1. Introduction
Opisthobranchs are marine molluscs in which the shell is reduced or completely absent. They move slowly and their soft-bodies often present bright, attractive coloration, with multihued geometric patterns [
1]. However, despite slow movements, the absence of physical attributes and gaudy livery, opisthobranchs have few documented predators. Field observations and proper ecological assays have demonstrated that they protect themselves by employing a series of defensive strategies including chemicals [
2]. These compounds that play a fundamental role for their survival are usually small molecules derived from the diet, which can be accumulated in specialized anatomical parts of the animal, resulting in their unpleasant taste. In some species the presence of the
de novo bio-synthesized defensive molecules has been clearly demonstrated [
3,
4]. The ability of opisthobranchs to select bioactive molecules from nature has resulted in an extraordinary library of compounds with intriguing framework, which are not found in their terrestrial counterparts and possess the potential as new pharmaceutical entities [
5].
In our ongoing studies on the chemical ecology of marine opisthobranchs, we have examined the chemical content of six specimens of
Aldisa andersoni collected off Muttom coast, India (Indian Ocean).
A. andersoni is a dorid nudibranch (order Nudibranchia, infraorder Anthobranchia, superfamily Doridoidea) only recorded from Sri Lanka coast (not far from the place of our collection). The animal presents an oval body, 20–30 mm in length and a series of rows of low tubercles on the blue dorsum. The tubercles are separated by black areas of pigment, the upper part of the skin is crossed by a bright yellow saddle behind the rhinophores and other yellow marks on the dorsum [
6]. These features appear to be indicative of aposematism (warning coloration), closely resembling the shape and the color pattern of unpalatable nudibranch species belonging to the genus
Phyllidia, which are known to contain toxic isonitrile molecules [
7]. Previous studies on different
Aldisa species from Pacific and Mediterranean areas resulted in the finding of unusual steroids. In particular, two feeding-deterrent sterols featuring cholic acid side chain have been reported from
Aldisa cooperi [
8], whereas an unusual 24-norchol-4-ene-3,22-dione has been isolated from
Aldisa smaragdina [
9]. It seems that the source of steroids in
Aldisa cooperi should be the sponge
Anthoarcuata graceae but the animal is able to modify the inactive cholestenone into two active compounds. On the other hand, the absence of the steroid isolated from
A. smaragdina in its prey, the sponge
Phorbas fictitius, suggests the modification of a suitable precursor or a biosynthetic origin.
The chemistry of
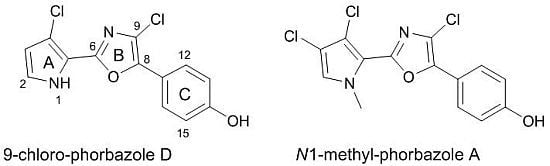
A. andersoni has not been studied previously. We found evidence of the presence of chlorinated phenyl pyrrolyloxazoles in the ether extract of the mollusc. These molecules were found to be mainly concentrated in the external part of the mollusc even though they were also detected in the internal glands. Two new molecules, 9-chloro-phorbazole D (
1) and
N1-methyl-phorbazole A (
2), co-occurring with the known related phorbazoles A, B and D have been isolated and characterized from this mollusc. Phorbazoles have been reported to date only from the sponge
Phorbas aff.
clathrata [
10,
11,
12]. The origin of these metabolites in
A. andersoni could be ascribed to a diet based on
Phorbas sponges although
de novo biosynthesis could not be excluded.
2. Results and Discussion
Six specimens of
A. andersoni were caught by scuba diving off Muttom coast (South India) during November 2009, were immediately frozen at −20 °C and transferred to the laboratory. Unfortunately no plausible prey was observed in the same area. Anatomical dissection of the body of nudibranchs was precluded due to the small size and the following procedure was used to obtain the distinct extracts of external and internal parts. First, the whole animals were immersed in acetone (20 mL) and submitted to ultrasound vibration for a few minutes. By this procedure only the metabolites present in the skin (external part) were extracted. The solvent was removed and the animals were crushed by a pestle and again treated with acetone (20 mL × 3), to extract the digestive gland contents (internal part). After evaporation of acetone, the aqueous residue of both the extracts was partitioned between Et
2O (50 mL × 4) and H
2O. The two ether extracts were analyzed by Thin Layer Chromatography (TLC) developed in the solvent system (CHCl
3/CH
3OH, 8:2). Both exhibited similar metabolite pattern with a series of UV-visible spots significantly more concentrated in the external extract. This extract was then directly submitted to RP-HPLC purification (MeOH/H
2O gradient) which resulted in the novel compounds
1 and
2 along with known phorbazoles A, B and D, that were identified by comparison of their spectroscopic data with the literature values [
10] (see
Experimental Section for details).
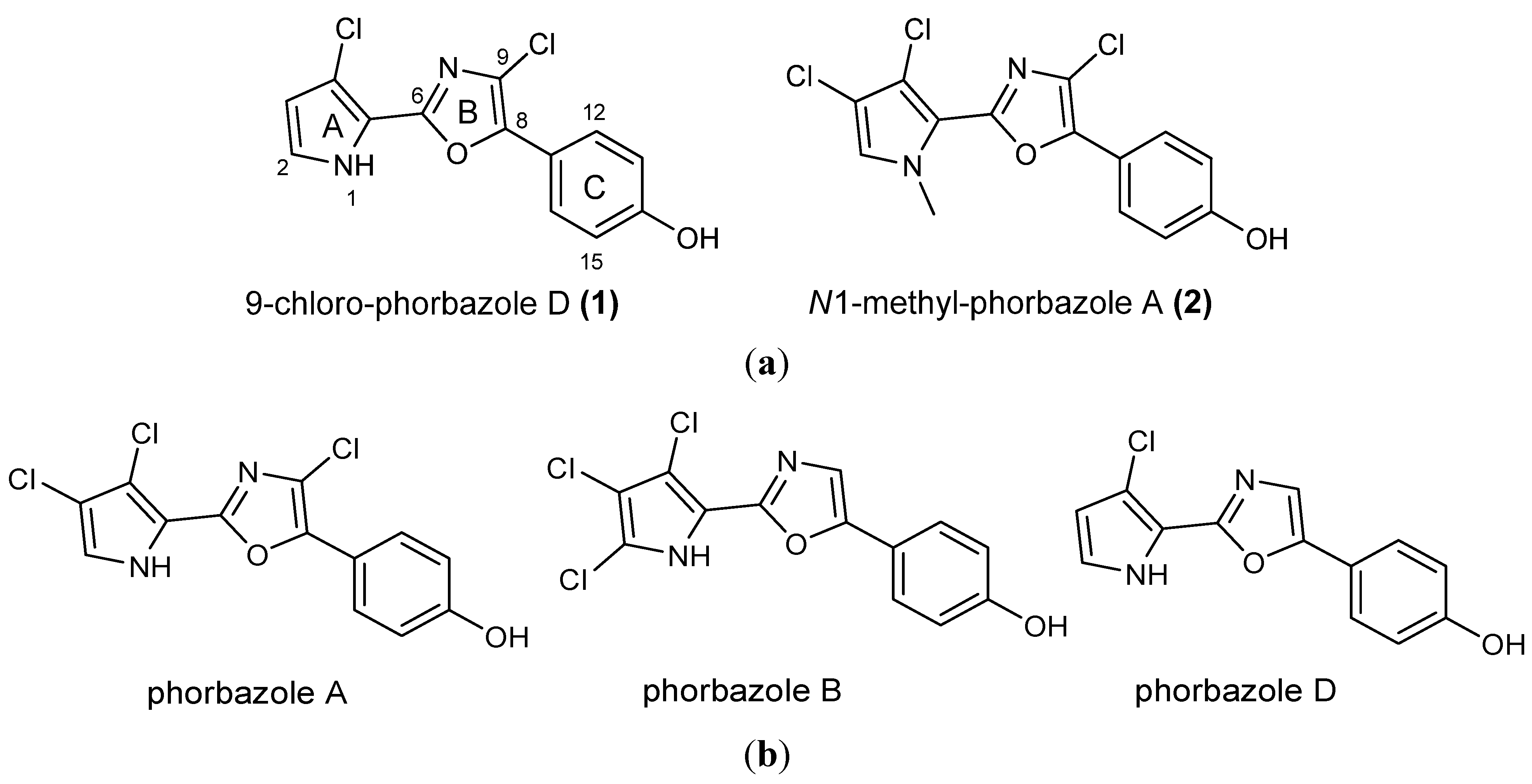
Analysis of NMR spectra of compounds 1 and 2 revealed a close structural relationship with known phorbazoles however indicating differences in the substitution pattern.
