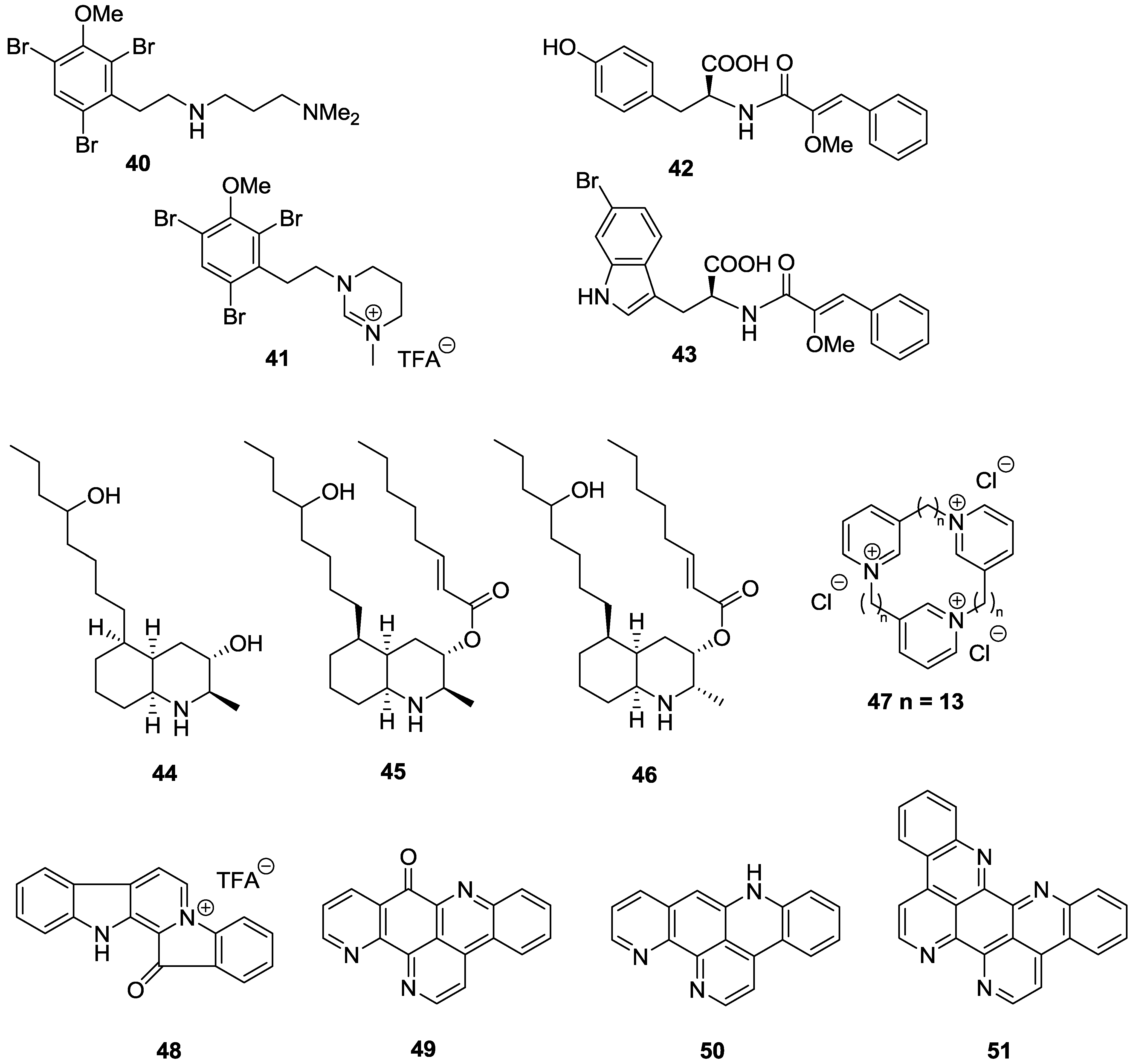

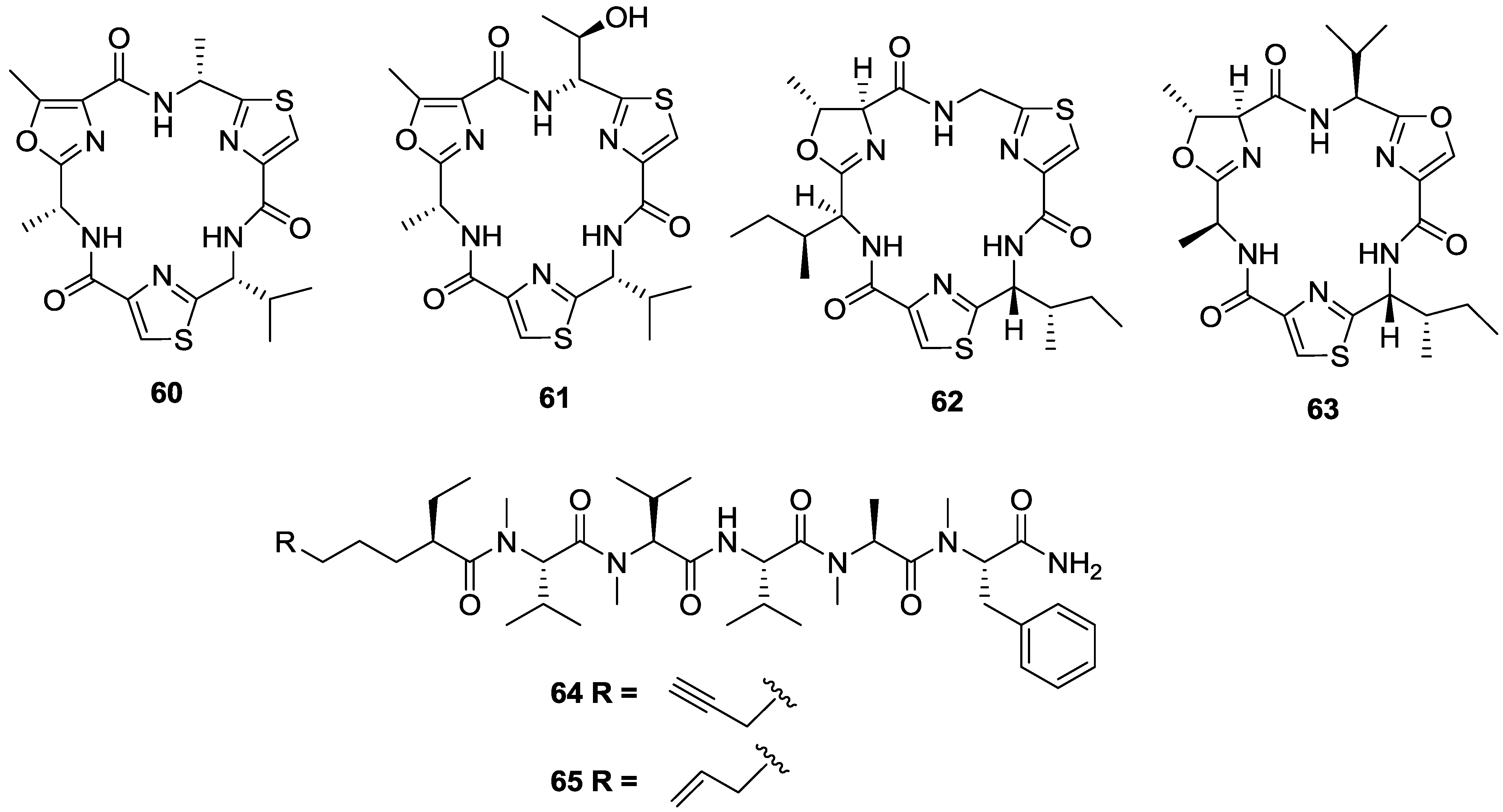
3. Conclusions
Conflicts of Interest
References
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the global burden of disease study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Murray, C.J.L.; Vos, T.; Lozano, R.; Naghavi, M.; Flaxman, A.D.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: A systematic analysis for the global burden of disease study 2010. Lancet 2012, 380, 2197–2223. [Google Scholar] [CrossRef]
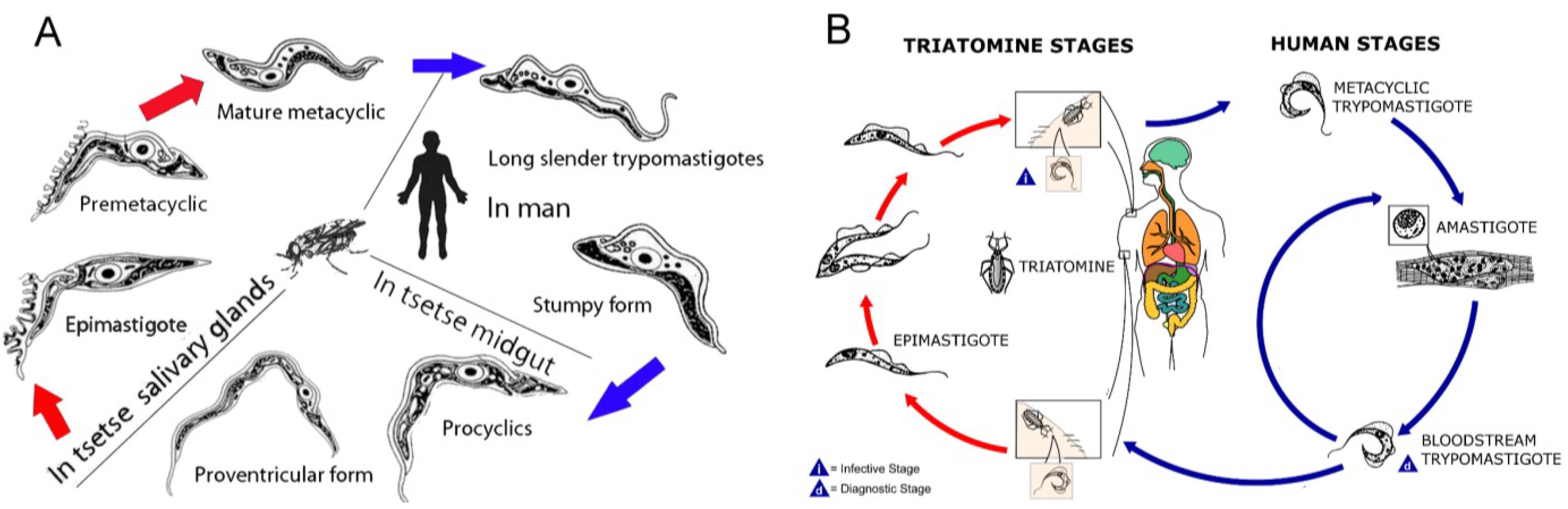
- Pepin, J.; Meda, H.A. The epidemiology and control of human African trypanosomiasis. Adv. Parasitol. 2001, 49, 71–132. [Google Scholar] [CrossRef]
- Zeledon, R.; Rabinovich, J.E. Chagas disease: An ecological appraisal with special emphasis on its insect vectors. Annu. Rev. Entomol. 1981, 26, 101–133. [Google Scholar] [CrossRef]
- Simarro, P.P.; Diarra, A.; Ruiz Postigo, J.A.; Franco, J.R.; Jannin, J.G. The human African trypanosomiasis control and surveillance programme of the World Health Organization 2000–2009: The way forward. PLoS Negl. Trop. Dis. 2011, 5, e1007. [Google Scholar] [CrossRef]
