Promoter Trapping in Microalgae Using the Antibiotic Paromomycin as Selective Agent

Abstract
:1. Introduction
2. Results
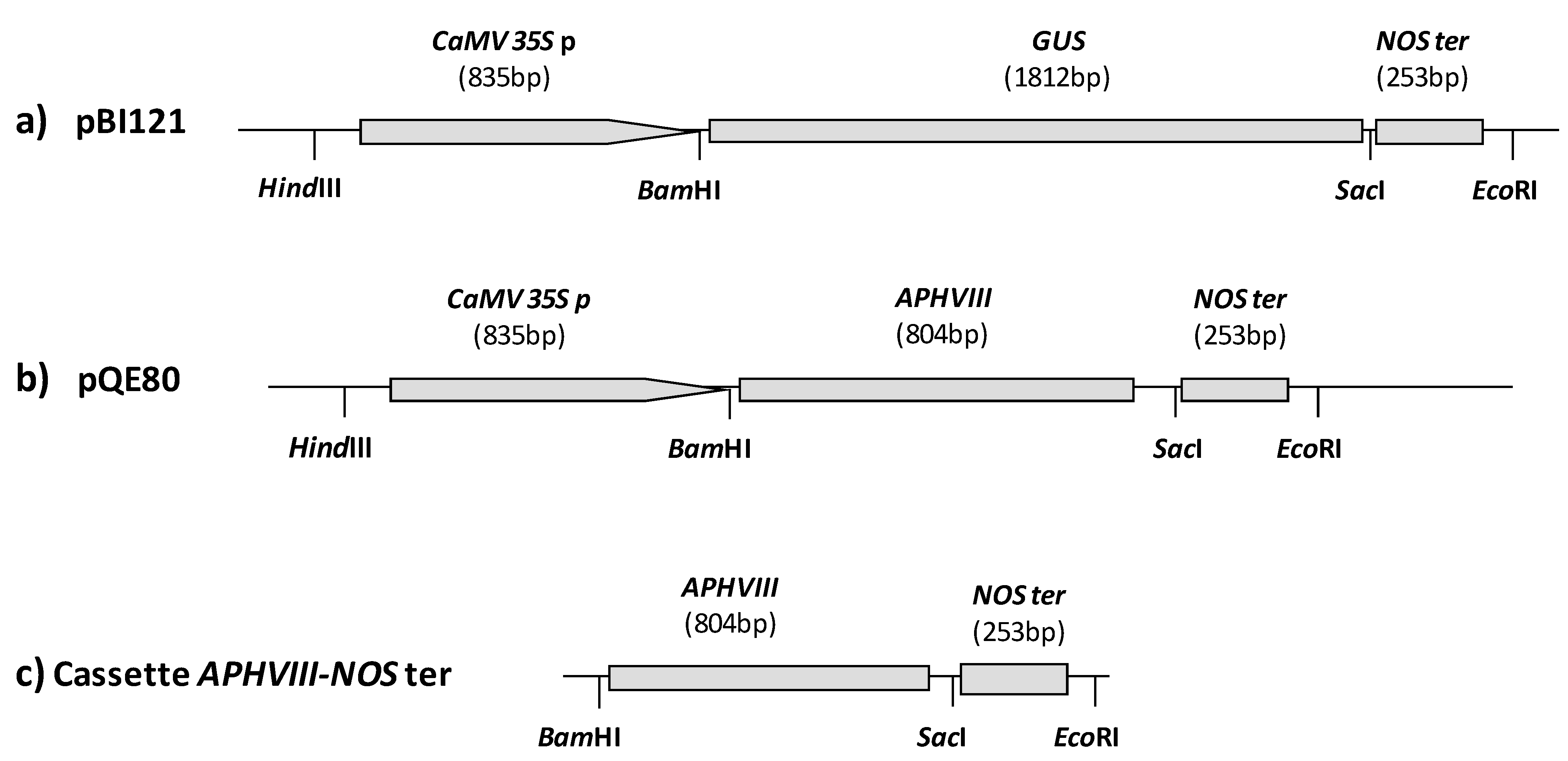
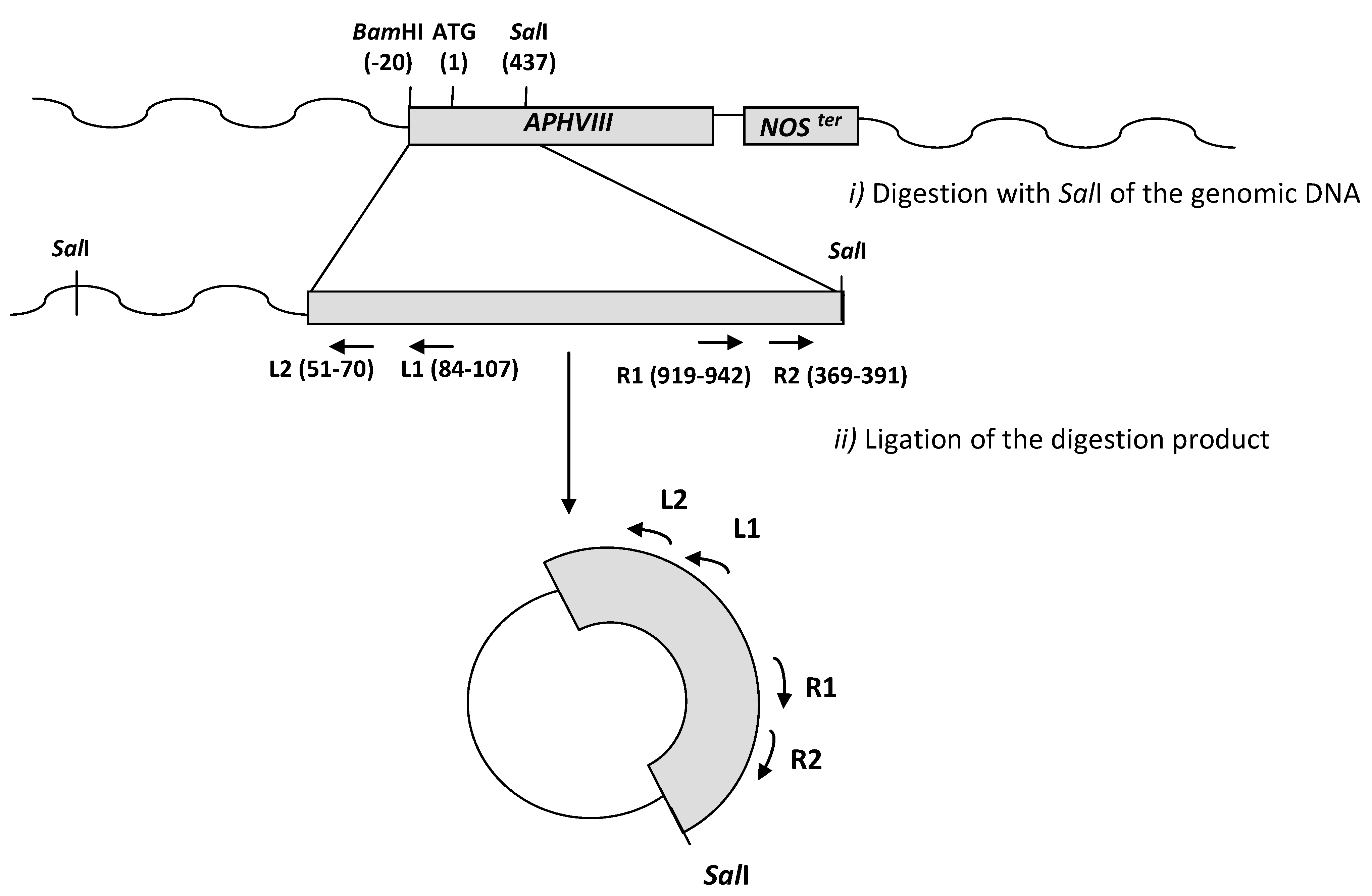
2.1. Construction of a Promoter-Less Cassette Suitable for Promoter Trapping in Microalgae
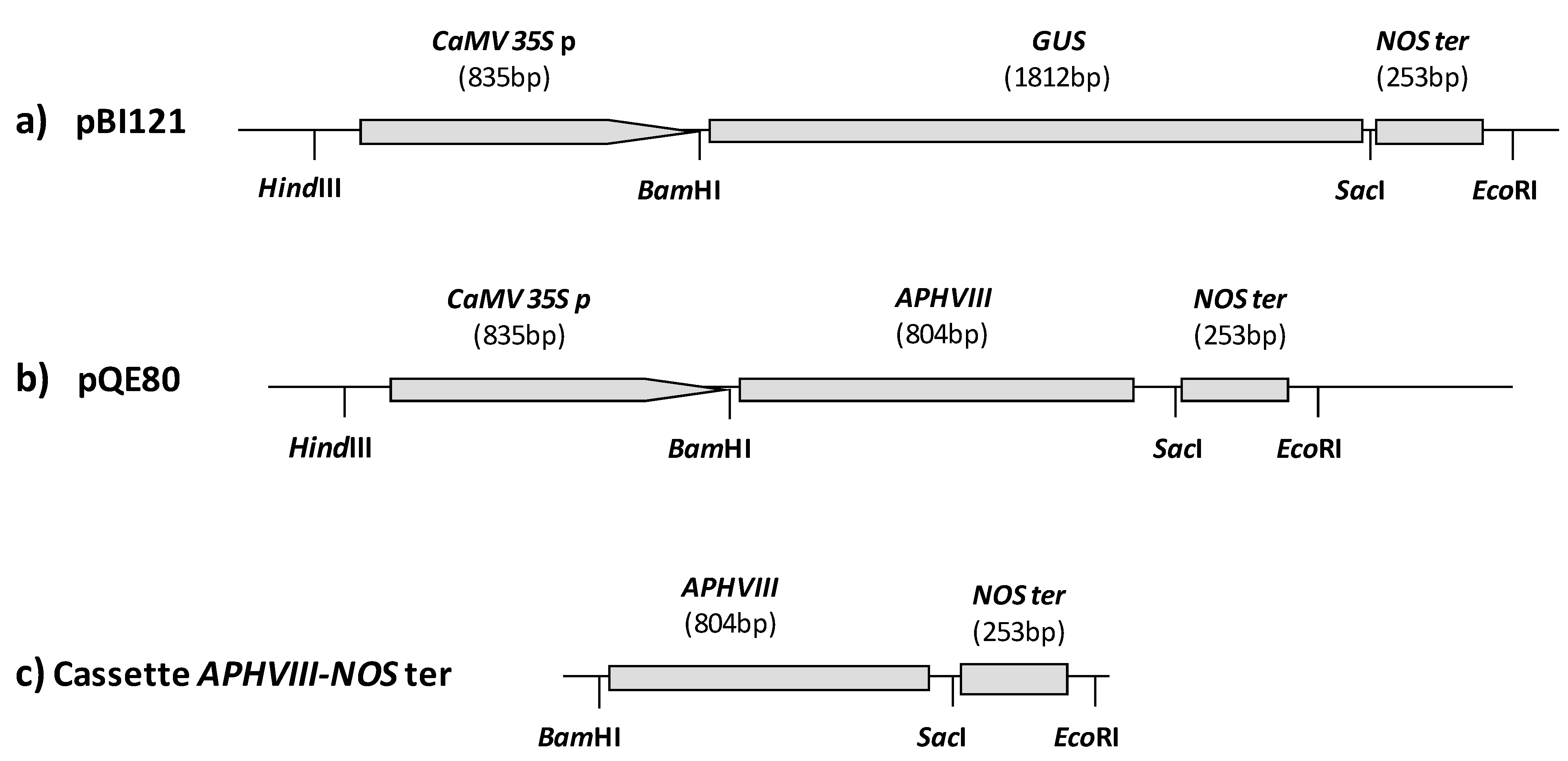
2.2. Transformation of C. reinhardtii with the APHVIII Promoter Trapping Construction and Analysis of Transformants

{kind=link}
{kind=link}
{kind=link}
| Transformation cassette | N of transformants per reaction | Transformation rate (transformants cell−1 μg−1 DNA) |
|---|---|---|
| Linearized pSI103 | 28 ± 5 (n = 3) | 1.8 × 10−6 |
| APHVIII-NOS ter cassette | 4 ± 2 (n = 20) | 2.6 × 10−7 |
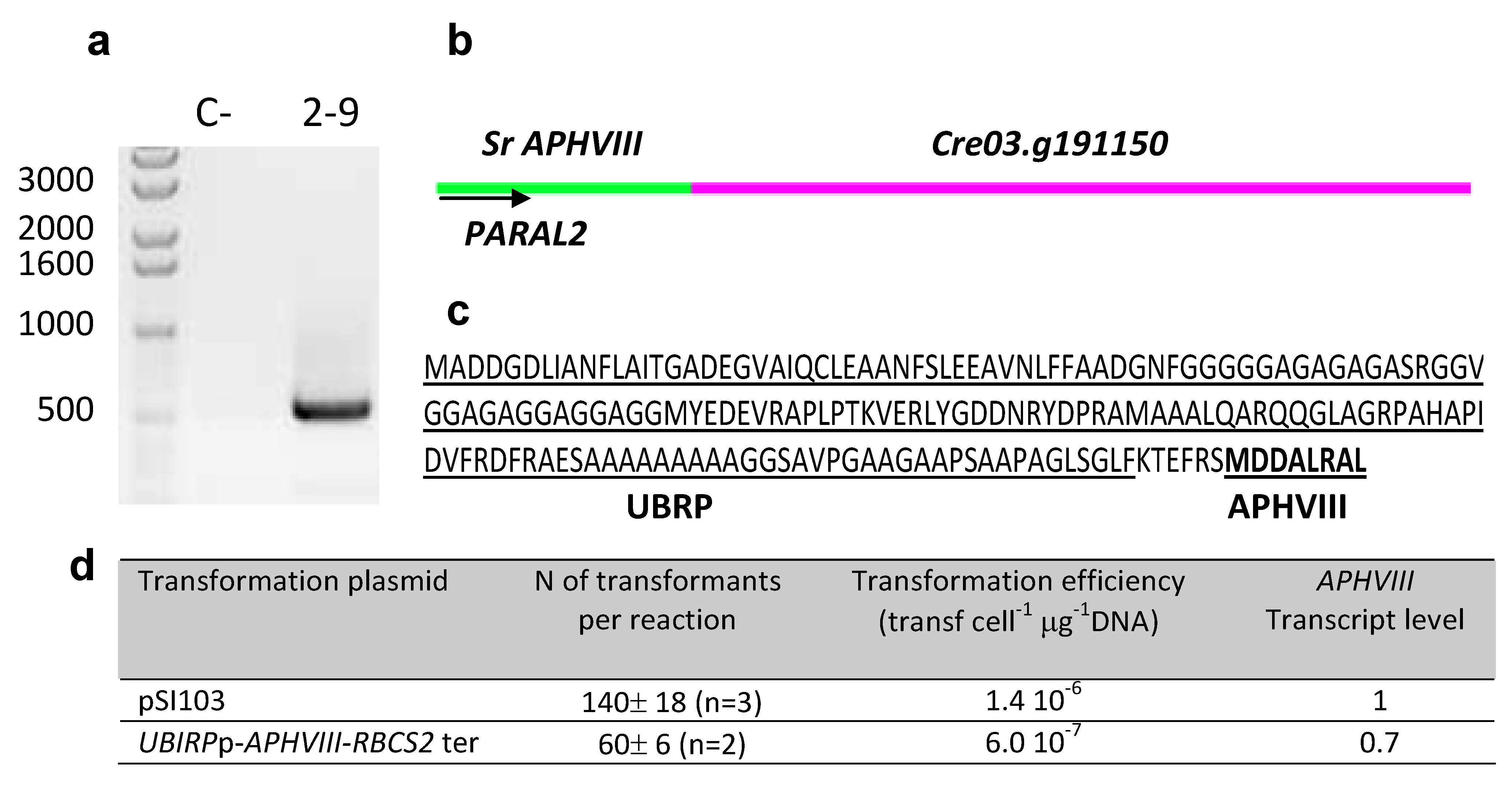
2.3. Molecular Characterization of the Selected Transformants
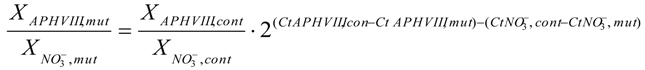
| Mutant | Insertion site | Nearest protein | Scheme of the detailed insertion site |
|---|---|---|---|
| 1–14 | 1:5437700 | No functional annotations for this locus | 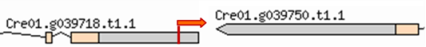 |
| (Cre01. g039718) | |||
| 2–6 | 3:7536446 | No functional annotations for this locus |  |
| (Cre03. g210150) | |||
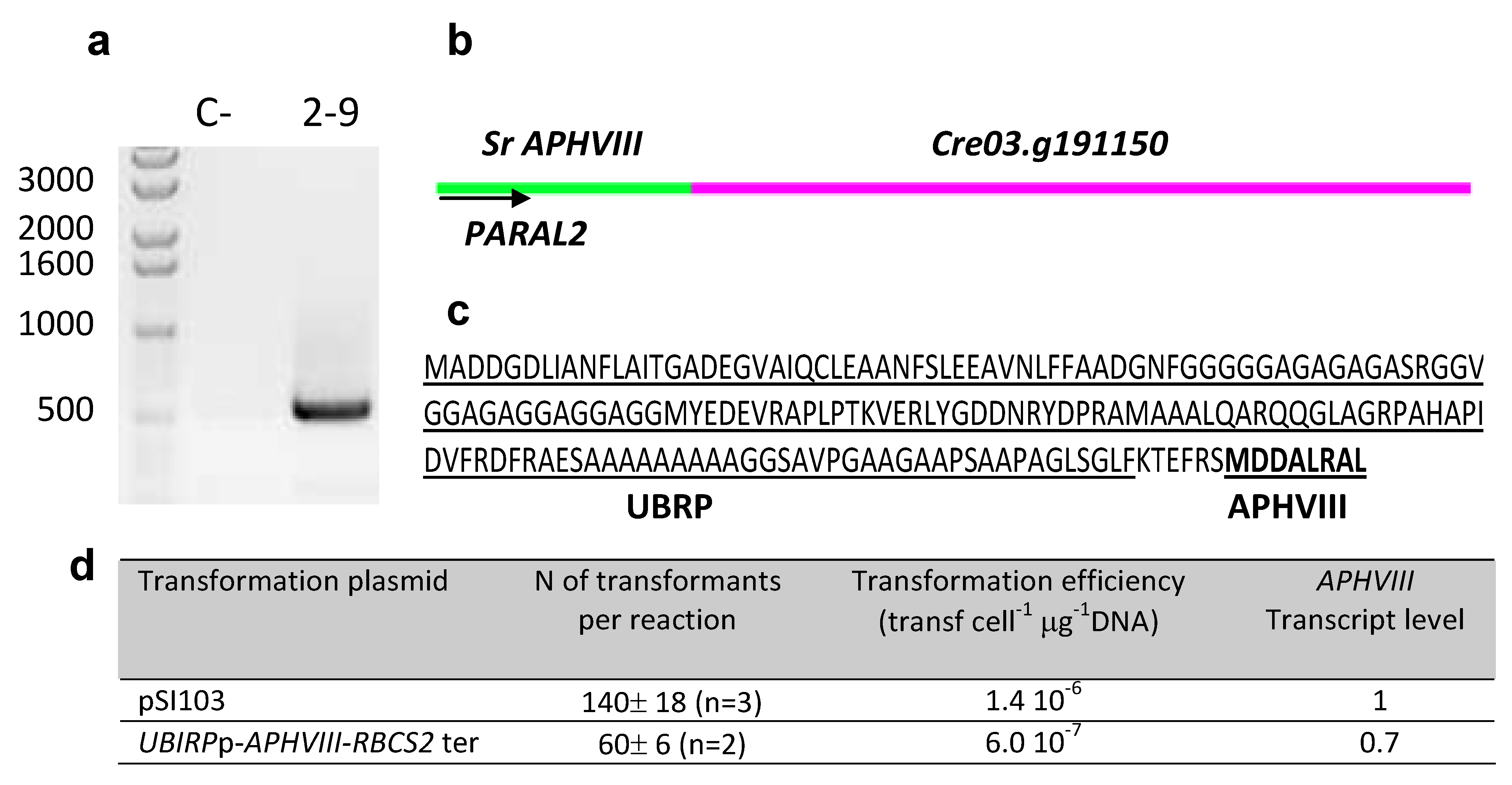
| 2–9 | 3:5019967 | Predicted Ubiquitine regulatory protein | 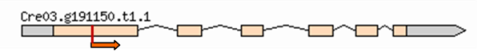 |
| (Cre03. g191150) | |||
| 3–4 | 2:6042257 | Ribulose bisphosphate carboxylase small chain | 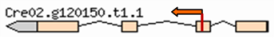 |
| (Cre02. g120150) | |||
| 3–8 | 11:6509508 | No functional annotations for this locus | 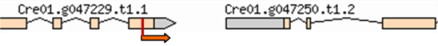 |
| (Cre01. g047250) | |||
| 4–5 | 2:8218509 | Dynein 1b light intermediate chain; D1bLIC |  |
| (Cre02. g135900) | |||
| 4–15 | 2:7024090 | Predicted dehydrogenase short chain |  |
| (Cre02. g128150) |
2.4. Testing the Promoter of the 2–9 Transformant
| Primer | Sequence (5′→3′) | Uses |
|---|---|---|
| APH8-FBAM | CGCCCTCCCCGGATCCGAAGAA | Amplification of the APHVIII gene from pSI103 plasmid |