Compound
1 had a molecular formula C
13H
8Cl
2N
2O
2 deduced by LCMS (
m/z 295 [M + H]
+) and HRESIMS (
m/z 316.9875 [M + Na]
+), that showed a characteristic cluster for two chlorine atoms. The
1H NMR spectrum of
1 exhibited only four sp
2 signals at δ 6.27 (1 H, d,
J = 2.8 Hz, H-3), 6.93 (2H, d,
J = 8.7 Hz, H-13 and H-15), 6.97 (1H, d,
J = 2.8 Hz, H-2) and 7.81 (2H, d,
J = 8.7 Hz, H-12 and H-16), according to two separated spin systems in the disubstituted pyrrole unit (ring A) and 1,4 disubstituted benzene ring (ring C). This implied that the two chlorine atoms were located in both the pyrrole and oxazole rings. Consistent with this, the
13C NMR spectrum contained 11 sp
2 carbon signals in the range of 111.8–159.6 ppm (
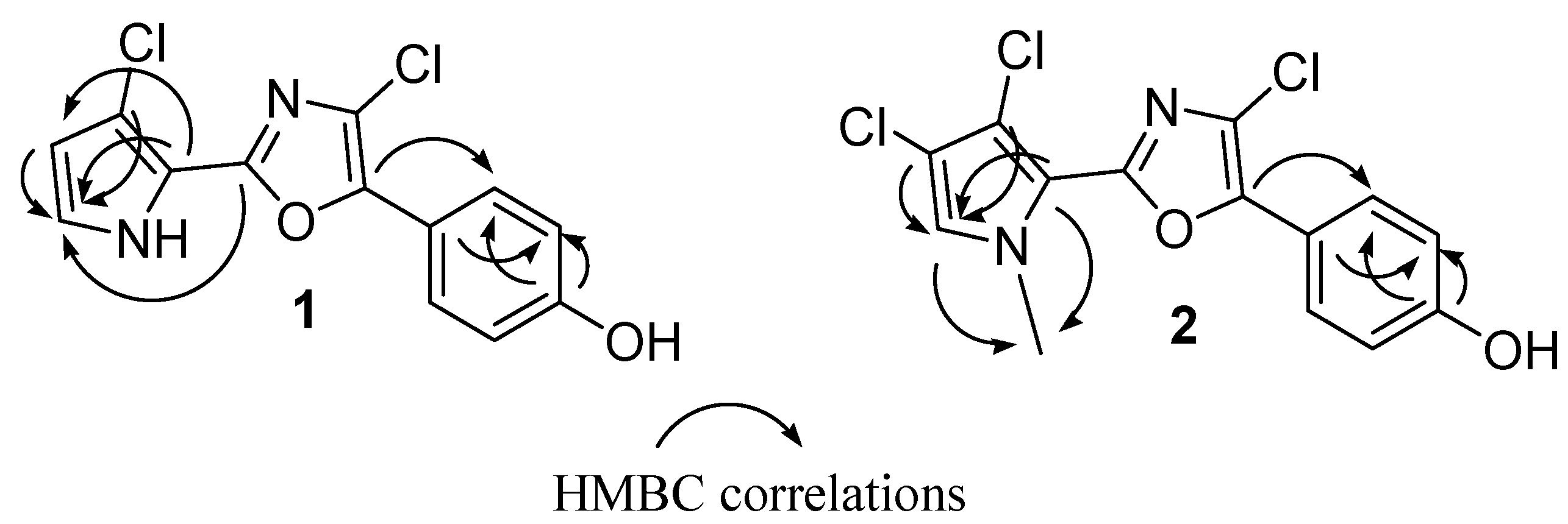
Table 1). The 2D NMR experiments aided us to fully characterize the molecule. In particular, a series of HMBC experiments, recorded with
J = 5 Hz and
J = 10 Hz, were crucial to attribute the quaternary carbons and to assign the structure as depicted in
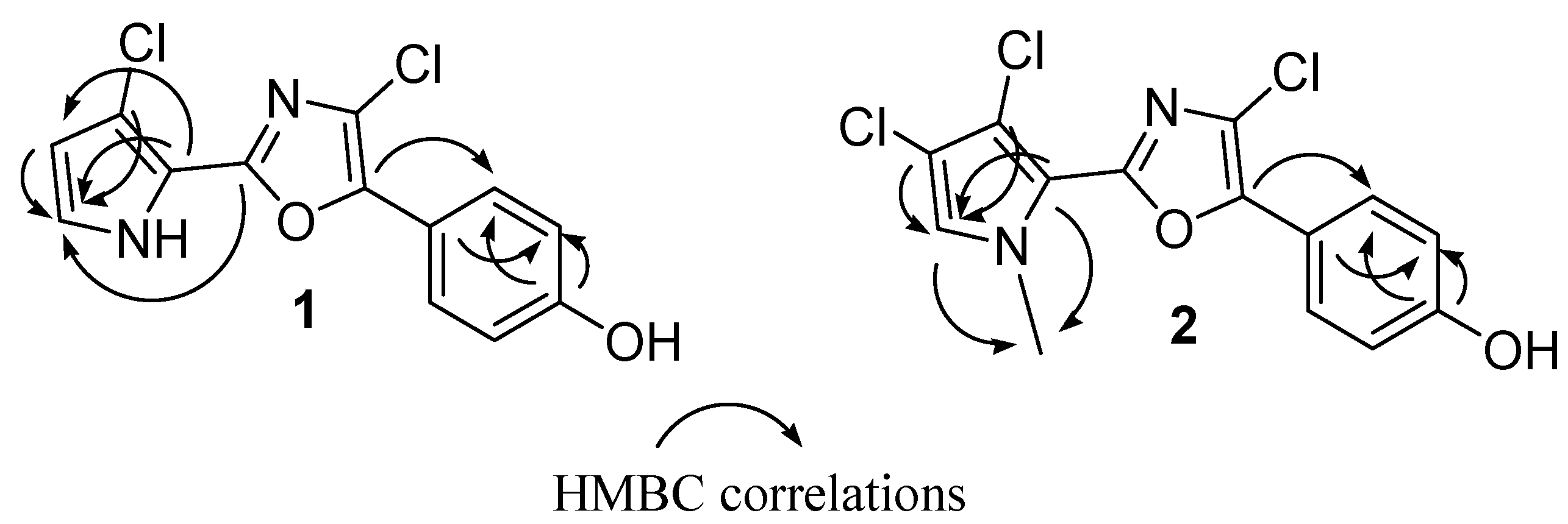
Figure 1. In fact, diagnostic correlations were observed between C-8 (δ
C 144.2) and H-12/H-16, and between C-6 (δ
C 153.7) to H-2 indicating the connection of the oxazole ring to both 1,4-disubstituted benzene and pyrrole. Expected HMBC correlations were also observed between C-5, C-4 and C-3 to H-2 (
Figure 2). Comparison of NMR data with those recorded in the same solvent for phorbazoles (see
Experimental section) confirmed the suggested structure. Compound
1 possessed one more chlorine atom on the oxazole ring with respect to phorbazole D, thus it was named 9-chloro-phorbazole D.
The ESIMS analysis of compound
2 showed peaks at
m/z 343 [M + H]
