Cyclisation Increases the Stability of the Sea Anemone Peptide APETx2 but Decreases Its Activity at Acid-Sensing Ion Channel 3


 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
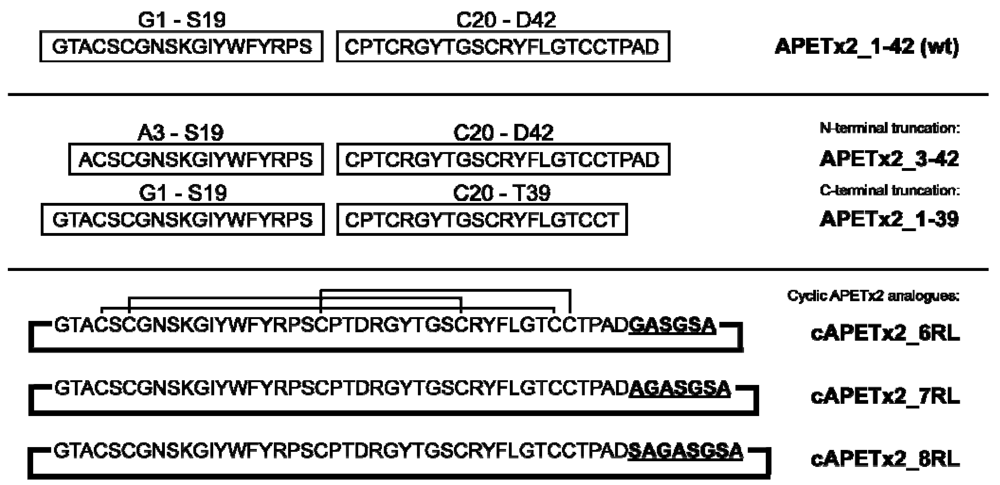
2.1. Peptide Design and Synthesis
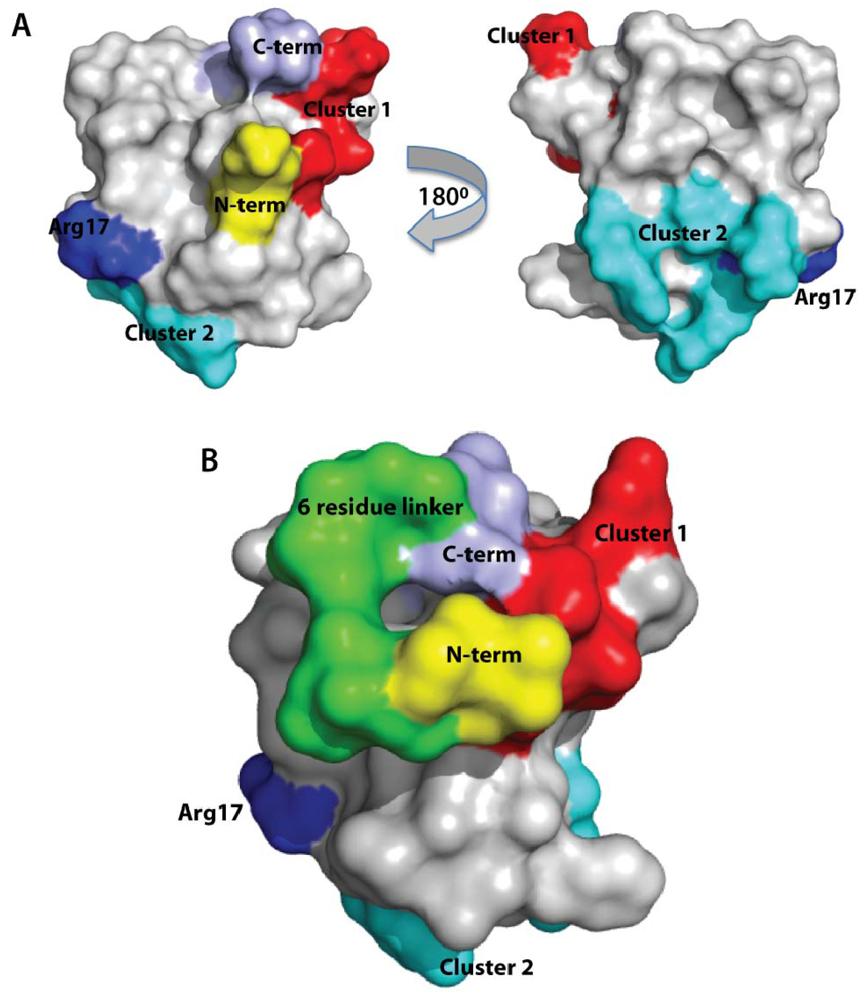
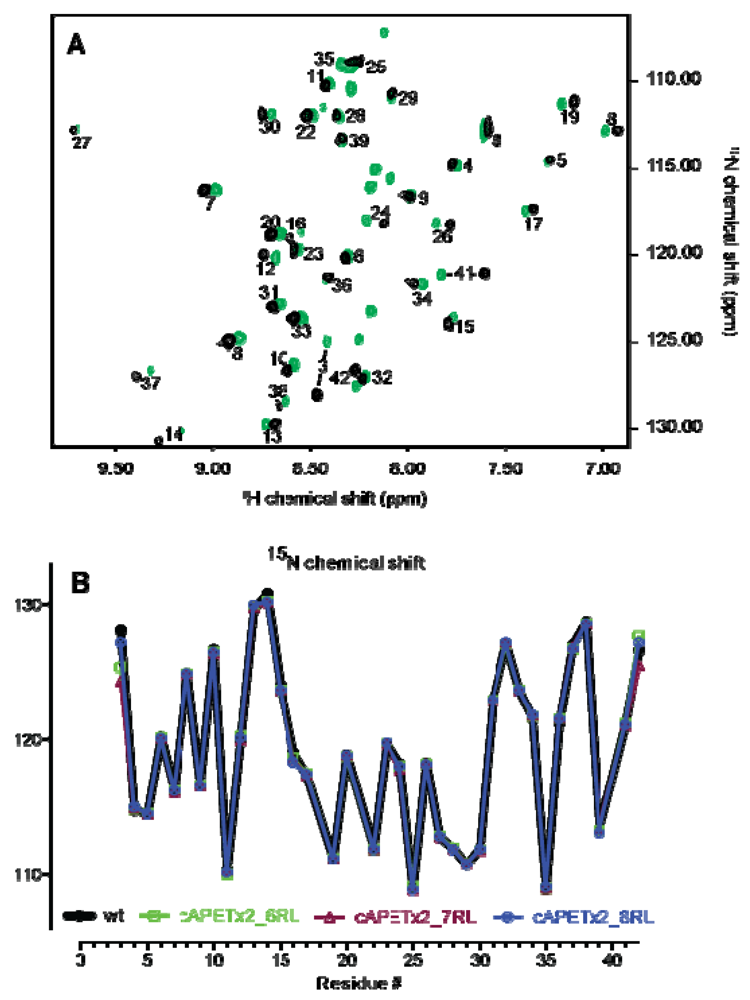
2.2. Confirmation of Structural Integrity by NMR

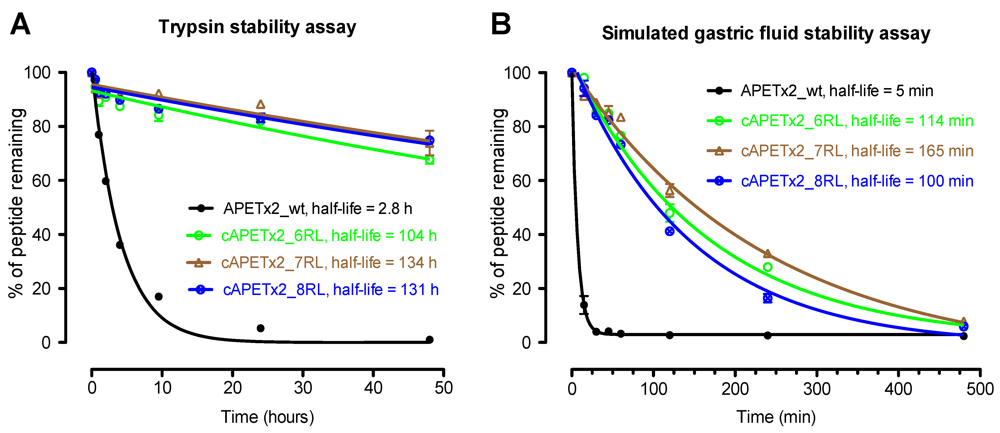
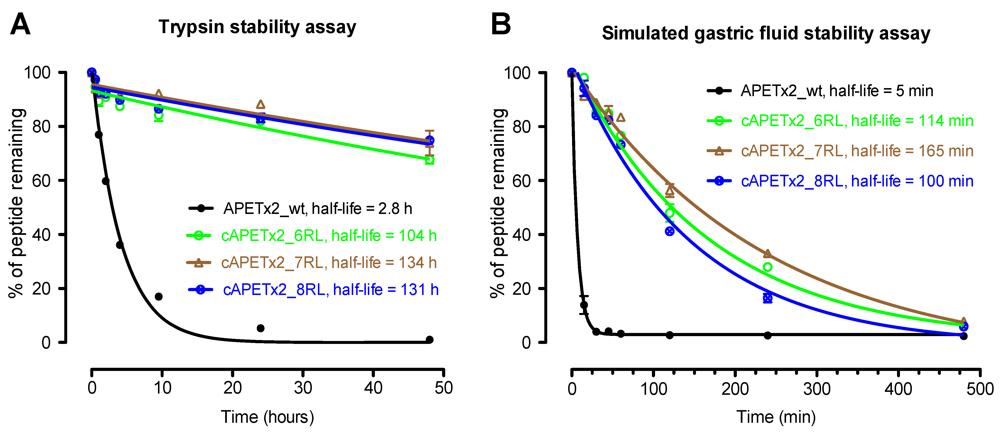
2.3. Biological Stability of Cyclic APETx2 Analogues
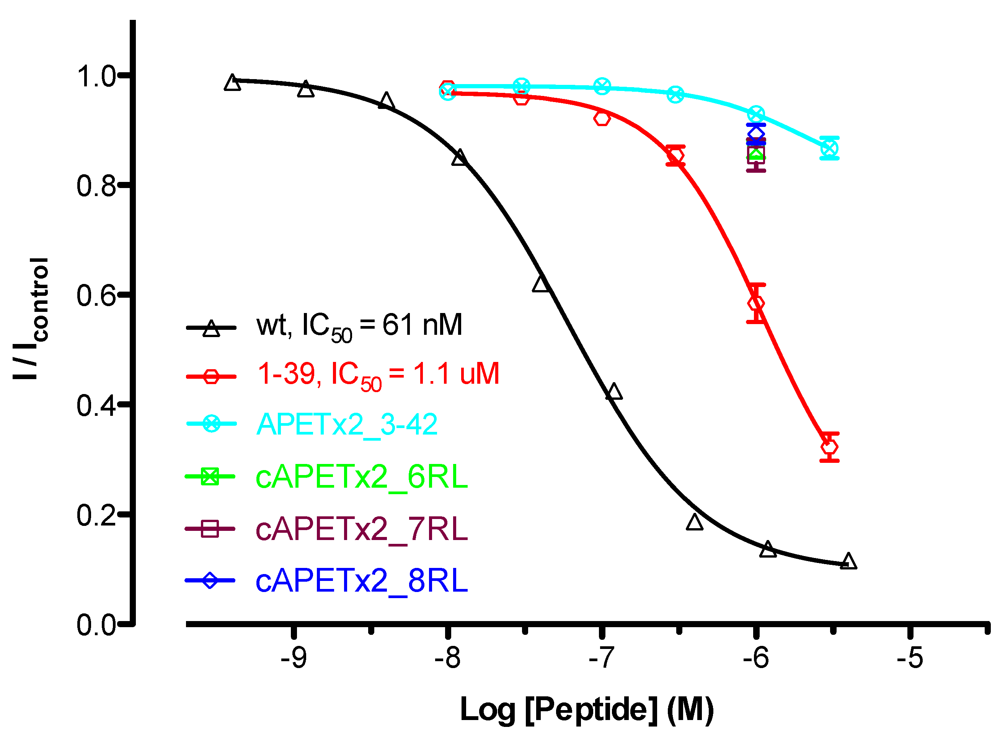
2.4. Activity of Cyclic APETx2_6-, 7- and 8-Residue Linker

2.5. Activity of APETx2 N- and C-Terminal Truncations
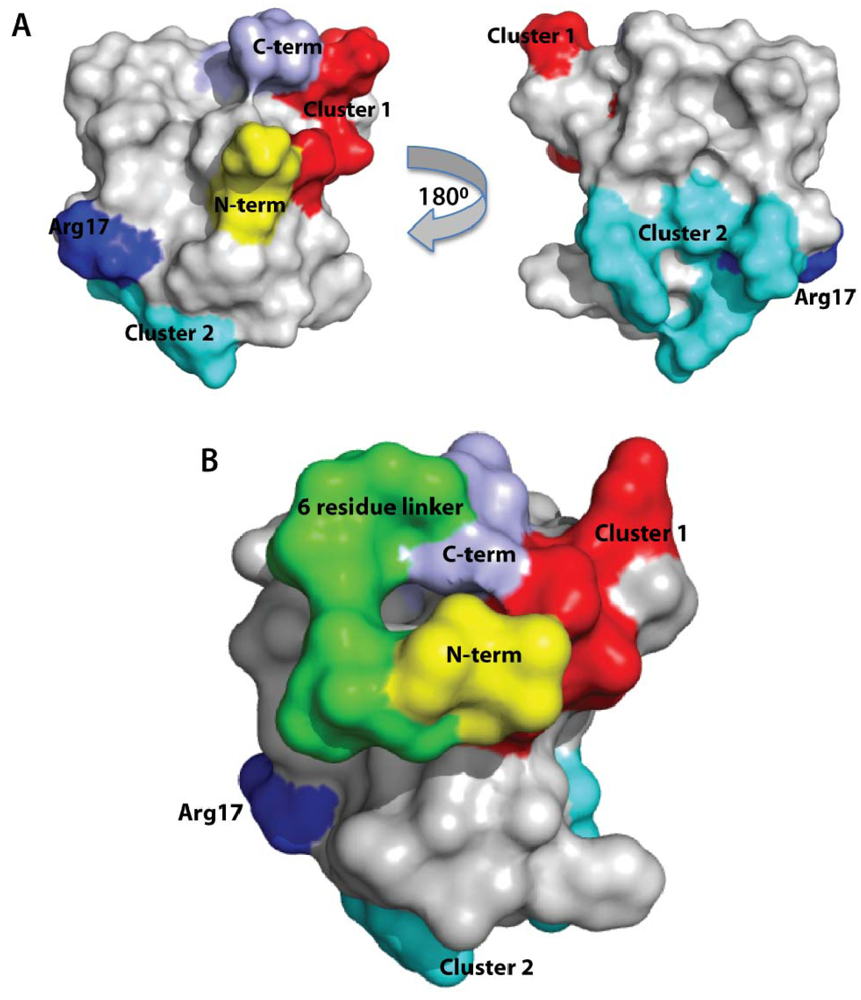
2.6. Discussion
3. Experimental Section
3.1. Materials
3.2. Peptide Production
3.2.1. Peptide Synthesis
3.2.2. RP-HPLC and Mass Spectrometry
3.2.3. Cyclisation and Oxidation of APETx2 Analogues
