Structural Analysis of a Heteropolysaccharide from Saccharina japonica by Electrospray Mass Spectrometry in Tandem with Collision-Induced Dissociation Tandem Mass Spectrometry (ESI-CID-MS/MS)

Abstract
:1. Introduction
2. Results and Discussion
2.1. Preparation of Fucoidan and Oligosaccharides

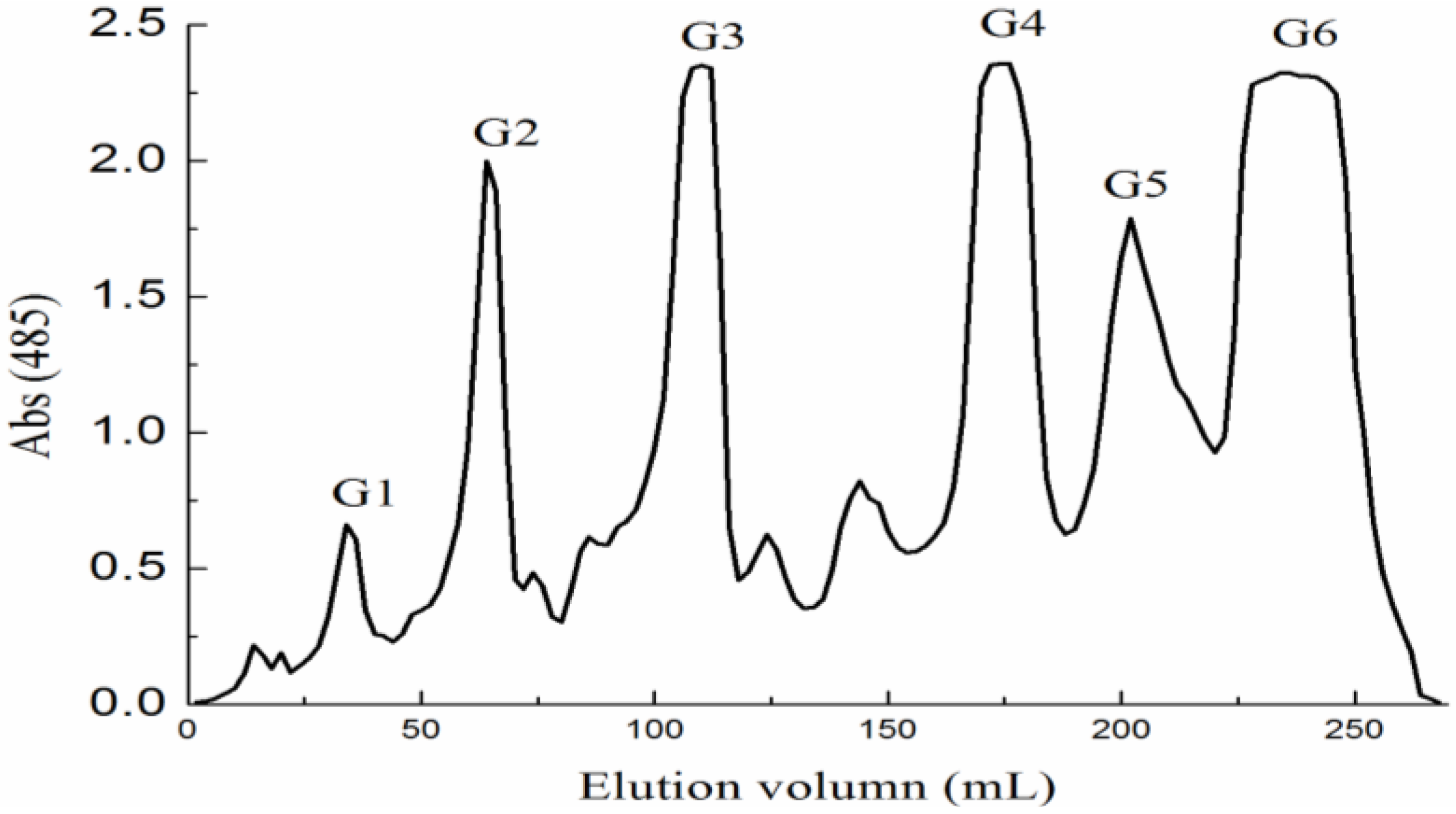{kind=link}
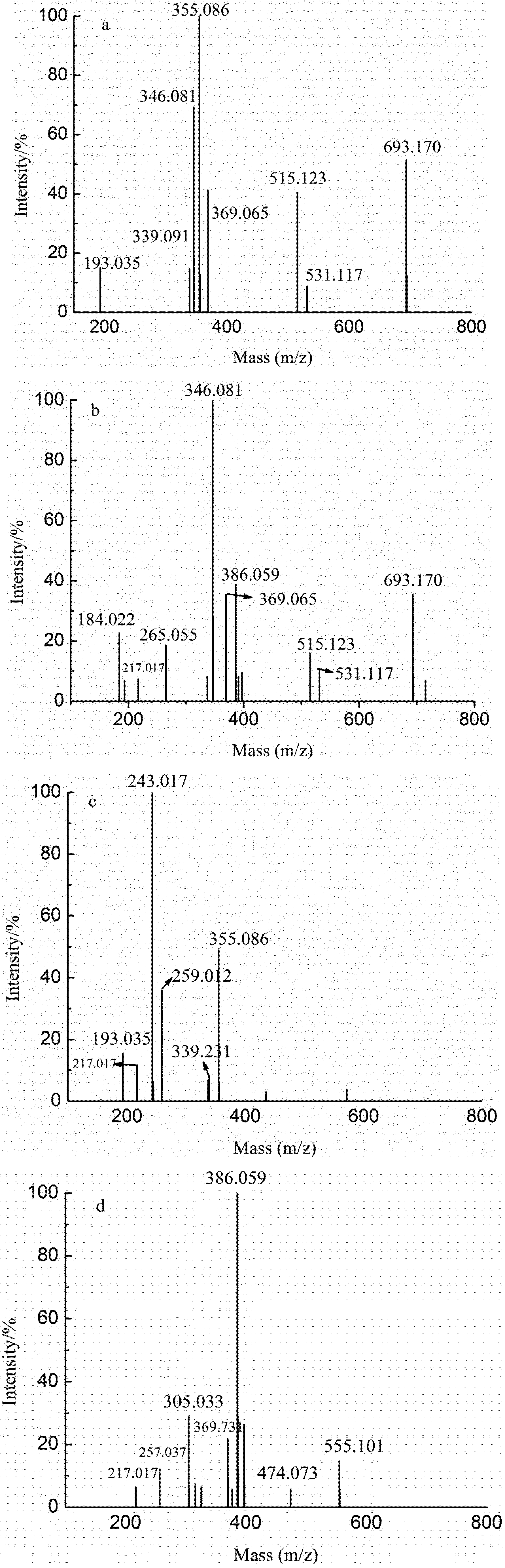{kind=link}
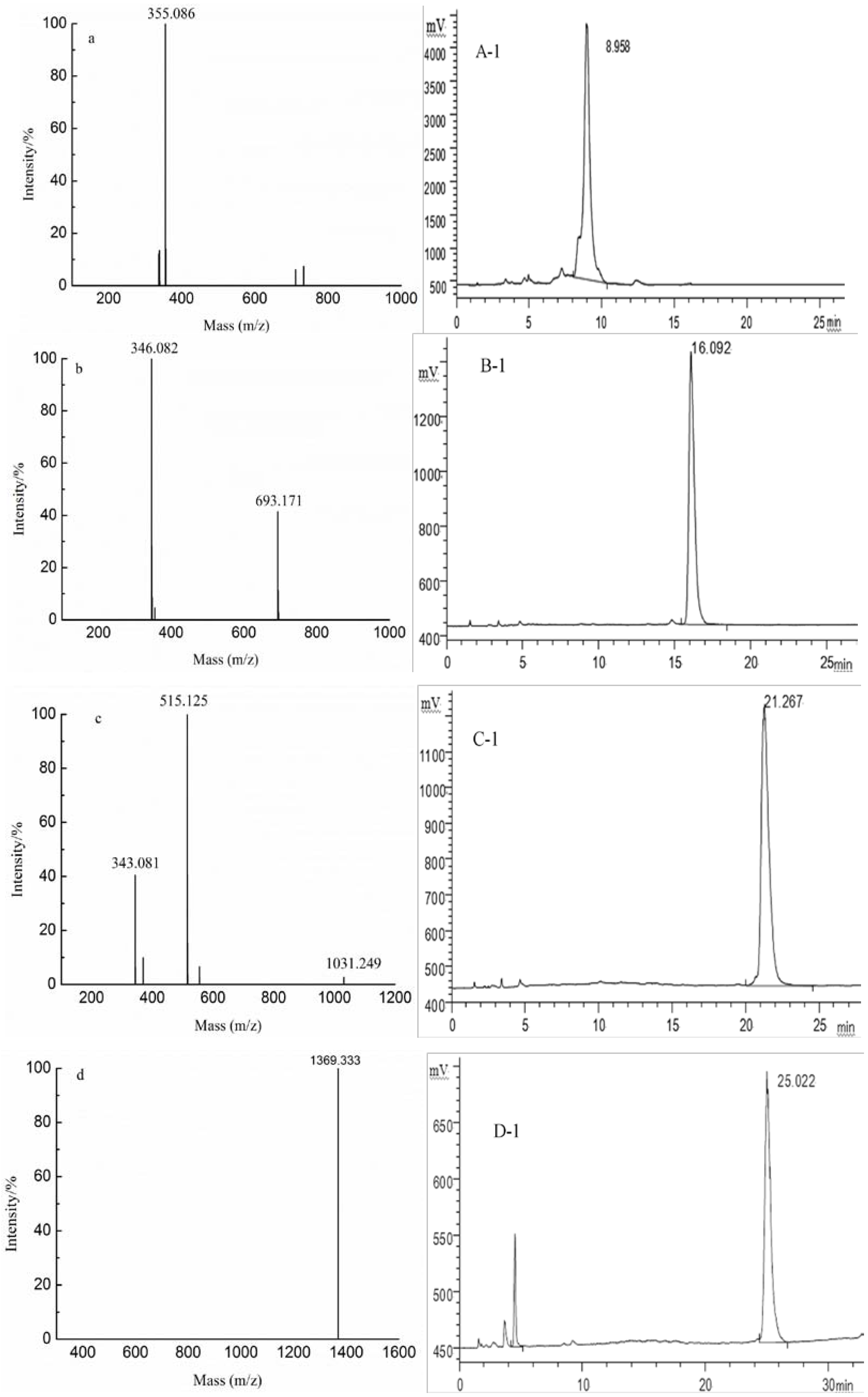{kind=link}