- Moncayo, A.; Silveira, A.C. Current epidemiological trends for Chagas disease in Latin America and future challenges in epidemiology, surveillance and health policy. Mem. Inst. Oswaldo Cruz 2009, 104 (Suppl. 1), 17–30. [Google Scholar]
- Apted, F.I.C.; Mulligan, H.W. Clinical manifestations and diagnosis of sleeping sickness. In The African Trypanosomiases; George Allen and Unwin LTD: London, UK, 1970; pp. 661–683. [Google Scholar]
- Atouguia, J.M.; Kennedy, P.G.E.; Davis, L.E. Neurological aspects of human African trypanosomiasis. In Infectious Diseases of the Nervous System; Davis, L.E., Kennedy, P.G.E., Eds.; Butterworth-Heinemann: Oxford, UK, 2000; pp. 321–372. [Google Scholar]
- Galfand, M. Transitory neurological signs in sleeping sickness. Trans. R. Soc. Trop. Med. Hyg. 1947, 41, 255–258. [Google Scholar] [CrossRef]
- Lundkvist, G.B.; Kristensson, K.; Bentivoglio, M. Why trypanosomes cause sleeping sickness. Physiology 2004, 19, 198–206. [Google Scholar] [CrossRef]
- Human African trypanosomiasis (sleeping sickness). World Health Organisation Fact Sheet 259. Available online: http://www.who.int/mediacentre/factsheets/fs259/en/ (accessed on 9 September 2013).