3.2.4. Formation of Truncation Analogues by Native Chemical Ligation
3.2.5. Concentration Determination
3.3. Nuclear Magnetic Resonance Spectroscopy
3.4. Stability Assays
3.4.1. Trypsin Stability Assay
3.4.2. Simulated Gastric Fluid Assay
3.5. Electrophysiological Recordings
4. Conclusions
Acknowledgments
References
- Lax, R. The Future of Peptide Development in the Pharmaceutical Industry. PharManufacturing: The International Peptide Review 2010, 10–15. [Google Scholar]
- King, G.F. Venoms as a platform for human drugs: Translating toxins into therapeutics. Expert Opin. Biol. Ther. 2011, 11, 1469–1484. [Google Scholar] [CrossRef]
- Thayer, A.M. Improving peptides. Chem. Eng. News 2011, 89, 13–20. [Google Scholar]
- Chiras, D.D. Nutrition and digestion. In Human Biology, 7th ed; Jones & Bartlett Learning: Mississanga, Canada, 2011; pp. 89–117. [Google Scholar]
- Clark, R.J.; Fischer, H.; Dempster, L.; Daly, N.L.; Rosengren, K.J.; Nevin, S.T.; Meunier, F.A.; Adams, D.J.; Craik, D.J. Engineering stable peptide toxins by means of backbone cyclization: Stabilization of the α-conotoxin MII. Proc. Natl. Acad. Sci. USA 2005, 102, 13767–13772. [Google Scholar]
- Linde, Y.; Ovadia, O.; Safrai, E.; Xiang, Z.; Portillo, F.P.; Shalev, D.E.; Haskell-Luevano, C.; Hoffman, A.; Gilon, C. Structure-activity relationship and metabolic stability studies of backbone cyclization and N-methylation of melanocortin peptides. Biopolymers 2008, 90, 671–682. [Google Scholar] [CrossRef]
- Adessi, C.; Frossard, M.J.; Boissard, C.; Fraga, S.; Bieler, S.; Ruckle, T.; Vilbois, F.; Robinson, S.M.; Mutter, M.; Banks, W.A.; et al. Pharmacological profiles of peptide drug candidates for the treatment of Alzheimer’s disease. J. Biol. Chem. 2002, 278, 13905–13911. [Google Scholar]