+ with the typical cluster due to the presence of three chlorine atoms. The HRESIMS spectrum indicated the molecular formula C
14H
9Cl
3N
2O
2 as deduced from the sodiated peak at
m/z 364.9634 [M + Na]
+. The NMR spectra of
2 (
Table 1) showed proton and carbon resonances very similar to those of phorbazole A (see
Experimental Section and [
10]), indicating the same substitution pattern in the heterocyclic rings. The only difference was in the presence of a 3H signal resonating at δ
H 4.00 (δ
C 38.4), which was attributed to a methyl linked at the pyrrole nitrogen. Thus compound
2 was suggested to be
N1-methyl-phorbazole A. Significant HMBC correlations were observed between the
N1-Me and both C-2 (δ
C 125.2) and C-5 (δ
C 115.3), in agreement with the depicted structure (
Figure 2). Treatment of phorbazole A with MeI (see
Experimental Section) gave a compound identical in all respects with compound
2, definitively confirming the structure.
Table 1.
1H (400/600 MHz) and 13C (75/150 MHz) NMR data a of compounds 1 and 2 in CD3OD.
Table 1.
1H (400/600 MHz) and 13C (75/150 MHz) NMR data a of compounds 1 and 2 in CD3OD.
| Position | 1 | 2 |
|---|
| δC mult. | δH (
J in Hz) | | δC mult. | δH (
J in Hz) |
|---|
| 2 | 122.8, CH | 6.97, d (2.8) | | 125.2, CH | 7.10, s |
| 3 | 111.8, CH | 6.27, d (2.8) | | 114.7, C | |
| 4 | 111.8, C | | | 112.4, C | |
| 5 | 115.8, C | | | 115.3, C | |
| 6 | 153.7, C | | | 152.3, C | |
| 8 | 144.2, C | | | 144.2, C | |
| 9 | 116.6, C | | | 117.9, C | |
| 11 | 119.4, C | | | 119.1, C | |
| 12,16 | 127.7, CH | 7.81, d (8.7) | | 127.8, CH | 7.81, d (8.7) |
| 13,15 | 116.5, CH | 6.93, d (8.7) | | 116.9, CH | 6.93, d (8.7) |
| 14 | 159.6, C | | | 159.8, C | |
| N1-Me | 122.8, CH | | | 38.4, CH3 | 4.00, s |
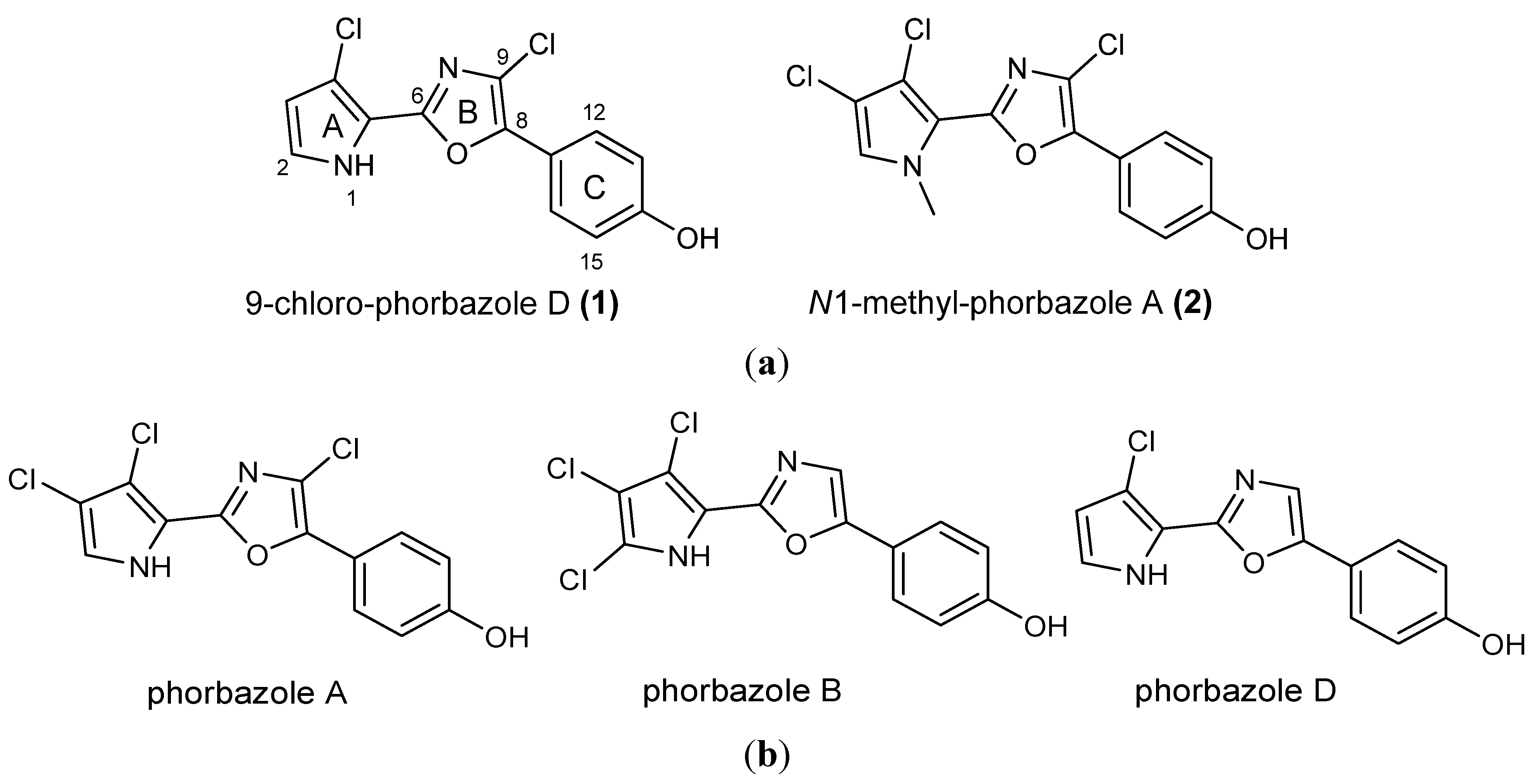
Figure 1.