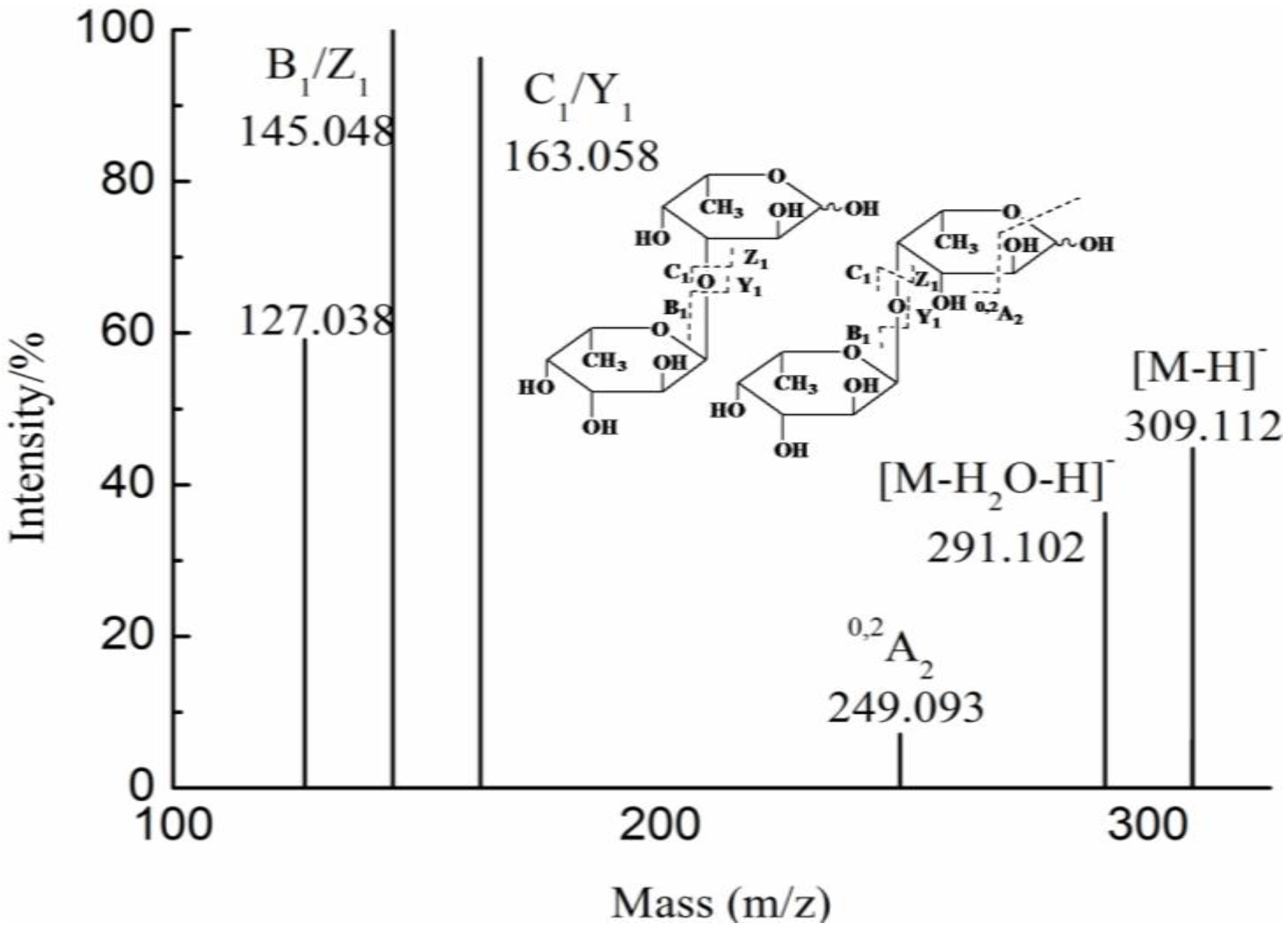{kind=link}
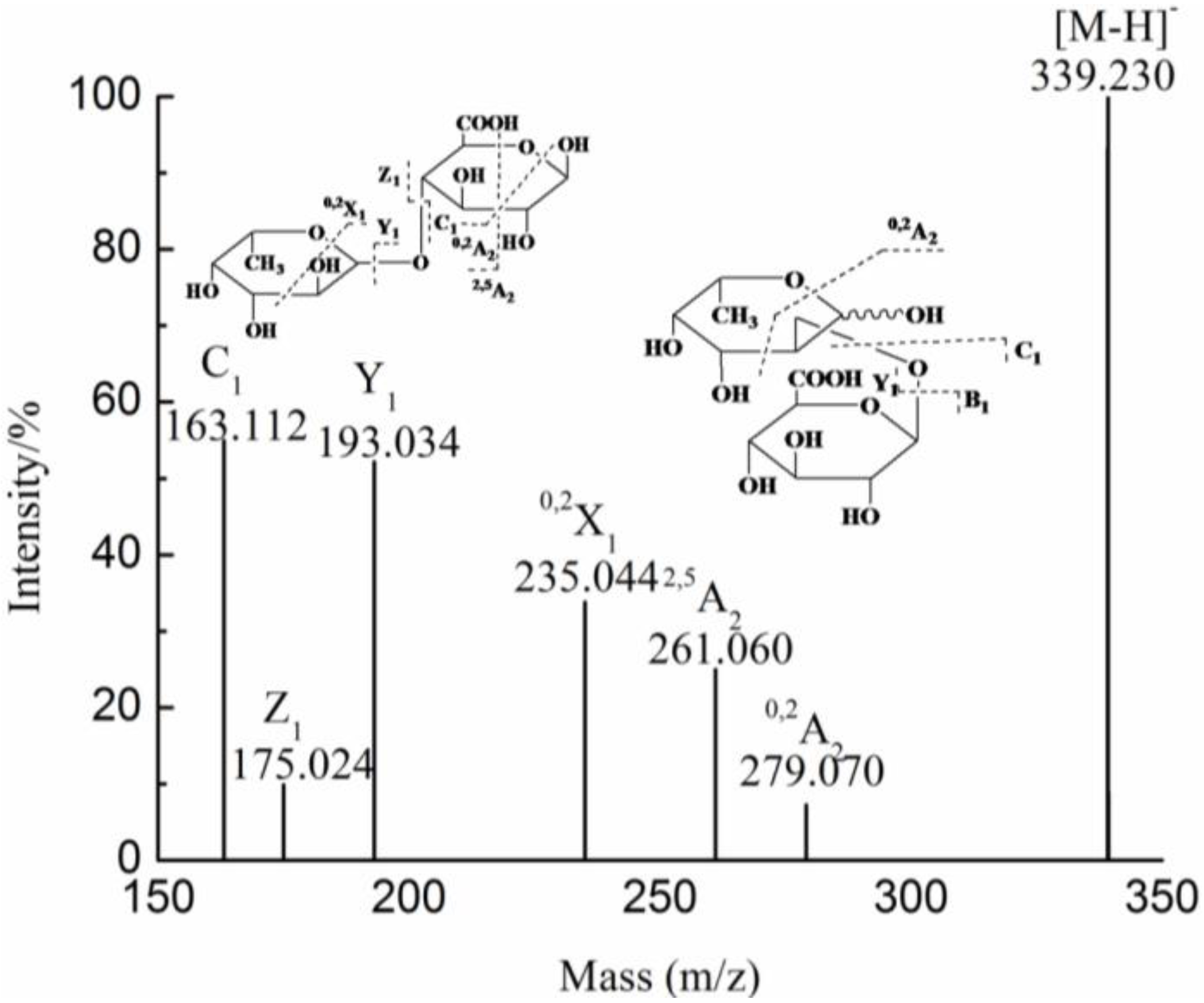{kind=link}
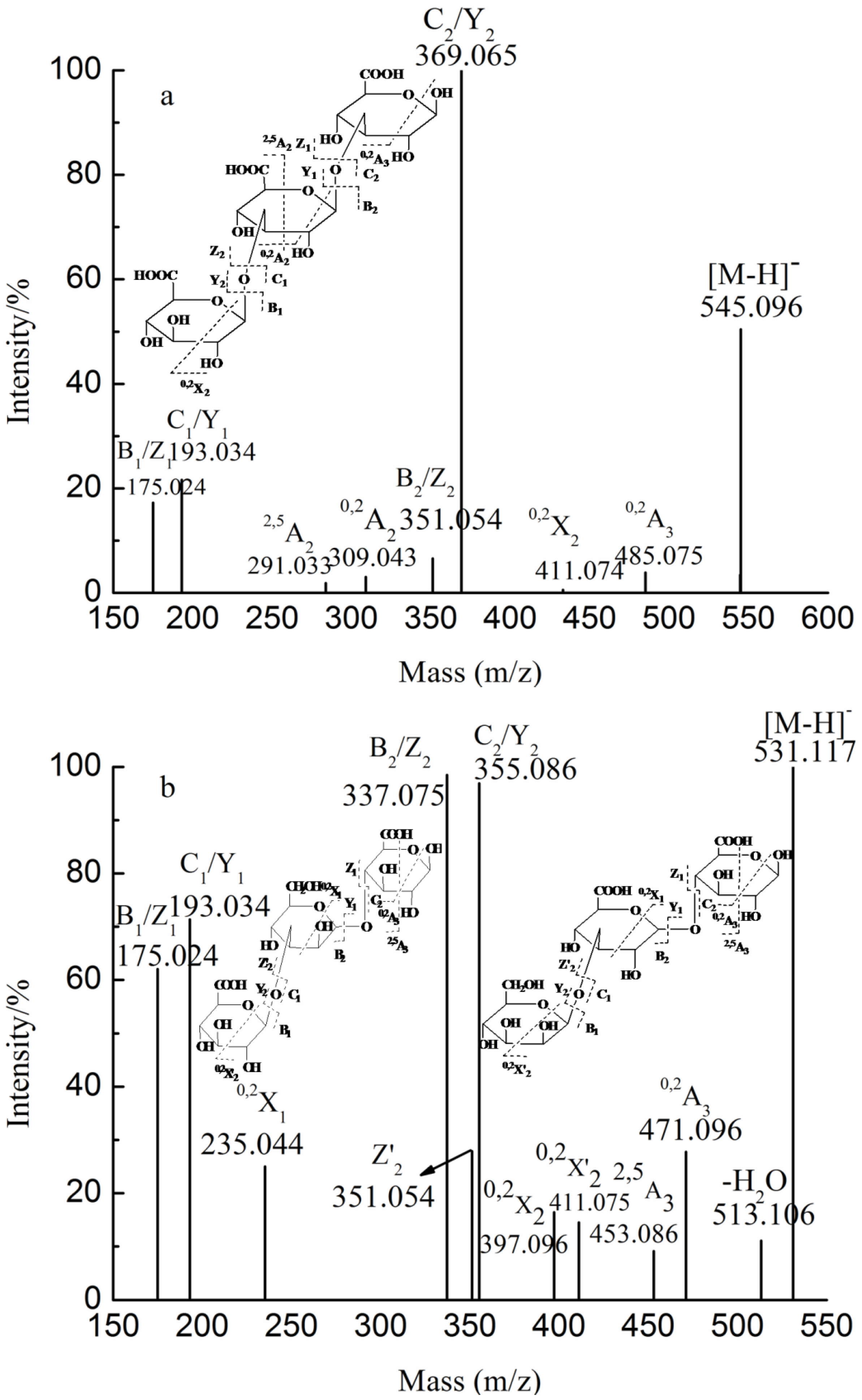{kind=link}
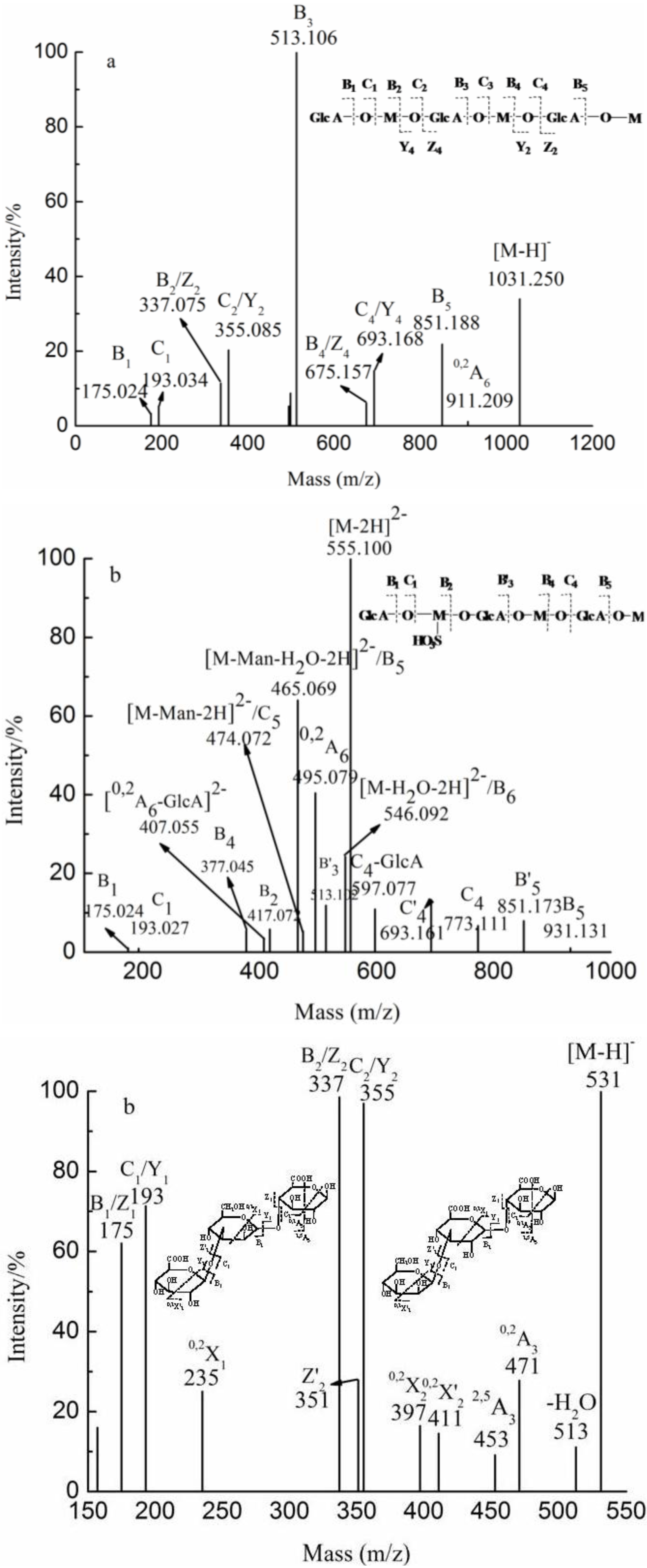{kind=link}
| Sample | Fuc (%) | U A (%) | SO4 (%) | Monosaccharides (molar ratio) | Mw | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fuc | Gal | Man | Glc A | Rha | Xyl | Glc | |||||
| F0.5 | 13.77 | 20.34 | 29.07 | 1 | 0.98 | 0.80 | 0.95 | 0.12 | 0.30 | 0.40 | 5954 |