- Lewis, R.J.; Garcia, M.L. Therapeutic potential of venom peptides. Nat. Rev. Drug Discov. 2003, 2, 790–802. [Google Scholar] [CrossRef]
- Saez, N.J.; Senff, S.; Jensen, J.E.; Er, S.Y.; Herzig, V.; Rash, L.D.; King, G.F. Spider-venom peptides as therapeutics. Toxins 2010, 2, 2851–2871. [Google Scholar] [CrossRef]
- Vetter, I.; Davis, J.L.; Rash, L.D.; Anangi, R.; Mobli, M.; Alewood, P.F.; Lewis, R.J.; King, G.F. Venomics: A new paradigm for natural products-based drug discovery. Amino Acids 2011, 40, 15–28. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Hasegawa, Y.; Honma, T.; Nagashima, Y.; Shiomi, K. Screening and cDNA cloning of Kv1 potassium channel toxins in sea anemones. Mar. Drugs 2010, 8, 2893–2905. [Google Scholar] [CrossRef]
- Diochot, S.; Baron, A.; Rash, L.D.; Deval, E.; Escoubas, P.; Scarzello, S.; Salinas, M.; Lazdunski, M. A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. EMBO J. 2004, 23, 1516–1525. [Google Scholar] [CrossRef]
- Sluka, K.A.; Winter, O.C.; Wemmie, J.A. Acid-sensing ion channels: A new target for pain and CNS diseases. Curr. Opin. Drug Discov. Devel. 2009, 12, 693–704. [Google Scholar]
- Li, W.G.; Xu, T.L. ASIC3 Channels in multimodal sensory perception. ACS Chem. Neurosci. 2011, 2, 26–37. [Google Scholar] [CrossRef]
- Deval, E.; Noël, J.; Lay, N.; Alloui, A.; Diochot, S.; Friend, V.; Jodar, M.; Lazdunski, M.; Lingueglia, E. ASIC3, a sensor of acidic and primary inflammatory pain. EMBO J. 2008, 27, 3047–3055. [Google Scholar] [CrossRef]
- Karczewski, J.; Spencer, R.H.; Garsky, V.M.; Liang, A.; Leitl, M.D.; Cato, M.J.; Cook, S.P.; Kane, S.; Urban, M.O. Reversal of acid-induced and inflammatory pain by the selective ASIC3 inhibitor, APETx2. Br. J. Pharmacol. 2010, 161, 950–960. [Google Scholar] [CrossRef]
- Theralpha, Product THA902. Available online: http://www.theralpha.com/index.php?page=tha902 (accessed on 4 May 2012).