(a) Structures of novel phorbazoles isolated from the external part of A. andersoni; (b) Structures of known phorbazoles co-occurring in the same extract.
Figure 1.
(a) Structures of novel phorbazoles isolated from the external part of A. andersoni; (b) Structures of known phorbazoles co-occurring in the same extract.
Figure 2.
Key HMBC correlations for phorbazoles 1 and 2.
Figure 2.
Key HMBC correlations for phorbazoles 1 and 2.
The presence of a very minor phorbazole compound was detected in the analyzed ethereal extract. This metabolite was isolated in very small amount which was insufficient for a detailed spectroscopic characterization. However, the
1H NMR and ESIMS spectra were recorded and indicated the presence of a structure in which all positions in both the oxazole and pyrrole rings were substituted with chlorine atoms. In fact, a protonated molecular peak at
m/z 363 [M + H]
+ with a cluster consistent with the presence of four chlorine atoms was observed in the ESIMS spectrum whereas the
1H NMR spectrum exhibited only two doublet signals due to the 4-hydroxyphenyl moiety (see
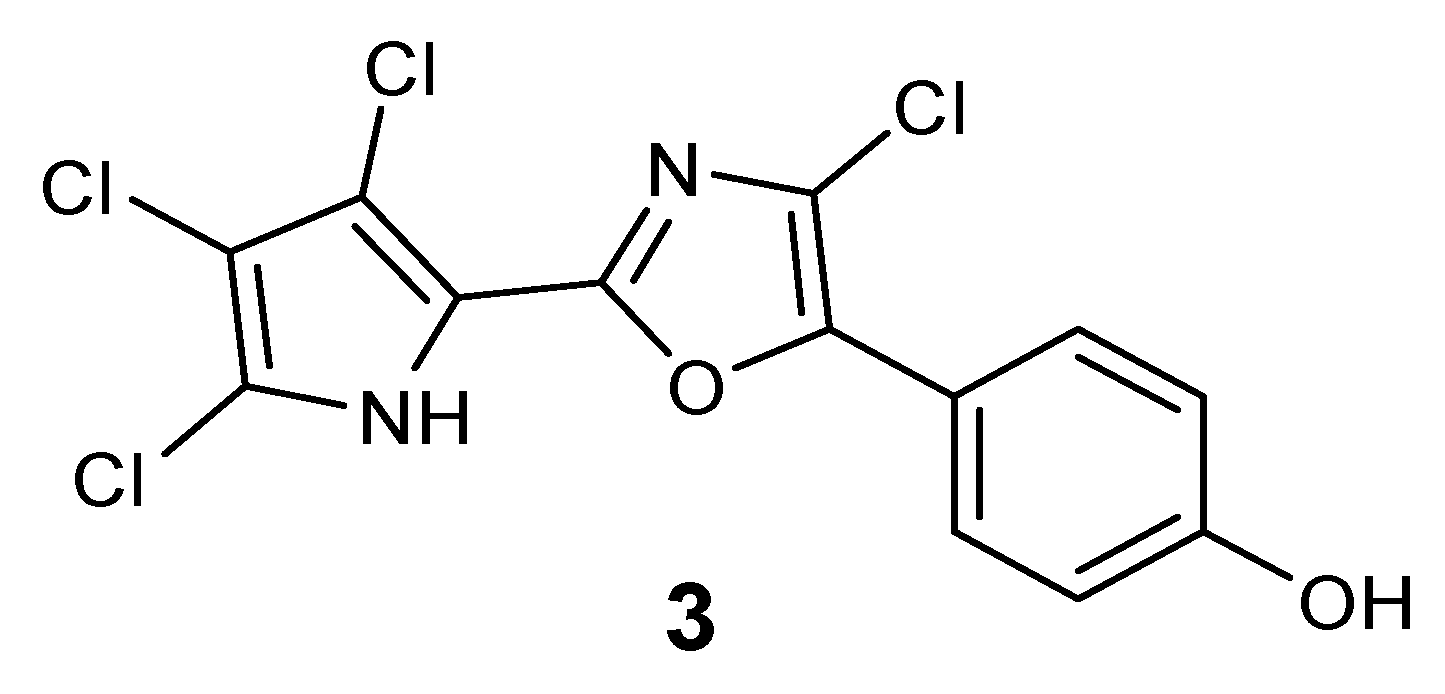
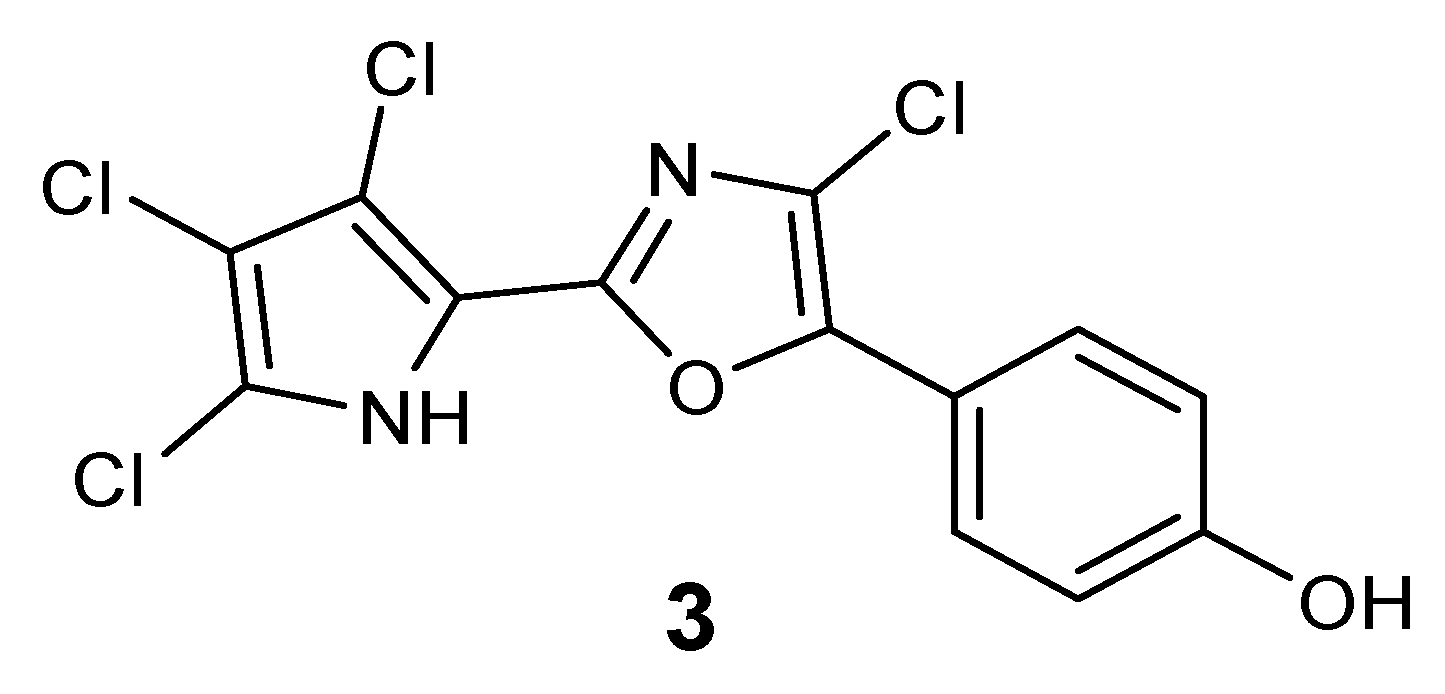
Experimental Section). The compound could be tentatively identified as 2-chloro-phorbazole A, as depicted in
Figure 3.
Figure 3.
Plausible structure of 2-chloro-phorbazole A (3).
Figure 3.
Plausible structure of 2-chloro-phorbazole A (3).
Phorbazole compounds were particularly concentrated in the most exposed external part of the nudibranch, suggesting their involvement in chemical defense. Selected phorbazoles were thus tested in the feeding deterrence assay (see
Experimental Section) against the trophic generalist shrimp
Palaemon elegans [
13]. Even though paucity of material prevented us from obtaining dose response curves for each compound, compound
1, compound
2 and phorbazole A resulted in significant feeding deterrence at a concentration of 1.0 mg/mL (
Figure 4). This finding supports that
A. andersoni, together with similarly colored phyllidiid nudibranchs, are members of a Müllerian mimetic circle in which different unpalatable species had an advantage from becoming similar to others [
14], sharing conspicuous visual signals that predators rapidly learn to avoid.
Preceding studies have indicated that marine invertebrates produce bioactive natural products that may be useful for developing new drugs. [
15] By exploring untapped geographical sources and/or novel groups of organisms one can maximize the search for new marine drugs to treat human diseases. Although immuno-modulatory activity on previously reported phorbazoles has been claimed by the authors [
10], no manuscript has appeared on the subject. In this regard, the