| APH8-RSAC | ACCCACGAGCTCCAACCCTACCC | |
| NRfor | GCGCTGCCCTCCGTCACCTTCC | Estimation of the number of NR genes by qPCR |
| NRre | CAGCCGCACGCCCGTCCAGTAG | |
| Parafor | GAGGATCTGGACGAGGAGCGGAA | Estimation of the number of APHVIII genes and the APHVIII transcript level by qPCR |
| Pararev | CCCTCAGAAGAACTCGTCCAACAGC | |
| qUbqL-for | GTACAGCGGCGGCTAGAGGCAC | Houskeeping gene for estimation of the APHVIII transcript level |
| qUbqL-rev | AGCGTCAGCGGCGGTTGCAGGTATCT | |
| ParaR 1 | GTGGAGGGTGGTGGGGACGAGAGG | Identification of the region upstream the marker gene by iPCR |
| ParaR 2 | GGTGTCCGTTCGATCGCAGTCTC | |
| ParaL1 | GCCCACCACCCCGAAGCCGATAAA | |
| ParaL2 | GGCCCCATCCTCCACAACAA | |
| UBIRP-F | GCTGCCCGCGACTGTGATGTA | Amplification and subcloning of the promoter identified in mutant 2-9 |
| UBIRP-R | GGGCCGCTGCTGCACCAAACGC | |
| UBIRP-FNotI | GGCGGCCGCGACTGTGATGTA | |
| UBIRP-RBstBI | GGTTCGAAGCTGCACCAAACGC |
3. Discussion
3.1. Choice of the Selectable Marker Gene for Promoter Trapping
3.2. Application to Other Microalgal Species
4. Experimental Section
4.1. Microorganisms and Culture Conditions
4.2. Nuclear Transformation of Chlamydomonas reinhardtii with the APHVIII Promoter-Less Cassette
4.3. Isolation of Genomic DNA
4.4. RNA Extraction and Reverse Transcription
4.5. Standard Polymerase Chain Reaction
4.6. Inverse PCR
4.7. Analysis of the Insertion Sites
4.8. Quantitative Real-Time PCR. Transcript Level Analysis and Determination of the Number of Copies of the APHVIII Gene Integrated in the Genome of Chlamydomonas
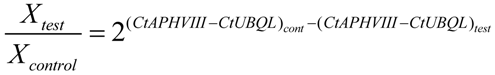
5. Conclusions
Acknowledgments
References
- Fernández, E.; Schnell, R.; Ranum, L.P.; Hussey, S.C.; Silflow, C.D.; Lefebvre, P.A. Isolation and characterization of the nitrate reductase structural gene of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 1989, 86, 6449–6453. [Google Scholar] [CrossRef]