- Jensen, J.E.; Durek, T.; Alewood, P.F.; Adams, D.J.; King, G.F.; Rash, L.D. Chemical synthesis and folding of APETx2, a potent and selective inhibitor of acid sensing ion channel 3. Toxicon 2009, 54, 56–61. [Google Scholar] [CrossRef]
- Chagot, B.; Escoubas, P.; Diochot, S.; Bernard, C.; Lazdunski, M.; Darbon, H. Solution structure of APETx2, a specific peptide inhibitor of ASIC3 proton-gated channels. Protein Sci. 2005, 14, 2003–2010. [Google Scholar]
- Clark, R.J.; Jensen, J.; Nevin, S.T.; Callaghan, B.P.; Adams, D.J.; Craik, D.J. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. Int. Ed. Engl. 2010, 49, 6545–6548. [Google Scholar]
- Kwan, A.H.; Mobli, M.; Gooley, P.R.; King, G.F.; Mackay, J.P. Macromolecular NMR spectroscopy for the non-spectroscopist. FEBS J. 2011, 278, 687–703. [Google Scholar]
- King, G.F.; Shih, Y.L.; Maciejewski, M.W.; Bains, N.P.; Pan, B.; Rowland, S.L.; Mullen, G.P.; Rothfield, L.I. Structural basis for the topological specificity function of MinE. Nat. Struct. Biol. 2000, 7, 1013–1017. [Google Scholar] [CrossRef]
- Schanda, P.; Brutscher, B. Very fast two-dimensional NMR spectroscopy for real-time investigation of dynamic events in proteins on the time scale of seconds. J. Am. Chem. Soc. 2005, 127, 8014–8015. [Google Scholar] [CrossRef]
- Anangi, R.; Chen, C.C.; Lin, Y.W.; Cheng, Y.R.; Cheng, C.H.; Chen, Y.C.; Chu, Y.P.; Chuang, W.J. Expression in Pichia pastoris and characterization of APETx2, a specific inhibitor of acid sensing ion channel 3. Toxicon 2010, 56, 1388–1397. [Google Scholar] [CrossRef]
- Lingueglia, E. Acid-sensing ion channels in sensory perception. J. Biol. Chem. 2007, 282, 17325–17329. [Google Scholar] [CrossRef]
- Deval, E.; Noël, J.; Gasull, X.; Delaunay, A.; Alloui, A.; Friend, V.; Eschalier, A.; Lazdunski, M.; Lingueglia, E. Acid-sensing ion channels in postoperative pain. J. Neurosci. 2011, 31, 6059–6066. [Google Scholar] [CrossRef]
- Blanchard, M.G.; Rash, L.D.; Kellenberger, S. Inhibition of voltage-gated Na(+) currents in sensory neurones by the sea anemone toxin APETx2. Br. J. Pharmacol. 2012, 165, 2167–2177. [Google Scholar]
- Norton, R.S. Structures of sea anemone toxins. Toxicon 2009, 54, 1075–1088. [Google Scholar] [CrossRef]
- Shiomi, K. Novel peptide toxins recently isolated from sea anemones. Toxicon 2009, 54, 1112–1118. [Google Scholar] [CrossRef]
- Lesner, A.; Łęgowska, A.; Wysocka, M.; Rolka, K. Sunflower trypsin inhibitor 1 as a molecular scaffold for drug discovery. Curr. Pharm. Des. 2011, 17, 4308–4317. [Google Scholar] [CrossRef]