in vitro growth inhibitory activity of phorbazoles has been investigated on a panel of five human cancer cell lines, which included two cell lines displaying actual sensitivity to pro-apoptotic stimuli,
i.e., the MCF-7 mammary adenocarcinoma [
16,
17] and the Hs683 oligodendroglioma [
17,
18] and three cell lines displaying various levels of resistance to pro-apoptotic stimuli,
i.e., the A549 non-small-cell lung cancer [
19] (NSCLC), the SKMEL-28 melanoma [
20] and the U373 glioblastoma [
17,
18]. The MTT colorimetric assay [
16,
17,
20] was employed to measure cell growth. The data obtained is illustrated in
Table 2.
Figure 4.
Palaemon elegans alimentary response. The zero concentration was defined as control, and significant differences in the consumption of treated vs. control pellets have been evaluated by one-tailed Fisher’s exact test (α = 0.05, n = 10 for each bar).
Figure 4.
Palaemon elegans alimentary response. The zero concentration was defined as control, and significant differences in the consumption of treated vs. control pellets have been evaluated by one-tailed Fisher’s exact test (α = 0.05, n = 10 for each bar).
Table 2.
Determination of the IC50 (µM) in vitro growth inhibitory concentration by means of the MTT colorimetric assay in five human cancer cell lines.
Table 2.
Determination of the IC50 (µM) in vitro growth inhibitory concentration by means of the MTT colorimetric assay in five human cancer cell lines.
| Compounds versus Cancer Cell Lines (IC50; µM) | 1 | 2 |
|---|
| (9-chloro-phorbazole D) | (N1-methyl-phorbazole A) |
|---|
| A549 (NSCLC) | 29 | 34 |
| MCF-7 (breast cancer) | 18 | 25 |
| SKMEL-28 (melanoma) | 22 | 29 |
| Hs683 (oligodendroglioma) | 25 | 25 |
| U373 (glioblastoma) | 19 | 19 |
| Mean ± SEM | 22 ± 2 | 26 ± 2 |
The data shows that compounds
1 and
2 display similar
in vitro growth inhibitory activity in the five human cancer cell lines under study and that the growth inhibitory activity of these two compounds is not modified as whether the cancer cells display actual sensitivity (MCF-7; Hs683) as opposed to various levels of resistance (A549; SKMEL-28; U373) to pro-apoptotic stimuli. The
in vitro growth inhibitory activity displayed by
1 and
2 are of the same range, or even higher, than those displayed by carboplatin [
21] and temozolomide [
22], which are the two compounds largely used to treat patients with aggressive cancers [
23,
24].
Computer-assisted phase contrast microscopy [
25,
26] (quantitative videomicroscopy) was then used to determine as to whether the
in vitro growth inhibitory activity observed for
2 (for which sufficient amounts were available) was related or not to the cytostatic
versus cytotoxic effects. The quantitative videomicroscopy analyses confirmed the
in vitro growth inhibitory activity of
N1-methyl-phorbazole A (
2) as initially revealed by the MTT colorimetric assay (
Table 2) and as illustrated below in
Figure 5.