- Rassi, A., Jr.; Rassi, A.; Marin-Neto, J.A. Chagas disease. Lancet 2010, 375, 1388–1402. [Google Scholar] [CrossRef]
- Rassi, A.; Rezende, J.M.; Luquetti, A.O. Clinical phases and forms of Chagas disease. In American Trypanosomiasis (Chagas Disease). One Hundred Years of Research, 1st ed.; Telleria, J., Tibayrenc, M., Eds.; Elsevier: Burlington, MA, USA, 2010; pp. 709–741. [Google Scholar]
- Munoz-Saravia, S.G.; Haberland, A.; Wallukat, G.; Schimke, I. Chronic Chagas heart disease: A disease on its way to becoming a worldwide health problem: Epidemiology, etiopathology, treatment, pathogenesis and laboratory medicine. Heart Fail. Rev. 2012, 17, 45–64. [Google Scholar] [CrossRef]
- Brun, R.; Blum, J.; Chappuis, F.; Burri, C. Human African trypanosomiasis. Lancet 2010, 375, 148–159. [Google Scholar] [CrossRef]
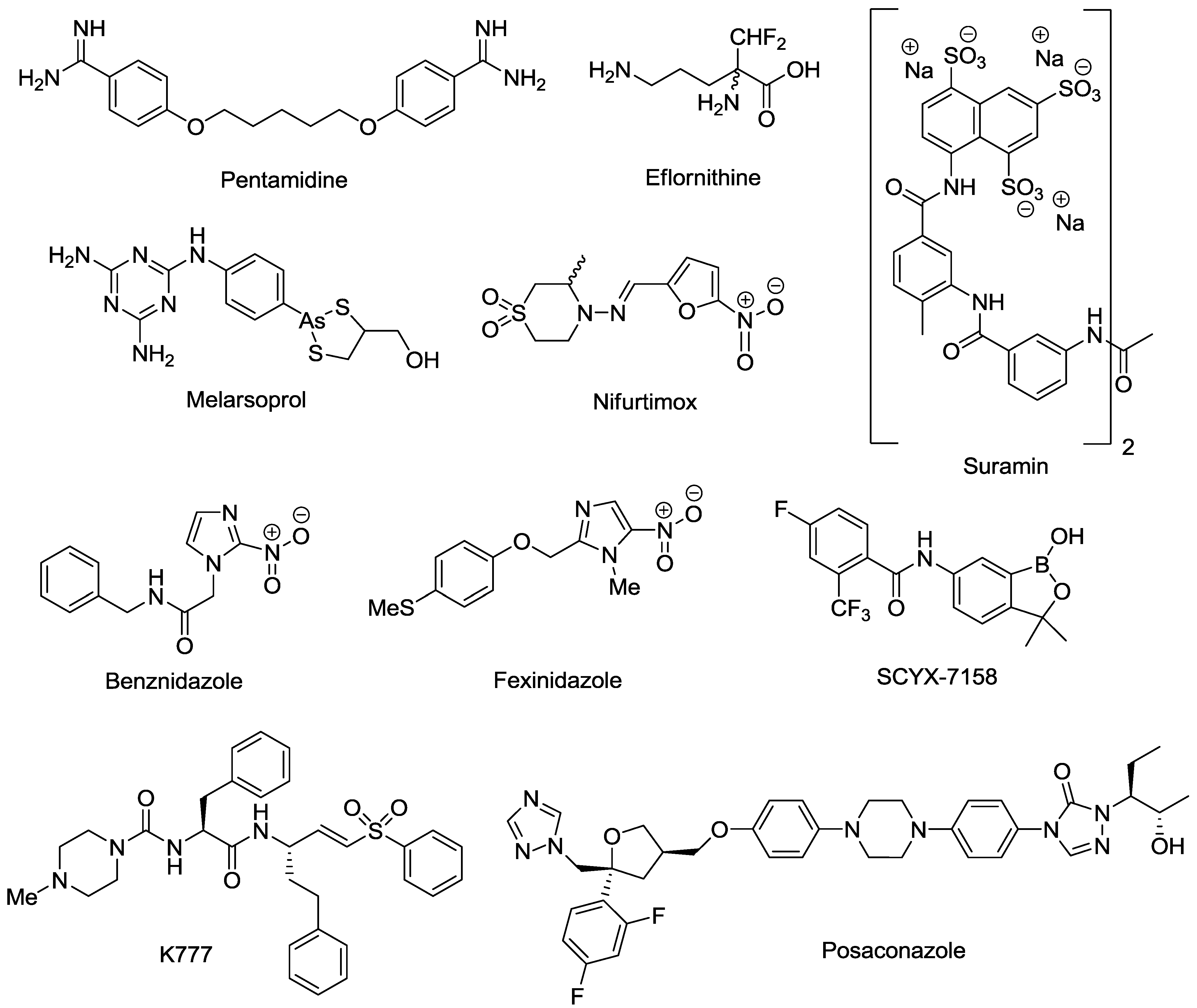
- Pepin, J.; Milord, F. The treatment of human African trypanosomiasis. Adv. Parasitol. 1994, 33, 1–47. [Google Scholar] [CrossRef]
- Milord, F.; Pepin, J.; Loko, L.; Ethier, L.; Mpia, B. Efficacy and toxicity of eflornithine for treatment of Trypanosoma brucei gambiense sleeping sickness. Lancet 1992, 340, 652–655. [Google Scholar] [CrossRef]
- Priotto, G.; Kasparian, S.; Ngouama, D.; Ghorashian, S.; Arnold, U.; Ghabri, S.; Karunakara, U. Nifurtimox-eflornithine combination therapy for second-stage Trypanosoma brucei gambiense sleeping sickness: A randomized clinical trial in Congo. Clin. Infect. Dis. 2007, 45, 1435–1442. [Google Scholar] [CrossRef]
- Priotto, G.; Kasparian, S.; Mutombo, W.; Ngouama, D.; Ghorashian, S.; Arnold, U.; Ghabri, S.; Baudin, E.; Buard, V.; Kazadi-Kyanza, S.; et al. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: A multicentre, randomised, phase III, non-inferiority trial. Lancet 2009, 374, 56–64. [Google Scholar] [CrossRef]
- Apt, W. Current and developing therapeutic agents in the treatment of Chagas disease. Drug Des. Devel. Ther. 2010, 4, 243–253. [Google Scholar] [CrossRef]
- Castro, J.A.; Diaz de Toranzo, E.G. Toxic effects of nifurtimox and benznidazole, two drugs used against American trypanosomiasis (Chagas disease). Biomed. Environ. Sci. 1988, 1, 19–33. [Google Scholar]
- Jackson, Y.; Alirol, E.; Getaz, L.; Wolff, H.; Combescure, C.; Chappuis, F. Tolerance and safety of nifurtimox in patients with chronic Chagas disease. Clin. Infect. Dis. 2010, 51, 69–75. [Google Scholar] [CrossRef]
- Hasslocher-Moreno, A.M.; Do Brasil, P.E.; De Sousa, A.S.; Xavier, S.S.; Chambela, M.C.; Sperandio Da Silva, G.M. Safety of benznidazole use in the treatment of chronic Chagas disease. J. Antimicrob. Chemother. 2012, 67, 1261–1266. [Google Scholar] [CrossRef]
- Kaiser, M.; Bray, M.A.; Cal, M.; Bourdin Trunz, B.; Torreele, E.; Brun, R. Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob. Agents Chemother. 2011, 55, 5602–5608. [Google Scholar] [CrossRef]
- Jacobs, R.T.; Nare, B.; Wring, S.A.; Orr, M.D.; Chen, D.; Sligar, J.M.; Jenks, M.X.; Noe, R.A.; Bowling, T.S.; Mercer, L.T.; et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl. Trop. Dis. 2011, 5, e1151. [Google Scholar] [CrossRef]
- Jacobs, R.T.; Nare, B.; Phillips, M.A. State of the art in African trypanosome drug discovery. Curr. Top. Med. Chem. 2011, 11, 1255–1274. [Google Scholar] [CrossRef]
- Barker, R.H., Jr.; Liu, H.; Hirth, B.; Celatka, C.A.; Fitzpatrick, R.; Xiang, Y.; Willert, E.K.; Phillips, M.A.; Kaiser, M.; Bacchi, C.J.; et al. Novel S-adenosylmethionine decarboxylase inhibitors for the treatment of human African trypanosomiasis. Antimicrob. Agents Chemother. 2009, 53, 2052–2058. [Google Scholar] [CrossRef]
- Bacchi, C.J.; Barker, R.H.; Rodriguez, A.; Hirth, B.; Rattendi, D.; Yarlett, N.; Hendrick, C.L.; Sybertz, E. Trypanocidal activity of 8-methyl-5′-[(Z)-4-aminobut-2-enyl](methylamino)adenosine (Genz-644131), an adenosylmethionine decarboxylase inhibitor. Antimicrob. Agents Chemother. 2009, 53, 3269–3272. [Google Scholar] [CrossRef]
- Price, H.P.; Menon, M.R.; Panethymitaki, C.; Goulding, D.; McKean, P.G.; Smith, D.F. Myristoyl-CoA: Protein N-myristoyltransferase, an essential enzyme and potential drug target in kinetoplastid parasites. J. Biol. Chem. 2003, 278, 7206–7214. [Google Scholar]
- Frearson, J.A.; Brand, S.; McElroy, S.P.; Cleghorn, L.A.T.; Smid, O.; Stojanovski, L.; Price, H.P.; Guther, M.L.S.; Torrie, L.S.; Robinson, D.A.; et al. N-myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature 2010, 464, 728–732. [Google Scholar] [CrossRef]
- Wyllie, S.; Oza, S.L.; Patterson, S.; Spinks, D.; Thompson, S.; Fairlamb, A.H. Dissecting the essentiality of the bifunctional trypanothione synthetase-amidase in Trypanosoma brucei using chemical and genetic methods. Mol. Microbiol. 2009, 74, 529–540. [Google Scholar] [CrossRef]
- Clayton, J. Chagas disease: Pushing through the pipeline. Nature 2010, 465, S12–S15. [Google Scholar] [CrossRef]
- A Study of the Use of Oral Posaconazole (POS) in the Treatment of Asymptomatic Chronic Chagas Disease. Clinical Trials. Available online: http://clinicaltrials.gov/show/NCT01377480 (accessed on 23 September 2013).