- Dunahay, T.G.; Jarvis, E.E.; Roessle, P.G. Genetic transformation of the diatoms Cyclotella cryptica and Navicula saprophila. J. Phycol. 1995, 31, 1004–1012. [Google Scholar]
- Apt, K.E.; Kroth-Pancic, P.G.; Grossman, A.R. Stable nuclear transformation of the diatom Phaeodactylum tricornutum. Mol. Gen. Genet. 1996, 252, 572–579. [Google Scholar]
- León-Bañares, R.; González-Ballester, D.; Galván, A.; Fernández, E. Transgenic microalgae as green cell- factories. Trends Biotechnol. 2004, 22, 45–52. [Google Scholar]
- Walker, T.L.; Collet, C.; Purton, S. Algal transgenics in the genomic era. J. Phycol. 2005, 41, 1077–1093. [Google Scholar] [CrossRef]
- León, R.; Fernández, E. Nuclear transformation of eukaryotic microalgae: Historical overview, achievementes and problems. Adv. Exp. Med. Biol. 2007, 616, 1–11. [Google Scholar] [CrossRef]
- Eichler-Stahlberg, A.; Weisheit, W.; Ruecker, O.; Heitzer, M. Strategies to facilitate transgene expression in Chlamydomonas reinhardtii. Planta 2009, 229, 873–883. [Google Scholar] [CrossRef]
- Beer, L.L.; Boyd, E.S.; Peters, J.W.; Posewith, M.C. Engineering algae for biohydrogen and biofuel production. Curr. Opin. Biotechnol. 2009, 20, 264–271. [Google Scholar]
- Harris, E.H. Chlamydomonas as a model organism. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2001, 52, 363–406. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, Y.T.; Cho, J.J.; Bae, J.H.; Hur, S.B.; Hwang, I.; Choi, T.J. Stable integration and functional expression of flounder growth hormone gene in transformed microalga, Chlorella ellipsoidea. Mar. Biotechnol. 2002, 4, 63–73. [Google Scholar] [CrossRef]
- Feng, S.; Xue, L.; Liu, H.; Lu, P. Improvement of efficiency of genetic transformation for Dunaliella salina by glass beads method. Mol. Biol. Rep. 2009, 36, 1433–1439. [Google Scholar] [CrossRef]
- Walker, T.L.; Becker, D.K.; Collet, C. Characterisation of the Dunaliella tertiolecta RbcS genes and their promoter activity in Chlamydomonas reinhardtii. Plant Cell. Rep. 2005, 23, 727–735. [Google Scholar] [CrossRef]