- Gould, A.; Ji, Y.; Aboye, T.L.; Camarero, J.A. Cyclotides, a novel ultrastable polypeptide scaffold for drug discovery. Curr. Pharm. Des. 2011, 17, 4294–4307. [Google Scholar] [CrossRef]
- Halai, R.; Callaghan, B.; Daly, N.L.; Clark, R.J.; Adams, D.J.; Craik, D.J. Effects of cyclization on stability, structure, and activity of α-conotoxin RgIA at the α9α10 nicotinic acetylcholine receptor and GABAB receptor. J. Med. Chem. 2011, 13, 6984–6992. [Google Scholar]
- Lovelace, E.S.; Armishaw, C.J.; Colgrave, M.L.; Wahlstrom, M.E.; Alewood, P.F.; Daly, N.L.; Craik, D.J. Cyclic MrIA: A stable and potent cyclic conotoxin with a novel topological fold that targets the norepinephrine transporter. J. Med. Chem. 2006, 49, 6561–6568. [Google Scholar]
- Schnölzer, M.; Alewood, P.; Jones, A.; Alewood, D.; Kent, S.B. In situ neutralization in Boc-chemistry solid phase peptide synthesis. Rapid, high yield assembly of difficult sequences. Int. J. Pept. Protein Res. 1992, 40, 180–193. [Google Scholar]
- Fu, T.J.; Abbott, U.R.; Hatzos, C. Digestibility of food allergens and nonallergenic proteins in simulated gastric fluid and simulated intestinal fluid-a comparative study. J. Agric. Food Chem. 2002, 50, 7154–7160. [Google Scholar] [CrossRef]
- Tugyi, R.; Uray, K.; Iván, D.; Fellinger, E.; Perkins, A.; Hudecz, F. Partial D-amino acid substitution: Improved enzymatic stability and preserved Ab recognition of a MUC2 epitope peptide. Proc. Natl. Acad. Sci. USA 2005, 102, 413–418. [Google Scholar]
- Samples Availability: Available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jensen, J.E.; Mobli, M.; Brust, A.; Alewood, P.F.; King, G.F.; Rash, L.D. Cyclisation Increases the Stability of the Sea Anemone Peptide APETx2 but Decreases Its Activity at Acid-Sensing Ion Channel 3. Mar. Drugs 2012, 10, 1511-1527. https://doi.org/10.3390/md10071511
Jensen JE, Mobli M, Brust A, Alewood PF, King GF, Rash LD. Cyclisation Increases the Stability of the Sea Anemone Peptide APETx2 but Decreases Its Activity at Acid-Sensing Ion Channel 3. Marine Drugs. 2012; 10(7):1511-1527. https://doi.org/10.3390/md10071511
Chicago/Turabian StyleJensen, Jonas E., Mehdi Mobli, Andreas Brust, Paul F. Alewood, Glenn F. King, and Lachlan D. Rash. 2012. "Cyclisation Increases the Stability of the Sea Anemone Peptide APETx2 but Decreases Its Activity at Acid-Sensing Ion Channel 3" Marine Drugs 10, no. 7: 1511-1527. https://doi.org/10.3390/md10071511
APA StyleJensen, J. E., Mobli, M., Brust, A., Alewood, P. F., King, G. F., & Rash, L. D. (2012). Cyclisation Increases the Stability of the Sea Anemone Peptide APETx2 but Decreases Its Activity at Acid-Sensing Ion Channel 3. Marine Drugs, 10(7), 1511-1527. https://doi.org/10.3390/md10071511