- Chen, C.K.; Leung, S.S.; Guilbert, C.; Jacobson, M.P.; McKerrow, J.H.; Podust, L.M. Structural characterization of CYP51 from Trypanosoma cruzi and Trypanosoma brucei bound to the antifungal drugs posaconazole and fluconazole. PLoS Negl. Trop. Dis. 2010, 4, e651. [Google Scholar] [CrossRef]
- Gunatilleke, S.S.; Calvet, C.M.; Johnston, J.B.; Chen, C.K.; Erenburg, G.; Gut, J.; Engel, J.C.; Ang, K.K.; Mulvaney, J.; Chen, S.; et al. Diverse inhibitor chemotypes targeting Trypanosoma cruzi CYP51. PLoS Negl. Trop. Dis. 2012, 6, e1736. [Google Scholar] [CrossRef]
- Lepesheva, G.I.; Villalta, F.; Waterman, M.R. Targeting Trypanosoma cruzi sterol 14alpha-demethylase (CYP51). Adv. Parasitol. 2011, 75, 65–87. [Google Scholar] [CrossRef]
- Soeiro Mde, N.; de Souza, E.M.; da Silva, C.F.; Batista Dda, G.; Batista, M.M.; Pavao, B.P.; Araujo, J.S.; Aiub, C.A.; da Silva, P.B.; Lionel, J.; et al. In vitro and in vivo studies of the antiparasitic activity of sterol 14alpha-demethylase (CYP51) inhibitor VNI against drug-resistant strains of Trypanosoma cruzi. Antimicrob. Agents Chemother. 2013, 57, 4151–4163. [Google Scholar] [CrossRef]
- Buckner, F.S. Sterol 14-demethylase inhibitors for Trypanosoma cruzi infections. Adv. Exp. Med. Biol. 2008, 625, 61–80. [Google Scholar] [CrossRef]
- Clayton, J. The promise of T. cruzi genomics. Nature 2010, 465, S16–S17. [Google Scholar] [CrossRef]
- Lisvane Silva, P.; Mantilla, B.S.; Barison, M.J.; Wrenger, C.; Silber, A.M. The uniqueness of the Trypanosoma cruzi mitochondrion: Opportunities to identify new drug target for the treatment of Chagas disease. Curr. Pharm. Des. 2011, 17, 2074–2099. [Google Scholar] [CrossRef]
- Soeiro, M.N.; de Castro, S.L. Trypanosoma cruzi targets for new chemotherapeutic approaches. Expert Opin. Ther. Targets 2009, 13, 105–121. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2013, 30, 237–323. [Google Scholar] [CrossRef]
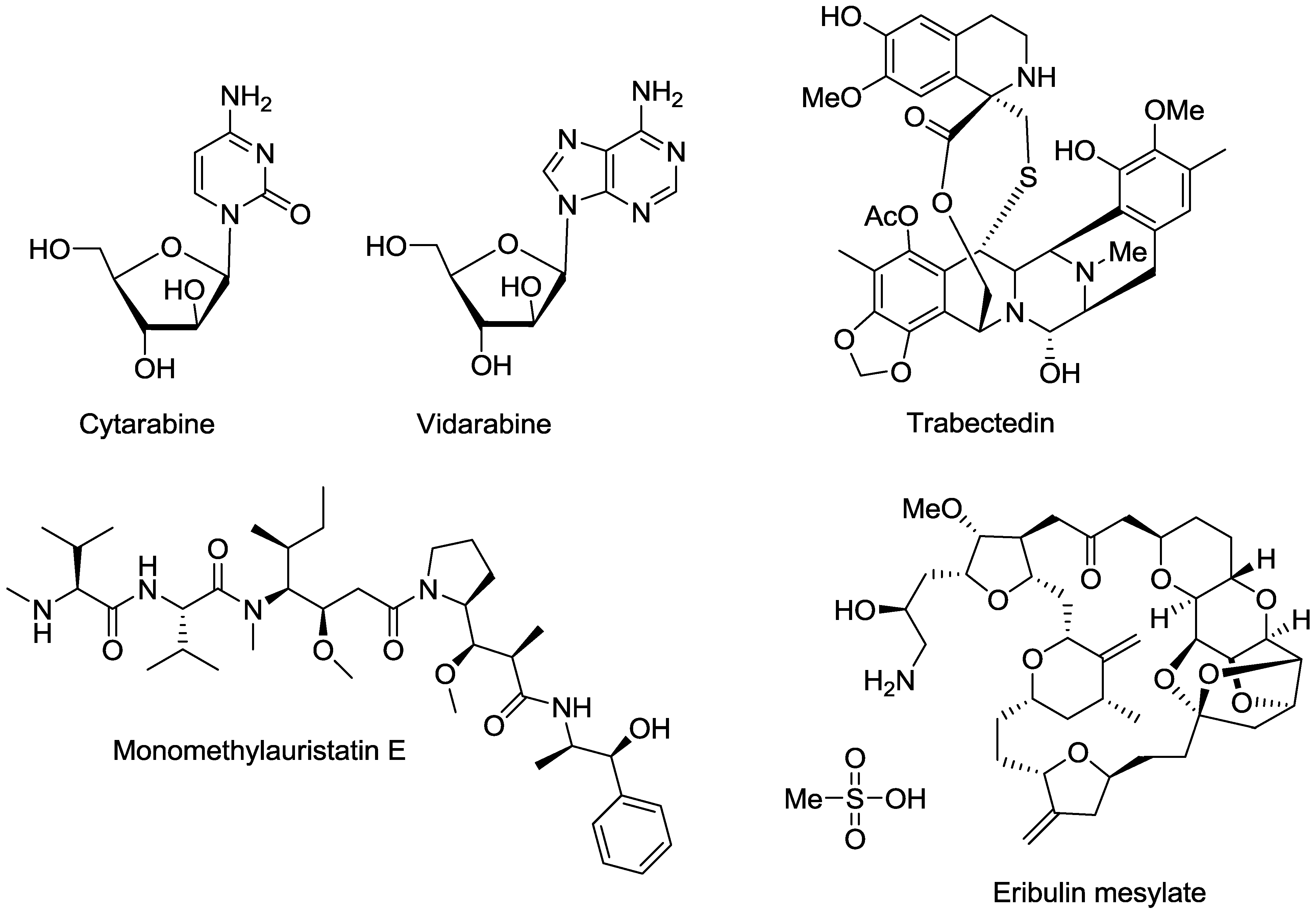
- Bergmann, W.; Feeney, R.J. The isolation of a new thymine pentoside from sponges. J. Am. Chem. Soc. 1950, 72, 2809–2810. [Google Scholar] [CrossRef]
- Swift, A.N. Contributions to the study of marine products. Component acids of lipids of sponges. J. Org. Chem. 1951, 16, 1206–1221. [Google Scholar] [CrossRef]
- O’Day, D.M.; Poirier, R.H.; Jones, D.B.; Elliott, J.H. Vidarabine therapy of complicated Herpes simplex keratitis. Am. J. Ophthalmol. 1976, 81, 642–649. [Google Scholar]
- Pavan-Langston, D.; Hess, F. Ocular and systemic antiviral activity of vidarabine. Compr. Ther. 1977, 3, 42–48. [Google Scholar]
- Mori, J.; Tsubokura, M.; Kami, M. Cytarabine dose for acute myeloid leukemia. N. Engl. J. Med. 2011, 364, 2166–2167. [Google Scholar] [CrossRef]
- Fox, B.W. Pharmacology and chemistry of some inhibitors of herpes replication. J. Antimicrob. Chemother. 1977, 3, 23–32. [Google Scholar] [CrossRef]
- Gedik, C.M.; Collins, A.R. The mode of action of 1-beta-d-arabinofuranosylcytosine in inhibiting DNA repair; New evidence using a sensitive assay for repair DNA synthesis and ligation in permeable cells. Mutat. Res. 1991, 254, 231–237. [Google Scholar] [CrossRef]
- Olivera, B.M.; Gray, W.R.; Zeikus, R.; McIntosh, J.M.; Varga, J.; Rivier, J.; De Santos, V.; Cruz, L.J. Peptide neurotoxins from fish-hunting cone snails. Science 1985, 230, 1338–1343. [Google Scholar]
- Miljanich, G.P. Ziconotide: Neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar] [CrossRef]
- Lovaza Drug Details. Food and Drug Administration Approved Products. Available online: http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.DrugDetails (accessed on 18 September 2013).
- Vascepa. Food and Drug Administration Orange Book: Approved drug products with therapeutic equivalence evaluations. Available online: http://www.accessdata.fda.gov/scripts/cder/ob/docs/obdetail.cfm?Appl_No=202057&TABLE1=OB_Rx (accessed on 18 September 2013).
- Strobel, C.; Jahreis, G.; Kuhnt, K. Survey of n-3 and n-6 polyunsaturated fatty acids in fish and fish products. Lipids Health Dis. 2012, 11, 144. [Google Scholar] [CrossRef]