- Neupert, J.; Shao, N.; Lu, Y.; Bock, R. Genetic transformation of the model green alga Chlamydomonas reinhardtii. Methods Mol. Biol. 2012, 847, 35–47. [Google Scholar]
- Dawson, H.N.; Burlingame, R.; Cannons, A.C. Stable Transformation of Chlorella: Rescue of nitrate reductase-deficient mutants with the nitrate reductase. Gene Curr. Microbiol. 1997, 35, 356–362. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, Z.; Gao, X.; Li, Q.; Zhang, Q.; Xu, Z. Expression of foreign genes in Dunaliella by electroporation. Mol. Biotechnol. 2005, 30, 185–192. [Google Scholar] [CrossRef]
- Walker, T.L.; Becker, D.K.; Dales, J.L.; Collet, C. Towards the development of a nuclear transformation system for Dunaliella tertiolecta. J. Appl. Phycol. 2005, 17, 363–368. [Google Scholar] [CrossRef]
- Steinbrenner, J.; Sandmann, G. Transformation of the Green Alga Haematococcus pluvialis with a phytoenedesaturase for accelerated astaxanthin biosynthesis. Appl. Environ. Microbiol. 2006, 72, 7477–7484. [Google Scholar] [CrossRef]
- Corellou, F.; Schwartz, C.; Motta, J.P.; Djouani-Tahri, B.; Sanchez, F.; Bouget, F.Y. Clocks in the green lineage: Comparative functional analysis of the circadian architecture of the picoeukaryote Ostreococcus. Plant Cell. 2009, 21, 3436–3449. [Google Scholar] [CrossRef]
- Kiliana, O.; Benemann, C.S.E.; Niyogi, K.K.; Vicka, B. High-efficiency homologous recombinationin the oil-producing alga Nannochloropsis sp. Proc. Natl. Acad. Sci. USA 2011, 108, 21265–21269. [Google Scholar]
- Radakovits, R.; Jinkerson, R.E.; Fuerstenberg, S.I.; Tae, H.; Settlage, R.E.; Boore, J.L.; Posewitz, M.C. Draft genome sequence and genetic transformation of the oleaginous alga Nannochloropis gaditana. Nat. Commun. 2012, 21, 686. [Google Scholar]
- Blanvillain, R.; Gallois, P. Promoter trapping system to study embryogenesis. Methods Mol. Biol. 2008, 427, 121–135. [Google Scholar]
- Springer, P.S. Gene traps tools for plant development and genomics. Plant Cells 2000, 12, 1007–1020. [Google Scholar]