| F1 | 54.84 | 7.3 | 32.26 | 1 | 0.36 | 0.13 | 0.10 | 0.02 | 0.04 | 0.10 | 8436 |
| F2 | 35.04 | 0.71 | 53.40 | 1 | 0.07 | 0.03 | 0.01 | 0 | 0 | 0.02 | 12586 |
| YF | - | - | - | 1 | 0.43 | 0.32 | 0.28 | 0.41 | 0 | 0.26 | - |
| YD-1 | - | - | - | 0 | 1.10 | 11.14 | 12.59 | 0.17 | 0 | 1.08 | - |
| YD-2 | - | - | - | 1 | 0.28 | 3.53 | 1.96 | 0 | 0 | 0.29 | - |
| YT | - | - | - | 0 | 0 | 8.21 | 9.30 | 0 | 0 | 1.48 | - |
| G5 | - | - | - | 1 | 0.11 | 0.02 | 0 | 0 | 0 | 0.07 | - |
| G6 | - | - | - | 1 | 0.04 | 0 | 0 | 0 | 0 | 0.05 | - |

| Samples | DP | Ions (charges) | m/z | Predicted structural compositions | |
|---|---|---|---|---|---|
| YF | 1 | 193.035(−1) | 193.035 | GlcA | |
| 2 | 355.086(−1) | 355.086 | GlcAMan | ||
| 2 | 369.065(−1) | 369.065 | GlcA2 | ||
| 2 | 339.091(−1) | 339.091 | GlcAFuc | ||
| 3 | 531.117(−1) | 531.117 | GlcA2Man | ||
| 4 | 346.081(−2) | 693.170 | GlcA2Man2 | ||