- Nestel, P.J.; Connor, W.E.; Reardon, M.F.; Connor, S.; Wong, S.; Boston, R. Suppression by diets rich in fish oil of very low-density lipoprotein production in man. J. Clin. Invest. 1984, 74, 82–89. [Google Scholar] [CrossRef]
- Sanders, T.A.B.; Sullivan, D.R.; Reeve, J.; Thompson, G.R. Triglyceride-lowering effect of marine polyunsaturates in patients with hypertriglyceridemia. Arteriosclerosis 1985, 5, 459–465. [Google Scholar] [CrossRef]
- Bordin, P.; Bodamer, O.A.F.; Venkatesan, S.; Gray, R.M.; Bannister, P.A.; Halliday, D. Effects of fish oil supplementation on apolipoprotein B100 production and lipoprotein metabolism in normolipidaemic males. Eur. J. Clin. Nutr. 1998, 52, 104–109. [Google Scholar]
- Madsen, L.; Rustan, A.C.; Vaagenes, H.; Berge, K.; Dyroy, E.; Berge, R.K. Eicosapentaenoic and docosahexaenoic acid affect mitochondrial and peroxisomal fatty acid oxidation in relation to substrate preference. Lipids 1999, 34, 951–963. [Google Scholar] [CrossRef]
- Davidson, M.H. Mechanisms for the hypotriglyceridemic effect of marine omega-3 fatty acids. Am. J. Cardiol. 2006, 98, 27–33. [Google Scholar] [CrossRef]
- Hirata, Y.; Uemura, D. Halichondrins—Antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar] [CrossRef]
- Kuznetsov, G.; Towle, M.J.; Cheng, H.S.; Kawamura, T.; TenDyke, K.; Liu, D.; Kishi, Y.; Yu, M.J.; Littlefield, B.A. Induction of morphological and biochemical apoptosis following prolonged mitotic blockage by halichondrin B macrocyclic ketone analog E7389. Cancer Res. 2004, 64, 5760–5766. [Google Scholar] [CrossRef]
- Jordan, M.A.; Kamath, K.; Manna, T.; Okouneva, T.; Miller, H.P.; Davis, C.; Littlefield, B.A.; Wilson, L. The primary antimitotic mechanism of action of the synthetic halichondrin E7389 is suppression of microtubule growth. Mol. Cancer Ther. 2005, 4, 1086–1095. [Google Scholar] [CrossRef]
- Dabydeen, D.A.; Burnett, J.C.; Bai, R.L.; Verdier-Pinard, P.; Hickford, S.J.H.; Pettit, G.R.; Blunt, J.W.; Munro, M.H.G.; Gussio, R.; Hamel, E. Comparison of the activities of the truncated halichondrin B analog NSC 707389 (E7389) with those of the parent compound and a proposed binding site on tubulin. Mol. Pharmacol. 2006, 70, 1866–1875. [Google Scholar] [CrossRef]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E. cAC10-vcMMAE, an anti-CD30–monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef]
- Pettit, G.R.; Kamano, Y.; Herald, C.L.; Tuinman, A.A.; Boettner, F.E.; Kizu, H.; Schmidt, J.M.; Baczynskyj, L.; Tomer, K.B.; Bontems, R.J. The isolation and structure of a remarkable marine animal antineoplastic constituent: Dolastatin 10. J. Am. Chem. Soc. 1987, 109, 6883–6885. [Google Scholar] [CrossRef]
- Deng, C.; Pan, B.; O’Connor, O.A. Brentuximab vedotin. Clin. Cancer Res. 2013, 19, 22–27. [Google Scholar] [CrossRef]
- Rinehart, K.L.; Holt, T.G.; Fregeau, N.L.; Stroh, J.G.; Keifer, P.A.; Sun, F.; Li, L.H.; Martin, D.G. Ecteinascidins 729, 743, 745, 759A, 759B, and 770: Potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4512–4515. [Google Scholar] [CrossRef]
- Zewail-Foote, M.; Hurley, L.H. Differential rates of reversibility of ecteinascidin 743-DNA covalent adducts from different sequences lead to migration to favored bonding sites. J. Am. Chem. Soc. 2001, 123, 6485–6495. [Google Scholar] [CrossRef]
- Takebayashi, Y.; Pourquier, P.; Zimonjic, D.B.; Nakayama, K.; Emmert, S.; Ueda, T.; Urasaki, Y.; Kanzaki, A.; Akiyama, S.; Popescu, N.; et al. Antiproliferative activity of ecteinascidin 743 is dependent upon transcription-coupled nucleotide-excision repair. Nat. Med. 2001, 7, 961–966. [Google Scholar] [CrossRef]
- Soares, D.G.; Escargueil, A.E.; Poindessous, V.; Sarasin, A.; De Gramont, A.; Bonatto, D.; Henriques, J.A.P.; Larsen, A.K. Replication and homologous recombination repair regulate DNA double-strand break formation by the antitumor alkylator ecteinascidin 743. Proc. Natl. Acad. Sci. USA. 2007, 104, 13062–13067. [Google Scholar] [CrossRef]
- Herrero, A.B.; Martin-Castellanos, C.; Marco, E.; Gago, F.; Moreno, S. Cross-talk between nucleotide excision and homologous recombination DNA repair pathways in the mechanism of action of antitumor trabectedin. Cancer Res. 2006, 66, 8155–8162. [Google Scholar] [CrossRef]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef]