- Haring, M.A.; Beck, C.F. A promoter trap for Chlamydomonas reinhardtii: Development of a gene cloning method using 5′ RACE-based probes. Plant J. 1997, 11, 1341–1348. [Google Scholar]
- Auchincloss, A.H.; Lorocha, A.I.; Rochaix, J.D. The argininosuccinatelyase gene of Chlamydomonas reinhardtii: Cloning of the cDNA and its characterization as a selectable shuttle marker. Mol. Gen. Genet. 1999, 261, 21–30. [Google Scholar] [CrossRef]
- Ohresser, M.; Matagne, R.F.; Loppes, R. Expression of the arylsuphatase reporter gene under the control of the NIT1 promoter in Chlamydomonas reinhartii. Curr. Genet. 1997, 31, 264–271. [Google Scholar] [CrossRef]
- Gónzalez-Ballester, D.; de Montaigu, A.; Higuera, J.J.; Galván, A.; Fernández, E. Functional genomics of the regulation of the nitrate assimilation pathway in Chlamydomonas. Plant Physiol. 2005, 137, 522–533. [Google Scholar]
- Dent, R.; Haglund, C.; Chin, B.L.; Kobayashi, M.C.; Niyogi, K.K. Functional genomics of eukaryotic photosynthesis using insertional mutagenesis of Chlamydomonas reinhardtii. Plant Physiol. 2005, 137, 545–556. [Google Scholar] [CrossRef]
- Pollock, S.V.; Pootakham, W.; Shibagaki, N.; Moseley, J.L.; Grossman, A.R. Insights into the acclimation of Chlamydomonas reinhardtii to sulfur deprivation. Photosynth. Res. 2005, 86, 475–489. [Google Scholar]
- Gónzalez-Ballester, D.; Pootakham, W.; Mus, F.; Yang, W.; Catalanotti, C.; Magneschi, L.; de Montaigu, A.; Higuera, J.J.; Prior, M.; Galván, A.; Fernandez, E.; Grossman, A.R. Reverse genetics in Chlamydomonas: A platform for isolating insertional mutants. Plant Methods 2011, 27, 24. [Google Scholar]
- Sizova, I.; Fuhrmann, M.; Hegemann, P. A Streptomyces rimosus aphVIII gene coding for a new type phosphotransferase provides stable antibiotic resistance to Chlamydomonas reinhardtii. Gene 2001, 277, 221–229. [Google Scholar]
- Lerche, K.; Hallmann, A. Stable nuclear transformation of Gonium pectoral. BMC Biotechnol. 2009, 9, 64–69. [Google Scholar] [CrossRef]