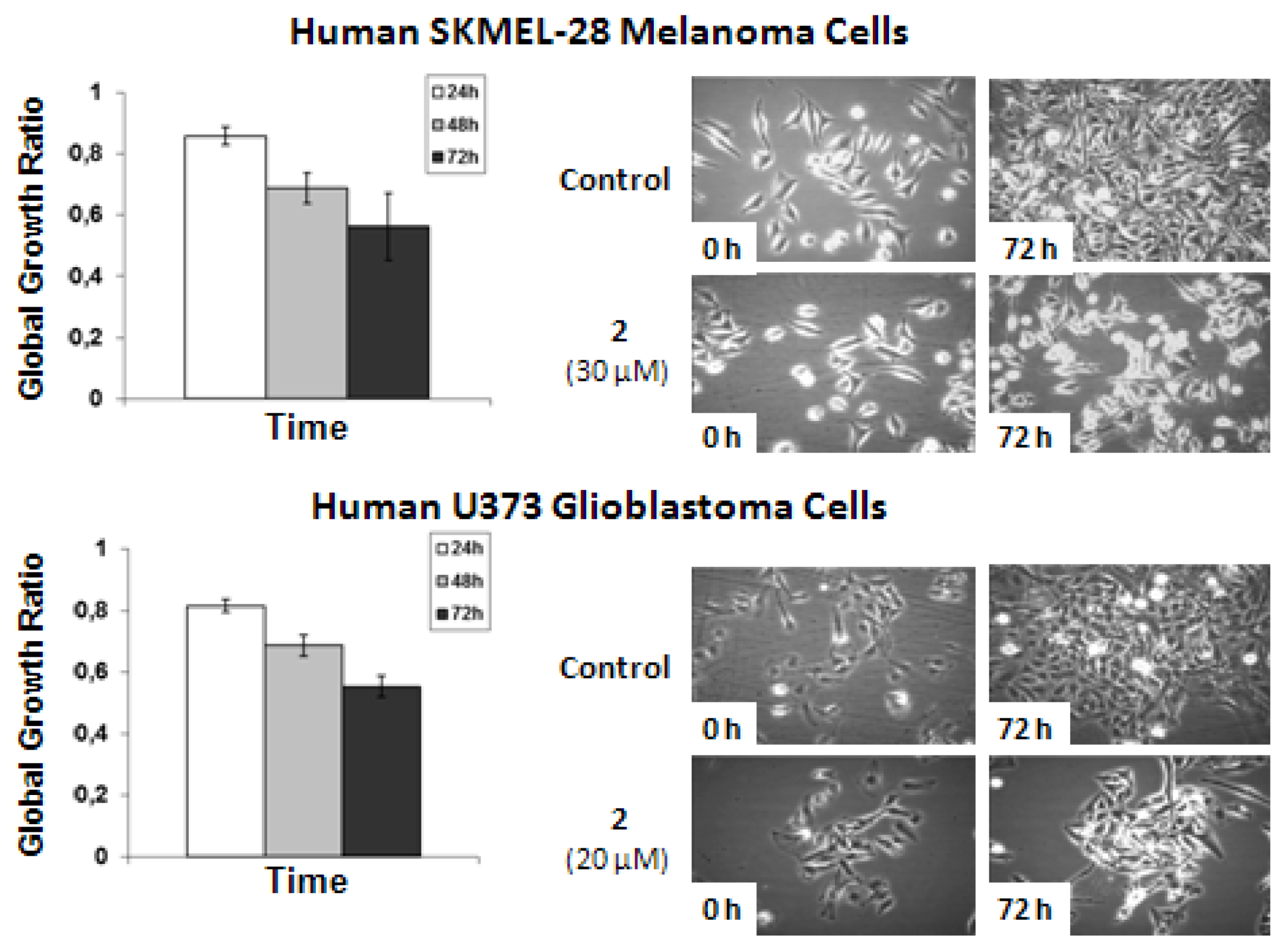
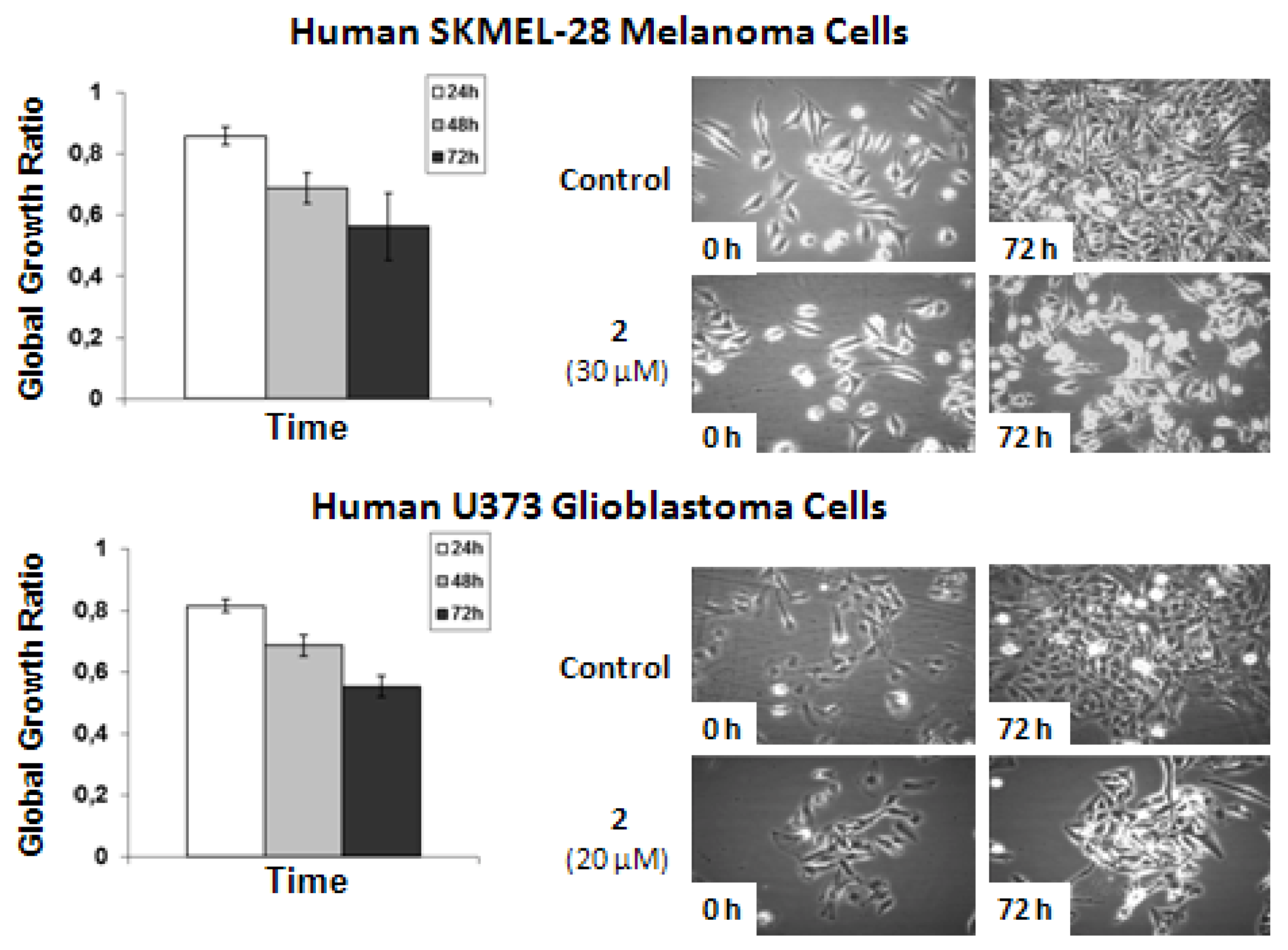
The data shows that
2-related
in vitro anticancer activity mainly relates to cytostatic effects in both human SKMEL-28 melanoma and U373 glioblastoma cells. The concentrations used to analyze the effects of
2 by means of quantitative videomicroscopy were those obtained by means of the MTT colorimetric assay,
i.e., 30 μM for SKMEL-28 melanoma cells and 20 μM for U373 glioblastoma cells (
Table 2). These MTT test-related IC
50 concentrations (
Table 2) also turned out to decrease by ~50% the global growth activity in U373 glioblastoma and SKMEL-28 melanoma cells under quantitative videomicroscopy monitoring (
Figure 5).