- Jones, A.J.; Avery, V.M. Whole-organism high-throughput screening against Trypanosoma brucei brucei. Exp. Opin. Drug Discov. 2013, 8, 495–507. [Google Scholar]
- Sykes, M.L.; Avery, V.M. Approaches to protozoan drug discovery: Phenotypic screening. J. Med. Chem. 2013, in press. [Google Scholar]
- Stevens, J.; Brisse, S. Systematics of trypanosomes of medical and veterinary importance. In The Trypanosomiases; Maudlin, I., Holmes, P.H., Miles, M.A., Eds.; CABI Publishing: Trowbridge, UK, 2004; pp. 1–23. [Google Scholar]
- Pink, R.; Hudson, A.; Mouries, M.A.; Bendig, M. Opportunities and challenges in antiparasitic drug discovery. Nat. Rev. Drug Discov. 2005, 4, 727–740. [Google Scholar] [CrossRef]
- Chennamaneni, N.K.; Arif, J.; Buckner, F.S.; Gelb, M.H. Isoquinoline-based analogs of the cancer drug clinical candidate tipifarnib as anti-Trypanosoma cruzi agents. Bioorg. Med. Chem. Lett. 2009, 19, 6582–6584. [Google Scholar] [CrossRef]
- Romanha, A.J.; Castro, S.L.; Soeiro Mde, N.; Lannes-Vieira, J.; Ribeiro, I.; Talvani, A.; Bourdin, B.; Blum, B.; Olivieri, B.; Zani, C.; et al. In vitro and in vivo experimental models for drug screening and development for Chagas disease. Mem. Inst. Oswaldo Cruz 2010, 105, 233–238. [Google Scholar] [CrossRef]
- Ennes-Vidal, V.; Menna-Barreto, R.F.; Santos, A.L.; Branquinha, M.H.; d’Avila-Levy, C.M. Effects of the calpain inhibitor MDL28170 on the clinically relevant forms of Trypanosoma cruzi in vitro. J. Antimicrob. Chemother. 2010, 65, 1395–1398. [Google Scholar] [CrossRef]
- Buckner, F.S.; Verlinde, C.L.; La Flamme, A.C.; Van Voorhis, W.C. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing beta-galactosidase. Antimicrob. Agents Chemother. 1996, 40, 2592–2597. [Google Scholar]
- Vickerman, K. Developmental cycles and biology of pathogenic trypanosomes. Br. Med. Bull. 1985, 41, 105–114. [Google Scholar]
- Da Silva, A.J.; Moser, M. Trypanosomiasis, American (Chagas disease, Trypanosoma cruzi). Center for Disease Control and Prevention: Public Health Image Library (PHIL). Available online: http://phil.cdc.gov/phil/details.asp (accessed on 14 October 2013).
- Nwaka, S.; Hudson, A. Innovative lead discovery strategies for tropical diseases. Nat. Rev. Drug Discov. 2006, 5, 941–955. [Google Scholar] [CrossRef]
- Dardonville, C.; Fernandez-Fernandez, C.; Gibbons, S.L.; Jagerovic, N.; Nieto, L.; Ryan, G.; Kaiser, M.; Brun, R. Antiprotozoal activity of 1-phenethyl-4-aminopiperidine derivatives. Antimicrob. Agents Chemother. 2009, 53, 3815–3821. [Google Scholar] [CrossRef]
- Jones, D.C.; Hallyburton, I.; Stojanovski, L.; Read, K.D.; Frearson, J.A.; Fairlamb, A.H. Identification of a κ-opioid agonist as a potent and selective lead for drug development against human African trypanosomiasis. Biochem. Pharmacol. 2010, 80, 1478–1486. [Google Scholar] [CrossRef]
- Sykes, M.L.; Baell, J.B.; Kaiser, M.; Chatelain, E.; Moawad, S.R.; Ganame, D.; Ioset, J.R.; Avery, V.M. Identification of compounds with anti-proliferative activity against Trypanosoma brucei brucei strain 427 by a whole cell viability based HTS campaign. PLoS Negl. Trop. Dis. 2012, 6, e1896. [Google Scholar] [CrossRef]
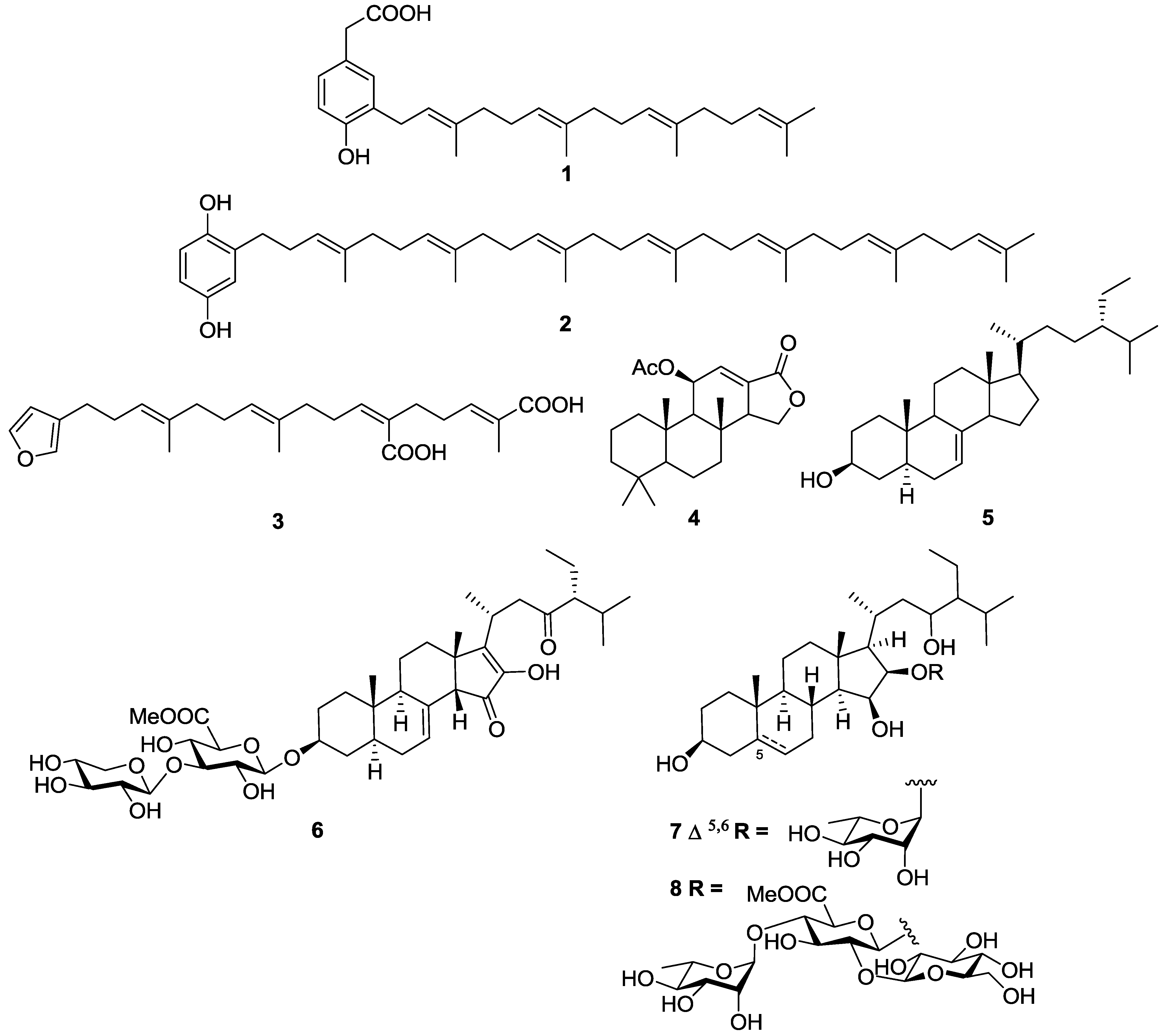
- Orhan, I.; Sener, B.; Kaiser, M.; Brun, R.; Tasdemir, D. Inhibitory activity of marine sponge-derived natural products against parasitic protozoa. Mar. Drugs 2010, 8, 47–58. [Google Scholar] [CrossRef]
- Tasdemir, D.; Topaloglu, B.; Perozzo, R.; Brun, R.; O’Neill, R.; Carballeira, N.M.; Zhang, X.; Tonge, P.J.; Linden, A.; Ruedi, P. Marine natural products from the Turkish sponge Agelas oroides that inhibit the enoyl reductases from Plasmodium falciparum, Mycobacterium tuberculosis and Escherichia coli. Bioorg. Med. Chem. 2007, 15, 6834–6845. [Google Scholar] [CrossRef]
- Regalado, E.L.; Tasdemir, D.; Kaiser, M.; Cachet, N.; Amade, P.; Thomas, O.P. Antiprotozoal steroidal saponins from the marine sponge Pandaros acanthifolium. J. Nat. Prod. 2010, 73, 1404–1410. [Google Scholar] [CrossRef]