| 693.170(−1) | |||||
| 6 | 515.123(−2) | 1031.249 | GlcA3Man3 | ||
| YD | YD-1 | 2 | 184.032(−2) | 369.065 | GlcA2 |
| 369.065(−1) | |||||
| 2 | 217.018(−2) | 435.030 | GlcAManSO3H | ||
| 3 | 265.055(−2) | 531.117 | GlcA2Man | ||
| 531.117(−1) | |||||
| 4 | 346.081(−2) | 693.170 | GlcA2Man2 | ||
| 693.170(−1) | |||||
| 4 | 386.059(−2) | 773.117 | GlcA2Man2SO3H | ||
| 6 | 515.123(−2) | 1031.249 | GlcA3Man3 | ||
| YD-2 | 1 | 193.035(−1) | 193.035 | GlcA | |
| 1 | 243.017(−1) | 243.017 | FucSO3H | ||
| 1 | 259.012(−1) | 259.012 | GalSO3H | ||
| 2 | 339.231(−1) | 339.231 | GlcAFuc | ||
| 2 | 355.086(−1) | 355.086 | GlcAMan | ||
| 2 | 217.072(−2) | 435.033 | GlcAManSO3H | ||
| YT | 2 | 217.072(−2) | 435.033 | GlcAManSO3H | |
| 3 | 257.037(−3) | 545.096 | GlcA3 | ||
| 3 | 305.033(−2) | 611.068 | GlcA2ManSO3H | ||
| 4 | 386.059(−2) | 773.118 | GlcA2Man2SO3H | ||
| 5 | 474.073(−2) | 949.147 | GlcA3Man2SO3H | ||