- Chen, P.Y.; Wang, C.K.; Soong, S.C.; To, K.Y. Complete sequence of binary vector pBI121 and its application in cloning T-DNA insertion from transgenic plants. Mol. Breed. 2003, 11, 287–293. [Google Scholar]
- Schroda, M.; Blöcker, D.; Beck, C.F. The HSP70A promoter as a tool for the improved expression of transgenes in Chlamydomonas. Plant J. 2000, 21, 121–131. [Google Scholar]
- Chlamydomonas reinhardtii Genome Database. Available online: http://www.phytozome.net/chlamy (accessed on 1 September 2012).
- León, R.; Couso, I.; Fernández, E. Metabolic engineering of ketocarotenoids biosíntesis in the unicelullarmicroalga Chlamydomonas reinhardtii. J. Biotechol. 2007, 130, 143–152. [Google Scholar]
- Yamamoto, Y.Y.; Tsuhara, Y.; Gohda, K.; Suzuki, K.; Matsui, K.M. Gene trapping of the Arabidopsis genome with a firefly luciferase reporter. Plant J. 2003, 35, 273–283. [Google Scholar] [CrossRef]
- Fuhrmann, M.; Oertel, W.; Hegemann, P. A synthetic gene coding for the green fluorescent protein (GFP) is a versatile reporter in Chlamydomonas reinhardtii. Plant J. 1999, 19, 353–361. [Google Scholar] [CrossRef]
- Fuhrmann, M.; Hausherr, A.; Ferbitz, L.; Schödl, T.; Heitze, M.; Hegemann, P. Monitoring dynamic expression of nuclear genes in Chlamydomonas reinhardtii by using a synthetic luciferase reporter gene. Plant Mol. Biol. 2004, 55, 869–881. [Google Scholar]
- Plesch, G.; Kamann, E.; Mueller-Roeber, B. Cloning of regulatory sequences mediating guard-cell-specific gene expression. Gene 2000, 249, 83–89. [Google Scholar]
- Liu, Y.G.; Whittier, R.F. Thermal asymmetric interlaced PCR: Automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics 1995, 25, 674–681. [Google Scholar] [CrossRef]
- González-Ballester, D.; de Montaigu, A.; Galván, A.; Fernández, E. Restriction enzyme site-directed amplification PCR: A tool to identify regions flanking a marker DNA. Anal. Biochem. 2005, 340, 330–335. [Google Scholar] [CrossRef]