- Regalado, E.L.; Jimenez-Romero, C.; Genta-Jouve, G.; Tasdemir, D.; Amade, P.; Nogueiras, C.; Thomas, O.P. Acanthifoliosides, minor steroidal saponins from the Caribbean sponge Pandaros acanthifolium. Tetrahedron 2011, 67, 1011–1018. [Google Scholar] [CrossRef]
- Kossuga, M.H.; Nascimento, A.M.; Reimao, J.Q.; Tempone, A.G.; Taniwaki, N.N.; Veloso, K.; Ferreira, A.G.; Cavalcanti, B.C.; Pessoa, C.; Moraes, M.O.; et al. Antiparasitic, antineuroinflammatory, and cytotoxic polyketides from the marine sponge Plakortis angulospiculatus collected in Brazil. J. Nat. Prod. 2008, 71, 334–339. [Google Scholar] [CrossRef]
- Feng, Y.J.; Davis, R.A.; Sykes, M.; Avery, V.M.; Camp, D.; Quinn, R.J. Antitrypanosomal cyclic polyketide peroxides from the Australian marine sponge Plakortis sp. J. Nat. Prod. 2010, 73, 716–719. [Google Scholar] [CrossRef]
- Chianese, G.; Fattorusso, E.; Scala, F.; Teta, R.; Calcinai, B.; Bavestrello, G.; Dien, H.A.; Kaiser, M.; Tasdemir, D.; Taglialatela-Scafati, O. Manadoperoxides, a new class of potent antitrypanosomal agents of marine origin. Org. Biomol. Chem. 2012, 10, 7197–7207. [Google Scholar] [CrossRef]
- Pimentel-Elardo, S.M.; Buback, V.; Gulder, T.A.M.; Bugni, T.S.; Reppart, J.; Bringmann, G.; Ireland, C.M.; Schirmeister, T.; Hentschel, U. New tetromycin derivatives with anti-trypanosomal and protease inhibitory activities. Mar. Drugs 2011, 9, 1682–1697. [Google Scholar] [CrossRef]
- Pontius, A.; Krick, A.; Kehraus, S.; Brun, R.; Konig, G.M. Antiprotozoal activities of heterocyclic-substituted xanthones from the marine-derived fungus Chaetomium sp. J. Nat. Prod. 2008, 71, 1579–1584. [Google Scholar] [CrossRef]
- Erdogan, I.; Sener, B.; Higa, T. Tryptophol, a plant auxin isolated from the marine sponge Ircinia spinulosa. Biochem. Syst. Ecol. 2000, 28, 793–794. [Google Scholar] [CrossRef]
- Martinez-Luis, S.; Gomez, J.F.; Spadafora, C.; Guzman, H.M.; Gutierrez, M. Antitrypanosomal alkaloids from the marine bacterium Bacillus pumilus. Molecules 2012, 17, 11146–11155. [Google Scholar] [CrossRef]
- Chan, S.T.S.; Pearce, A.N.; Page, M.J.; Kaiser, M.; Copp, B.R. Antimalarial β-carbolines from the New Zealand ascidian Pseudodistoma opacum. J. Nat. Prod. 2011, 74, 1972–1979. [Google Scholar] [CrossRef]
- Scala, F.; Fattorusso, E.; Menna, M.; Taglialatela-Scafati, O.; Tierney, M.; Kaiser, M.; Tasdemir, D. Bromopyrrole alkaloids as lead compounds against protozoan parasites. Mar. Drugs 2010, 8, 2162–2174. [Google Scholar] [CrossRef]
- Cafieri, F.; Fattorusso, E.; Taglialatela-Scafati, O. Novel bromopyrrole alkaloids from the sponge Agelas dispar. J. Nat. Prod. 1998, 61, 122–125. [Google Scholar] [CrossRef]
- Cafieri, F.; Fattorusso, E.; Mangoni, A.; Taglialatelascafati, O. Longamide and 3,7-dimethylisoguanine, 2 novel alkaloids from the marine sponge Agelas longissima. Tetrahedron Lett. 1995, 36, 7893–7896. [Google Scholar]
- Aiello, A.; D’Esposito, M.; Fattorusso, E.; Menna, M.; Muller, W.E.G.; Perovic-Ottstadt, S.; Schroder, H.C. Novel bioactive bromopyrrole alkaloids from the Mediterranean sponge Axinella verrucosa. Bioorg. Med. Chem. 2006, 14, 17–24. [Google Scholar] [CrossRef]
- Walker, R.P.; Faulkner, D.J.; Van Engen, D.; Clardy, J. Sceptrin, an antimicrobial agent from the sponge Agelas sceptrum. J. Am. Chem. Soc. 1981, 103, 6772–6773. [Google Scholar] [CrossRef]
- Wu, H.; Nakamura, H.; Kobayashi, J.; Kobayashi, M.; Ohizumi, Y.; Hirata, Y. Structures of agelasines, diterpenes having a 9-methyladeninium chromophore isolated from the Okinawan marine sponge Agelas nakamurai hoshino. Bull. Chem. Soc. Jpn. 1986, 59, 2495–2504. [Google Scholar] [CrossRef]
- Vik, A.; Proszenyak, A.; Vermeersch, M.; Cos, P.; Maes, L.; Gundersen, L.L. Screening of agelasine D and analogs for inhibitory activity against pathogenic protozoa; Identification of hits for visceral leishmaniasis and Chagas disease. Molecules 2009, 14, 279–288. [Google Scholar] [CrossRef]
- Davis, R.A.; Sykes, M.; Avery, V.M.; Camp, D.; Quinn, R.J. Convolutamines I and J, antitrypanosomal alkaloids from the bryozoan Amathia tortusa. Bioorg. Med. Chem. 2011, 19, 6615–6619. [Google Scholar] [CrossRef]
- Feng, Y.J.; Davis, R.A.; Sykes, M.L.; Avery, V.M.; Quinn, R.J. Iotrochamides A and B, antitrypanosomal compounds from the Australian marine sponge Iotrochota sp. Bioorg. Med. Chem. Lett. 2012, 22, 4873–4876. [Google Scholar] [CrossRef]