| 6 | 369.731(−3) | 1111.201 | GlcA3Man3SO3H | ||
| 555.101(−2) | |||||
| G1 | 8 | 1369.333(−1) | 1369.333 | GlcA4Man4 | |
| G2 | 6 | 343.081(−3) | 1031.249 | GlcA3Man3 | |
| 515.125(−2) | |||||
| 1031.249(−1) | |||||
| G3 | 4 | 346.082(−2) | 693.173 | GlcA2Man2 | |
| 693.173(−1) | |||||
| G4 | 2 | 355.086(−1) | 355.086 | GlcAMan | |
2.2. Analysis of the Oligosaccharides of All Fractions by ESI-MS
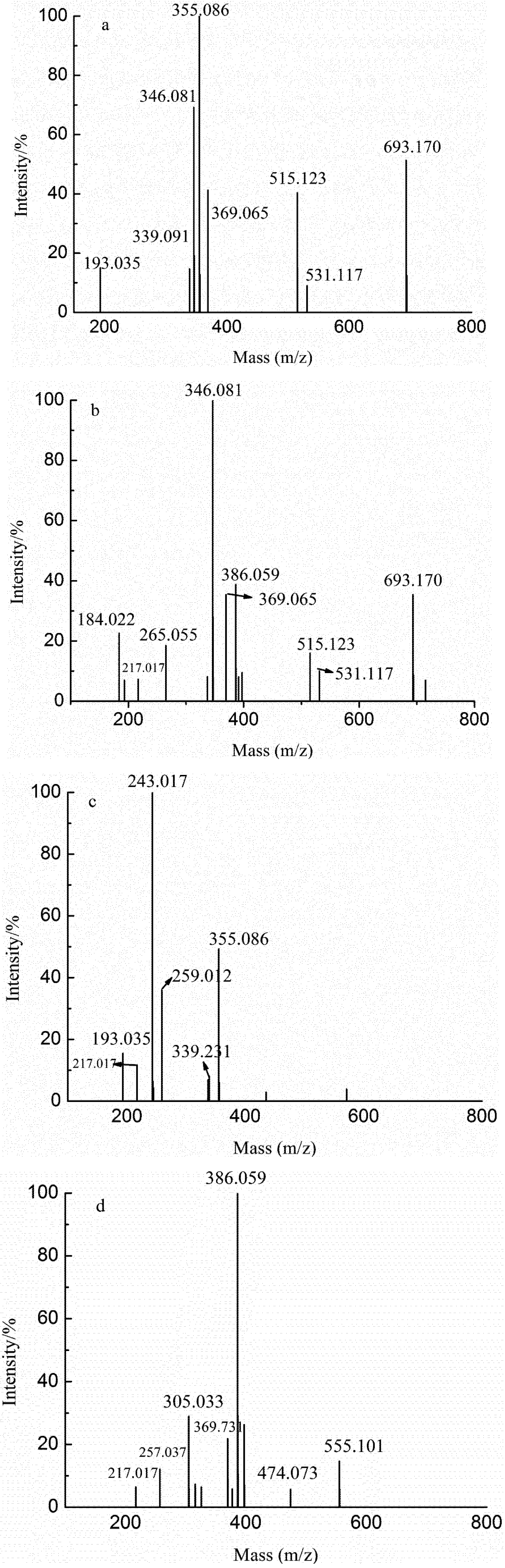
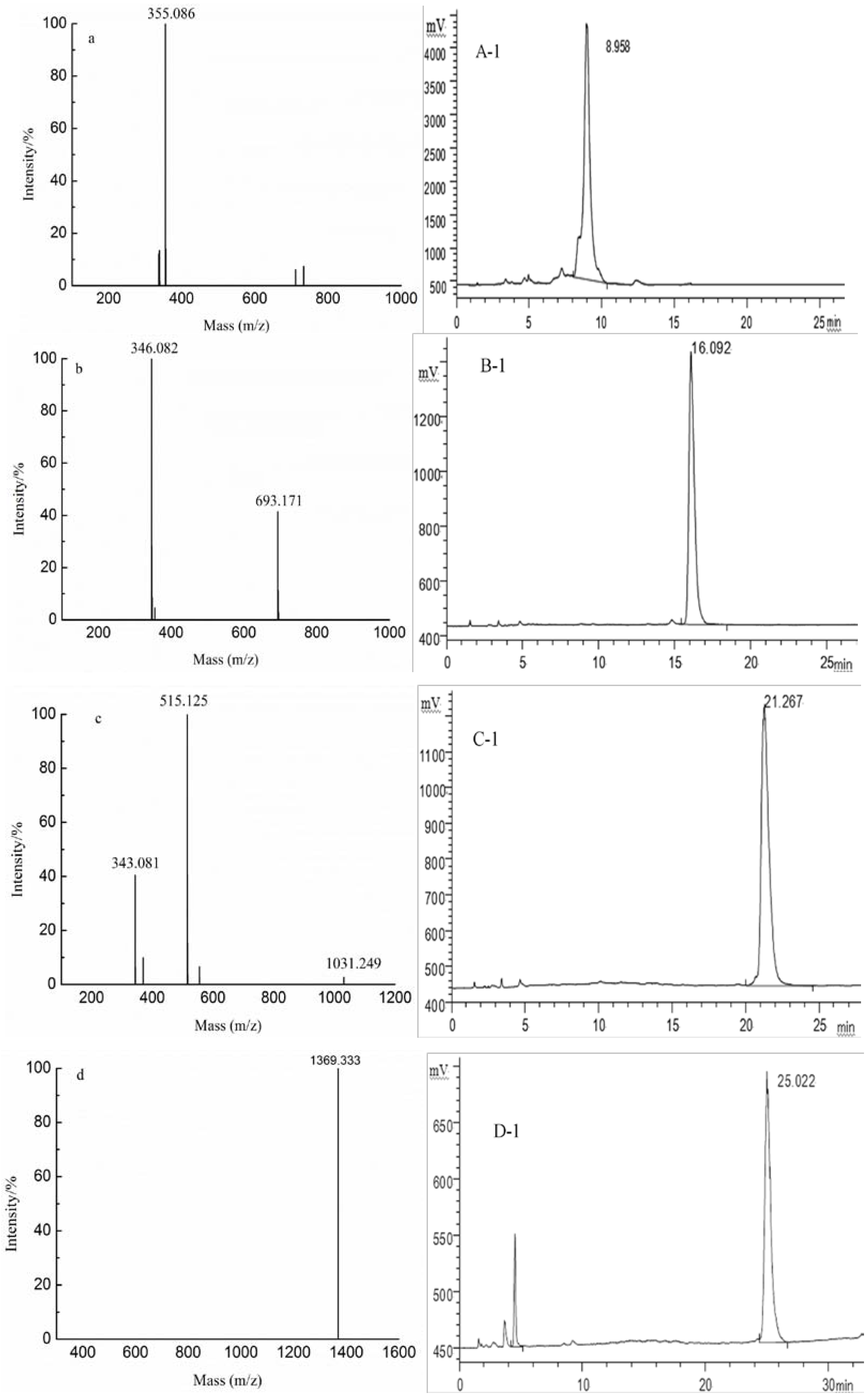