Figure 5.
Quantitative videomicroscopy analyses of 2-induced effects on SKMEL-28 melanoma and U373 glioblastoma cells.
Figure 5.
Quantitative videomicroscopy analyses of 2-induced effects on SKMEL-28 melanoma and U373 glioblastoma cells.
3. Experimental Section
3.1. General Experimental Procedures
Optical rotations were measured using a Jasco DIP 370 digital spectropolarimeter. IR spectra were measured on a Biorad FTS 155 FTIR spectrophotometer. 1D- and 2D-NMR spectra were recorded on Bruker Avance-400 (400.13 MHz) and Bruker DRX-600 equipped with TXI CryoProbeTM in CD3OD and DMSO-d6 (shifts are referenced to the solvent signal at δ 3.34 and 2.50, respectively) and 13C NMR spectra were recorded on Bruker DPX-300 (75 MHz) and Bruker DRX-600 (150 MHz) spectrometers (δ values are reported to CD3OD, 49.0 ppm and to DMSO-d6 at 39.5 ppm). HRESIMS measurements were carried out on Micromass Q-TOF micro; HPLC Shimadzu liquid chromatograph LC-10AD was equipped with an UV SPD-10A wavelength detector and performed with a semipreparative column RP-18 (Supelco, 250 mm × 46 mm, 5 μm); TLC plates (KieselGel 60 F254) were from Merck. Solvents for chromatography were HPLC grade hence were used without further purification.
3.2. Sample Collection
Six specimens of Aldisa andersoni were collected in November 2009 from the Indian Ocean by G.V. and transferred to the laboratory. The nudibranchs (average size 25 mm) were identified by Dr. Juan Lucas Cervera (University of Cadiz).
3.3. Extraction and Purification
The frozen molluscs A. andersoni were treated, in order to obtain extracts of the mantle and the internal glands, separately. The external extract was obtained by simply soaking the whole animals in acetone bath (20 mL) for few minutes, whereas the internal extract was obtained by grinding the animal after sonication. The extracts were concentrated in vacuo and the aqueous residues were then fractionated between H2O and Et2O (4 × 50 mL). The Et2O portions were evaporated under reduced pressure to give 15 mg from the external and 62 mg from the internal parts of the nudibranchs. The external part extract was directly purified by C18 reversed-phase HPLC using a Supelco RP-18 column (250 × 4.60 mm) with a linear gradient from MeOH/H2O 70:30 to MeOH 100% in 40 min (flow 1 mL/min) to afford in order of decreasing polarity phorbazole D (tR 7.4 min, 0.7 mg), 9-chloro-phorbazole D (1) (tR 11.8 min, 4.9 mg), phorbazole A (tR 19.2 min, 4.0 mg), phorbazole B (tR 21.0 min, 2.5 mg), N1-methyl-phorbazole A (2) (tR 28.9 min, 1.1 mg), and 2-chloro-phorbazole A (3) (tR 31.2 min, <0.5 mg).
3.4. 9-Chloro-phorbazole D (1)
Yellow powder (32.7%); UV (MeOH) λ
max (log ε) 208 (6.90), 256 (6.80), 334 (7.01); IR (liquid film) ν
max 1614, 1506, 1449, 1406, 1278, 1176, 837 cm
−1;
1H NMR and
13C NMR CD
3OD, see
Table 1,
1H NMR in DMSO-
d6: δ
H 7.73 (d,
3JH,H = 8.4 Hz, 2H, H-12 and H-16), δ
H 7.07 (d,
3JH,H = 2.9 Hz, 1H, H-2), δ
H 6.94 (d,
3JH,H = 8.4 Hz, 2H, H-13 and H-15), δ
H 6.31 (d,
3JH,H = 2.9 Hz, 1H, H-3);
13C NMR in DMSO-
d6: δ
C 158.3 (C-14), δ
C 151.7 (C-6), δ
C 142.1 (C-8), δ
C 126.2 (C-12 and C-16), δ
C 122.5 (C-2), δ
C 122.3 (C-9), δ
C 116.9 (C-11), δ
C 116.0 (C-13 and C-15), δ
C 114.7 (C-5), δ
C 113.2 (C-4), δ
C 110.6 (C-3). HRESIMS [M + Na]
+ m/z 316.9875 (calcd for C
13H
8Cl
2N
2O
2, 316.9861).
3.5. N1-Methyl-phorbazole A (2)
Yellow powder (7.3%);
1H NMR and
13C NMR in CD
3OD, see
Table 1,
1H NMR in DMSO-
d6: δ
H 7.71 (d,
3JH,H = 8.8 Hz, 2H, H-12 and H-16), δ
H 7.42 (s, H-2), δ
H 6.94 (d,
3JH,H = 8.8 Hz, 2H, H-13 and H-15), δ
H 3.92 (s, 1-
N-Me);
13C NMR in DMSO-
d6: δ
C 157.6 (C-14), δ
C 151.3 (C-6), δ
C 142.3 (C-8), δ
C 126.1 (C-12 and C-16), δ
C 124.2 (C-2), δ
C 122.4 (C-9), δ
C 116.1 (C-11), δ
C 115.7 (C-13 and C-15), δ
C 115.3 (C-5), δ
C 111.2 (C-4), δ
C 109.0 (C-3), δ
C 37.5 (
N1-Me). HRESIMS [M + Na]
+ m/z 364.9634 (calcd for C
14H
9Cl
3N
2O
2, 364.9627).