- Wright, A.D.; Goclik, E.; Koenig, G.M.; Kaminsky, R. Lepadins D–F: Antiplasmodial and antitrypanosomal decahydroquinoline derivatives from the tropical marine tunicate Didemnum sp. J. Med. Chem. 2002, 45, 3067–3072. [Google Scholar] [CrossRef]
- Volk, C.A.; Kock, M. Viscosamine: The first naturally occurring trimeric 3-alkyl pyridinium alkaloid. Org. Lett. 2003, 5, 3567–3569. [Google Scholar] [CrossRef]
- Rodenko, B.; Al-Salabi, M.I.; Teka, I.A.; Ho, W.; El-Sabbagh, N.; Ali, J.A.M.; Ibrahim, H.M.S.; Wanner, M.J.; Koomen, G.; De Koning, H.P. Synthesis of marine-derived 3-alkylpyridinium alkaloids with potent antiprotozoal activity. ACS Med. Chem. Lett. 2011, 2, 901–906. [Google Scholar] [CrossRef]
- Kirsch, G.; Konig, G.M.; Wright, A.D.; Kaminsky, R. A new bioactive sesterterpene and antiplasmodial alkaloids from the marine sponge Hyrtios cf. erecta. J. Nat. Prod. 2000, 63, 825–829. [Google Scholar] [CrossRef]
- Feng, Y.J.; Davis, R.A.; Sykes, M.L.; Avery, V.M.; Carroll, A.R.; Camp, D.; Quinn, R.J. Antitrypanosomal pyridoacridine alkaloids from the Australian ascidian Polysyncraton echinatum. Tetrahedron Lett. 2010, 51, 2477–2479. [Google Scholar] [CrossRef]
- Watts, K.R.; Ratnam, J.; Ang, K.H.; Tenney, K.; Compton, J.E.; McKerrow, J.; Crews, P. Assessing the trypanocidal potential of natural and semi-synthetic diketopiperazines from two deep water marine-derived fungi. Bioorg. Med. Chem. 2010, 18, 2566–2574. [Google Scholar]
- Linington, R.G.; Gonzalez, J.; Urena, L.D.; Romero, L.I.; Ortega-Barría, E.; Gerwick, W.H. Venturamides A and B: Antimalarial constituents of the Panamanian marine cyanobacterium Oscillatoria sp. J. Nat. Prod. 2007, 70, 397–401. [Google Scholar] [CrossRef]
- Portmann, C.; Blom, J.F.; Kaiser, M.; Brun, R.; Juttner, F.; Gademann, K. Isolation of aerucyclamides C and D and structure revision of microcyclamide 7806A: Heterocyclic ribosomal peptides from Microcystis aeruginosa PCC 7806 and their antiparasite evaluation. J. Nat. Prod. 2008, 71, 1891–1896. [Google Scholar] [CrossRef]
- Sanchez, L.M.; Knudsen, G.M.; Helbig, C.; De Muylder, G.; Mascuch, S.M.; Mackey, Z.B.; Gerwick, L.; Clayton, C.; McKerrow, J.H.; Linington, R.G. Examination of the mode of action of the almiramide family of natural products against the kinetoplastid parasite Trypanosoma brucei. J. Nat. Prod. 2013, in press. [Google Scholar]
- Verlinde, C.L.; Hannaert, V.; Blonski, C.; Willson, M.; Perie, J.J.; Fothergill-Gilmore, L.A.; Opperdoes, F.R.; Gelb, M.H.; Hol, W.G.; Michels, P.A. Glycolysis as a target for the design of new anti-trypanosome drugs. Drug Resist. Updat. 2001, 4, 50–65. [Google Scholar] [CrossRef]
- Fattorusso, C.; Persico, M.; Calcinai, B.; Cerrano, C.; Parapini, S.; Taramelli, D.; Novellino, E.; Romano, A.; Scala, F.; Fattorusso, E.; et al. Manadoperoxides A–D from the Indonesian sponge Plakortis cfr. simplex. Further insights on the structure-activity relationships of simple 1,2-dioxane antimalarials. J. Nat. Prod. 2010, 73, 1138–1145. [Google Scholar] [CrossRef]
- El-Seedi, H.R.; El-Barbary, M.A.; El-Ghorab, D.M.H.; Bohlin, L.; Borg-Karlson, A.K.; Goransson, U.; Verpoorte, R. Recent insights into the biosynthesis and biological activities of natural xanthones. Curr. Med. Chem. 2010, 17, 854–901. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jones, A.J.; Grkovic, T.; Sykes, M.L.; Avery, V.M. Trypanocidal Activity of Marine Natural Products. Mar. Drugs 2013, 11, 4058-4082. https://doi.org/10.3390/md11104058
Jones AJ, Grkovic T, Sykes ML, Avery VM. Trypanocidal Activity of Marine Natural Products. Marine Drugs. 2013; 11(10):4058-4082. https://doi.org/10.3390/md11104058
Chicago/Turabian StyleJones, Amy J., Tanja Grkovic, Melissa L. Sykes, and Vicky M. Avery. 2013. "Trypanocidal Activity of Marine Natural Products" Marine Drugs 11, no. 10: 4058-4082. https://doi.org/10.3390/md11104058
APA StyleJones, A. J., Grkovic, T., Sykes, M. L., & Avery, V. M. (2013). Trypanocidal Activity of Marine Natural Products. Marine Drugs, 11(10), 4058-4082. https://doi.org/10.3390/md11104058