- Tonooka, Y.; Fujishima, M. Comparison and critical evaluation of PCR-mediated methods to walk along the sequence of genomic DNA. Appl. Microbiol. Biotechnol. 2009, 85, 37–43. [Google Scholar] [CrossRef]
- De Montaigu, A.; Sanz-Luque, E.; Macias, M.I.; Galván, A.; Fernández, E. Transcriptional regulation of CDP1 and CYG56 is required for proper NH4+ sensing in Chlamydomonas. J. Exp. Bot. 2011, 62, 1425–1437. [Google Scholar]
- Fischer, N.; Rochaix, J.D. The flanking regions of PsaDdrive efficient gene expression in the nucleus of the green alga Chlamydomonas reinhardtii. Mol. Genet. Genomics 2001, 265, 888–894. [Google Scholar] [CrossRef]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Maréchal-Drouard, L.; et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 2007, 318, 245–250. [Google Scholar]
- Bowler, C.; Allen, A.E.; Badger, J.H.; Grimwood, J.; Jabbari, K.; Kuo, A.; Maheswari, U.; Martens, C.; Maumus, F.; Otillar, R.P.; et al. The Phaeodactylum genome reveals the evolutionary history of diatom genome. Nature 2008, 456, 239–244. [Google Scholar]
- Derelle, E.; Ferraz, C.; Rombauts, S.; Rouzé, P.; Worden, A.Z.; Robbens, S.; Partensky, F.; Degroeve, S.; Echeynié, S.; Cooke, R.; et al. Moreau, genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proc. Natl. Acad. Sci. USA 2006, 103, 11647–11652. [Google Scholar]
- Worden, A.Z.; Lee, J.-H.; Mock, T.; Rouzé, P.; Simmons, M.P.; Aerts, A.L.; Allen, A.E.; Cuvelier, M.L.; Derelle, E.; Everett, M.V.; et al. Green evolution and dynamic adaptations revealed by genomes of the marine Picoeukaryotes Micromonas. Science 2009, 324, 268–272. [Google Scholar]
- The Nuclear Genome Sequencing Project of Microalgae. Available online: http://genome.jgi-psf.org/programs/plants/plant-projects.jsf (accessed on 1 September 2012).