3.6. Methylation of Phorbazole A
An aliquot of phorbazole A (1 mg) was treated with 1 mL of MeI and Na2CO3 (12 mg) in 500 μL of anhydrous acetone stirring at r.t. for 3 h. As reaction reached the completion, the acetone was removed under reduced pressure, and the reaction mixture was loaded into a SiO2 column eluting with CHCl3/MeOH 8:2, to obtain 0.8 mg of a compound identical with the natural compound 2 by NMR and MS.
3.7. 2-Chloro-phorbazole A (3)
LCMS (m/z 363 [M + H]+); 1H NMR in CD3OD: δH 7.82 (d, 3JH,H = 8.7 Hz, 2H, H-12 and H-16), δH 6.94 (d, 3JH,H = 8.7 Hz, 2H, H-13 and H-15).
3.8. NMR Data of Phorbazole A in CD3OD
1H NMR (recorded at 600 MHz) δH 7.80 (d, 3JH,H = 8.7, 2H, H-12 and H-16), δH 7.08 (s, 1H, H-2), δH 6.92 (d, 3JH,H = 8.7, 2H, H-13 and H-15); 13C NMR δC 159.7 (C-14, C), δC 152.8 (C-6, C), δC 144.6 (C-8, C), δC 127.8 (C-12 and C-16, CH), δC 124.4 (C-9, C), δC 120.2 (C-2, CH), δC 119.1 (C-5 and C-11, C), δC 116.8 (C-13 and C-15, CH), δC 113.9 (C-3 or C-4, C), δC 113.6 (C-4 or C-3, C).
3.9. NMR Data of Phorbazole B in CD3OD
1H NMR (recorded at 600 MHz) δH 7.51 (d, 3JH,H = 8.0, 2H, H-12 and H-16), δH 7.29 (s, 1H, H-9), δH 6.82 (d, 3JH,H = 8.0, 2H, H-13 and H-15); 13C NMR δC 159.3 (C-14, C), δC 152.1 (C-8, C), δC 155.5 (C-6, C), δC 126.8 (C-12 and C-16, CH), δC 121.1 (C-9, C), δC 120.6 (C-11, C), δC 117.8 (C-5 and C-2, C), δC 116.8 (C-13 and C-15, CH), δC 111.9 (C-3 and C-4, C).
3.10. NMR Data of Phorbazole D in CD3OD
1H NMR (recorded at 600 MHz) δH 7.64 (d, 3JH,H = 8.0, 2H, H-12 and H-16), δH 7.36 (s, 1H, H-9), δH 6.95 (d, 3JH, H = 2.0 Hz, 1H, H-2), δH 6.90 (d, 3J = 8.0, 2H, H-13 and H-15), δH 6.26 (d, 3JH,H = 2.0 Hz, 1H, H-3); 13C NMR δC 159.5 (C-14, C), δC 155.2 (C-6, C), δC 152.2 (C-8, C), δC 126.2 (C-12 and C-16, CH), δC 121.8 (C-2, CH), δC 120.5 (C-9, CH), δC 119.8 (C-11, C), δC 116.3 (C-13 and C-15, CH), δC 111.1 (C-3, CH), C-4 and C-5 n.d.
3.11. Feeding-Deterrence Assay
The compounds were tested for their feeding deterrence activity against the generalist shrimp
Palaemon elegans (Rathke, 1837). Assays were performed as described in Mollo [
13], by using food pellets treated with compounds
1 and
2, and phorbazole A, at concentrations of 1.0 mg/mL. Each pure compound, dissolved with 0.5 mL of acetone to give the desired final concentration, was added to a mixture of lyophilized squid mantle, alginate, purified sea sand and mixed. After evaporation of the solvent, distilled water and a drop of red food color were added to make a final volume of 1.0 mL. The paste was hardened into a 0.25 M CaCl
2 solution (two minutes) and cut into 10 mm long strips. Accordingly, controls were prepared with acetone only (zero concentration). Shrimps were collected along the coast of Pozzuoli, Italy, and habituated to the control food for a week before experiments. After three days of total fasting, ten randomly picked shrimps were assayed as a series of individual replicates for each concentration and the control (
n = 10 for each series). Shrimps were placed individually into plastic beakers filled with seawater. A colored food strip was given to each shrimp, and shrimps were not re-used. Control and treatments were carried out in parallel. The presence of a red spot visible by transparency in the gastric mill and the stomach of the shrimps after 30 min was considered as an acceptance of the food, while the absence of the spot gave a rejection response. Statistical analysis between treatments and controls was performed using the one-tailed Fisher-Exact test, which is traditionally used with relatively small samples, with α = 0.05 as significant level. After the experiments, the shrimps were returned in field to the same location they were collected.
3.12. Determination of in Vitro Anticancer Activity
The histological types and origins of the five cancer cell lines that were used for the MTTcolorimetric assay are detailed in
Table 2. The cells were cultured in RPMI (Lonza, Verviers, Belgium) medium supplemented with 10% heat inactivated foetal calf serum (Lonza). All culture media were supplemented with 4 mM glutamine, 100 µg/mL gentamicin, and 200 U/mL penicillin and 200 µg/mL streptomycin (Lonza). The overall growth level of the human cancer cell lines was determined using a colorimetric MTT (3-[4,5-dimethylthiazol-2yl]-diphenyl tetrazolium bromide, Sigma, Belgium) assay as detailed previously [
16,
17,
20,
21]. Six replicates of each experimental condition were performed.. Thus, this procedure enables the concentration of
1 and
2 that decreased by 50% the growth of each cell line after having cultured it with the compound of interest for 72 h (the IC
50 index in μM) to be determined.
3.13. Computer-Assisted Phase Contrast Microscopy (Quantitative Videomicroscopy)
The direct visualization of compound-induced effects in terms of cytotoxicity
versus cytostaticity in human SKMEL-28 melanoma and U373 glioblastoma cells was achieved as detailed in literature [
25,
26]. Briefly, the quantitative videomicroscopy approach enables an image of the bottom of the seeded flask to be taken every four minutes over the course of a 72 h-observation period. Thus, 1080 digitized images are available for each experimental condition, which were run in triplicates. A global growth ratio (the GGR index) was calculated for
2, resulting in a value that can be directly compared to the MTT assay-determined IC
50 value (
Table 2). First, the global growth (GG) is calculated in each control and in each treated condition by dividing the number of cells on the last image by the number of cells on the first image. The GGR index was thus obtained for
2 by dividing the GG values calculated for treated SKMEL-28 or U373 cells by the GG values calculated for the control (the data are presented as mean ± SEM values). For example, a GGR value of 0.5 for
2 at a given experimental time means that this compound decreased by 50% (1.0 − 0.5 = 0.5) the global growth of the considered cancer cell population as compared to control.



 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}