- Loppes, R.; Radoux, M.; Ohresser, M.C.; Matagne, R.F. Transcriptional regulation of the Nia1 gene encoding nitrate reductase in Chlamydomonas reinhardtii: Effects of various environmental factors on the expression of a reporter gene under the control of the Nia1 promoter. Plant Mol. Biol. 1999, 41, 701–711. [Google Scholar]
- Harris, E.H. ChlamydomonasSourcebook: Introduction to Chlamydomonas and Its Laboratory Use, 2nd; Stern, D., Witman, G., Eds.; Academic Press: San Diego, CA, USA, 2009; Volume 1. [Google Scholar]
- Sambrook, J.; Russell, D.W. Molecular Cloning, a Laboratory Manual, 3rd ed; Cold Spring Harbour Laboratory Press: Cold Spring Harbour, New York, NY, USA, 2001. [Google Scholar]
- Kindle, K.L. High-frequency nuclear transformation of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 1990, 87, 1228–1232. [Google Scholar] [CrossRef]
- Ingham, D.J.; Beer, S.; Money, S.; Hansen, G. Quantitative real-time PCR assay for determining transgene copy number in transformed plants. Biotechniques 2001, 31, 132-134, 136-140. [Google Scholar]
- Samples Availability: Available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vila, M.; Díaz-Santos, E.; De la Vega, M.; Rodríguez, H.; Vargas, Á.; León, R. Promoter Trapping in Microalgae Using the Antibiotic Paromomycin as Selective Agent. Mar. Drugs 2012, 10, 2749-2765. https://doi.org/10.3390/md10122749
Vila M, Díaz-Santos E, De la Vega M, Rodríguez H, Vargas Á, León R. Promoter Trapping in Microalgae Using the Antibiotic Paromomycin as Selective Agent. Marine Drugs. 2012; 10(12):2749-2765. https://doi.org/10.3390/md10122749
Chicago/Turabian StyleVila, Marta, Encarnación Díaz-Santos, Marta De la Vega, Herminia Rodríguez, Ángeles Vargas, and Rosa León. 2012. "Promoter Trapping in Microalgae Using the Antibiotic Paromomycin as Selective Agent" Marine Drugs 10, no. 12: 2749-2765. https://doi.org/10.3390/md10122749
APA StyleVila, M., Díaz-Santos, E., De la Vega, M., Rodríguez, H., Vargas, Á., & León, R. (2012). Promoter Trapping in Microalgae Using the Antibiotic Paromomycin as Selective Agent. Marine Drugs, 10(12), 2749-2765. https://doi.org/10.3390/md10122749