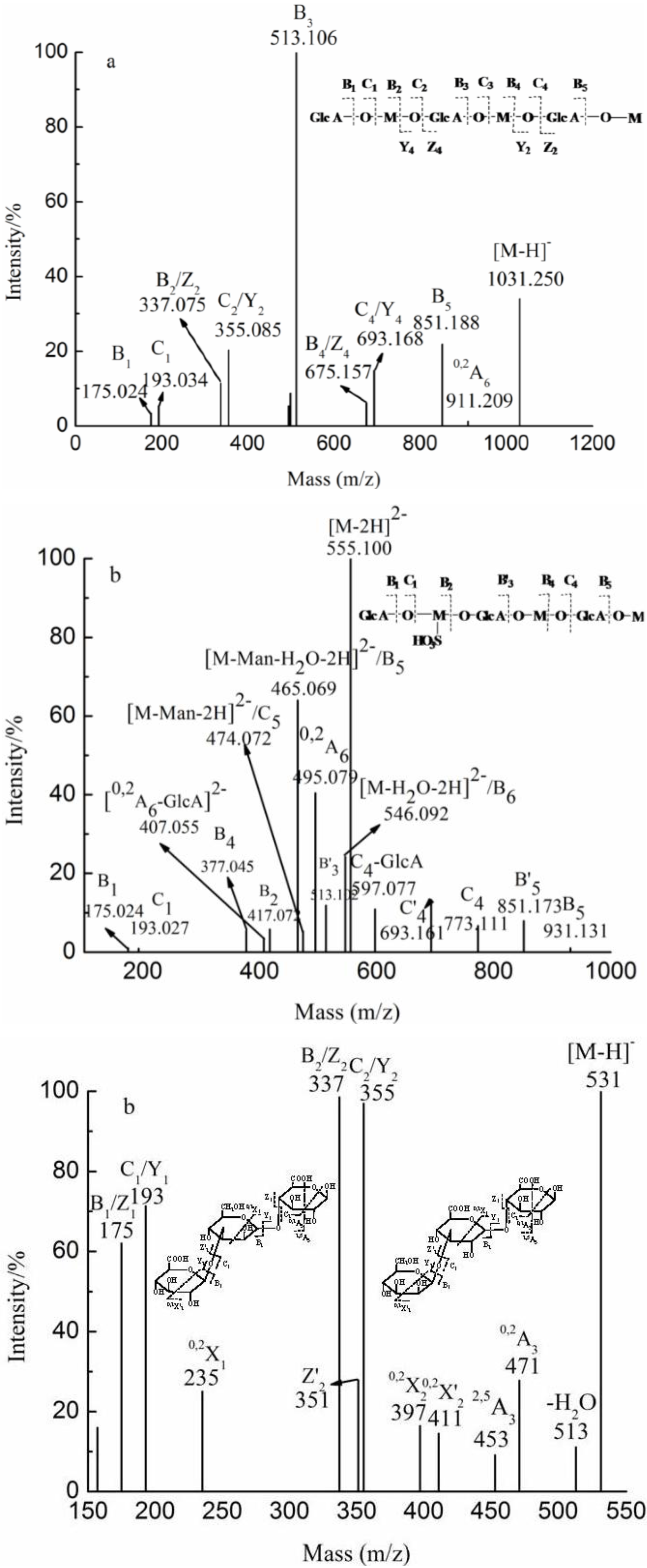
2.3. Analysis of the Structural Features of the Fractions by ESI-CID-MS/MS
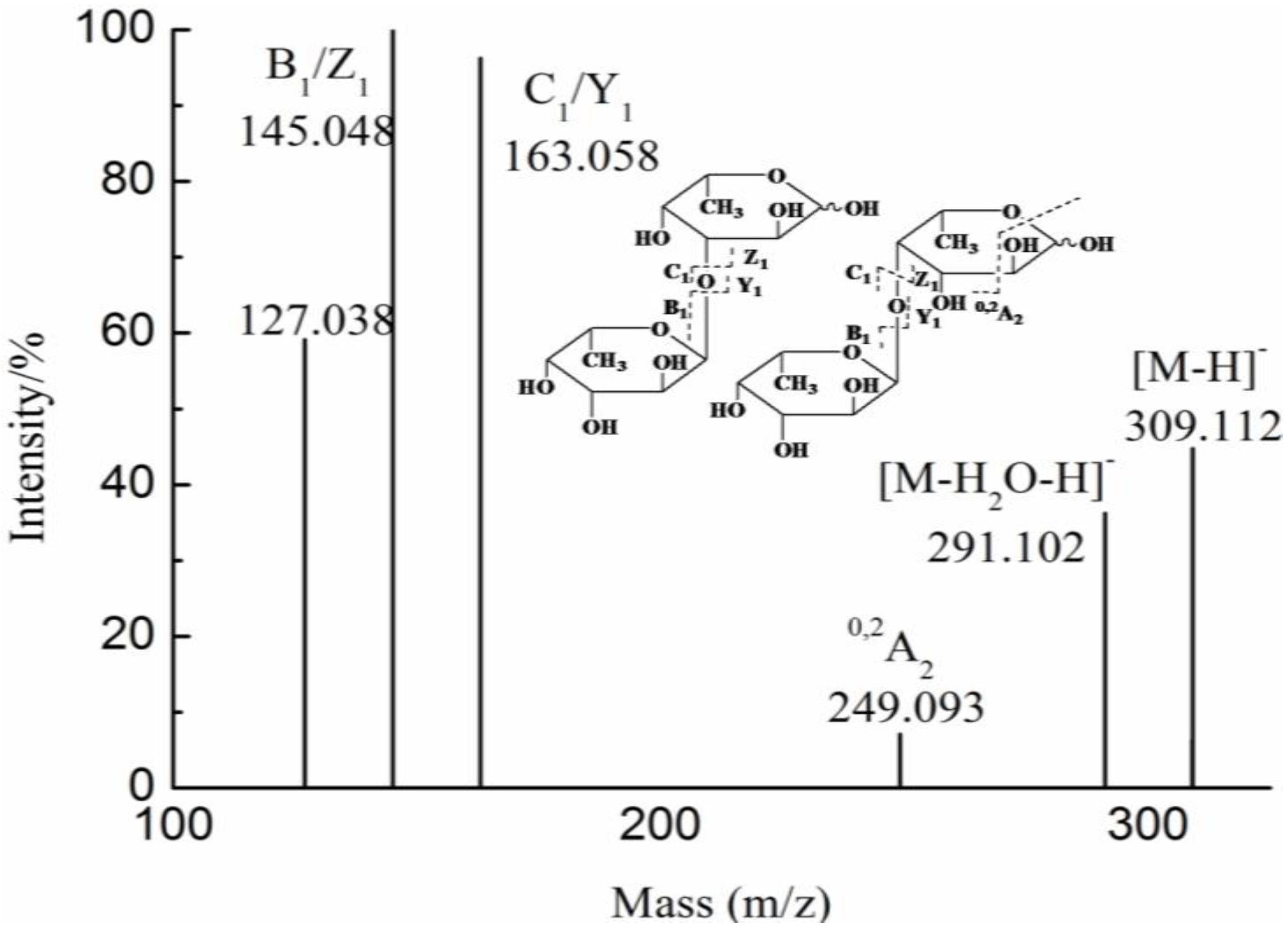
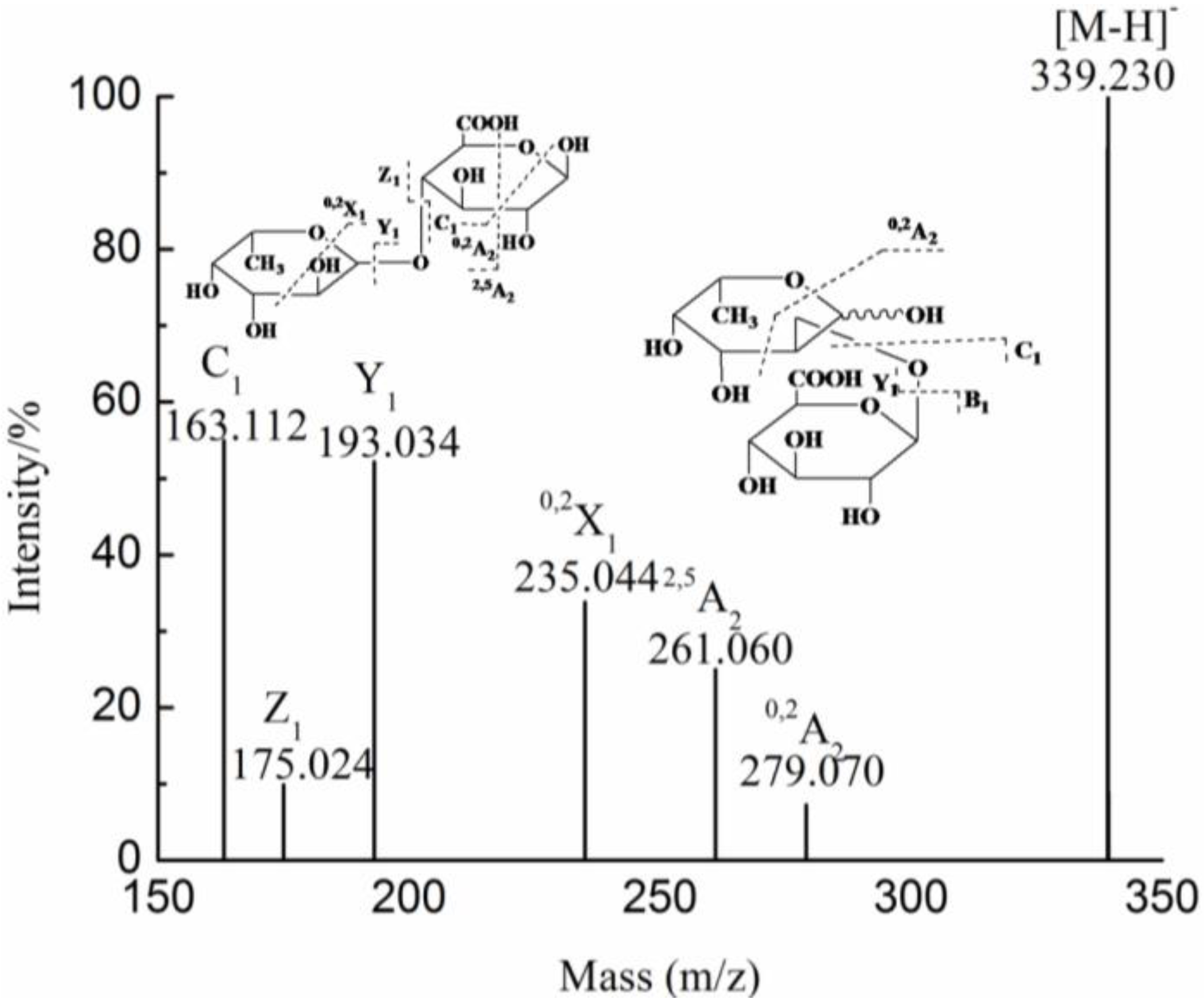
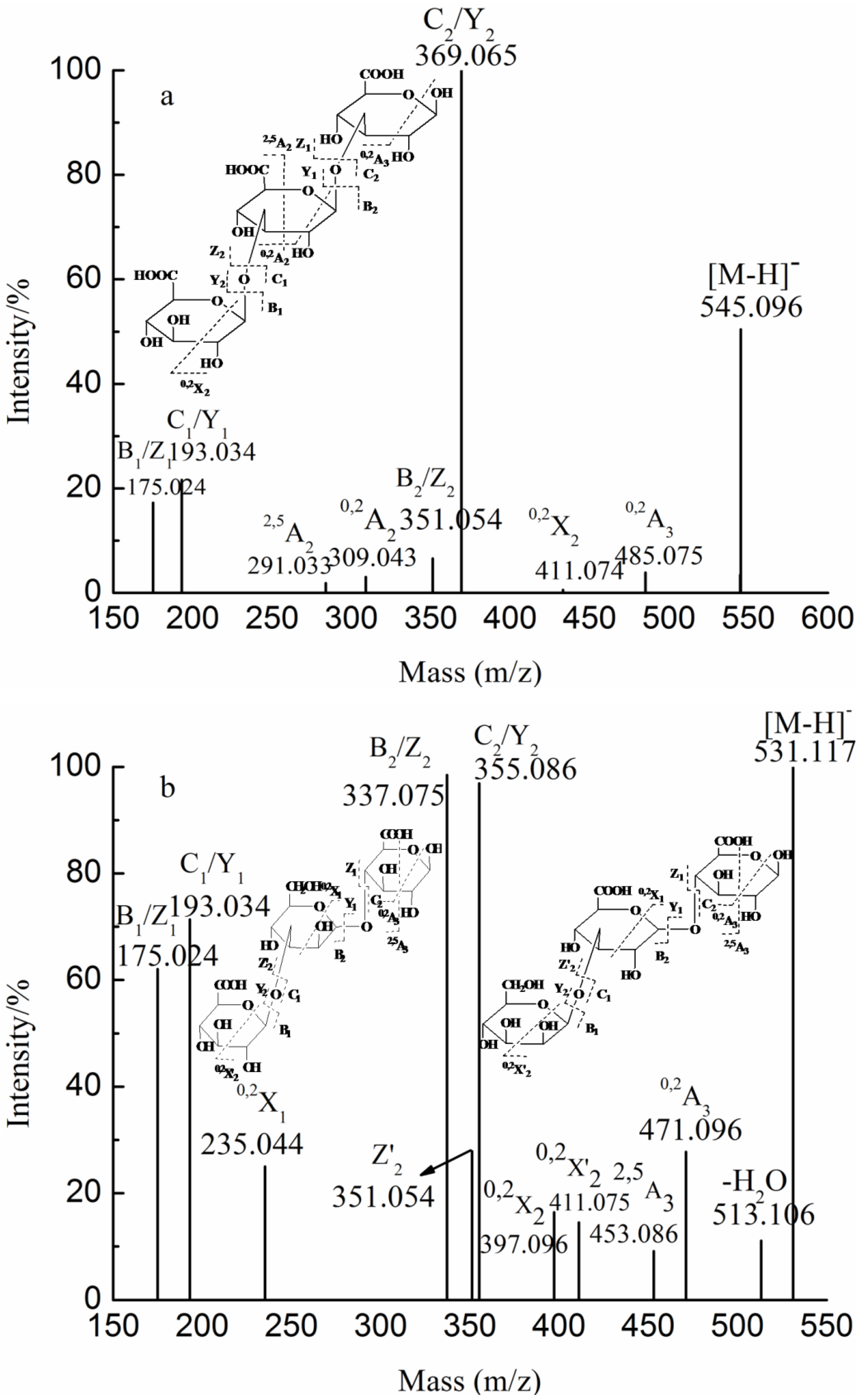
3. Experimental Section
3.1. Materials
3.2. Preparation and Purification of Fucoidans
3.3. Preparation and Purification of Oligosaccharides from F0.5
3.4. Composition Analysis
3.5. MS Analysis of Oligosaccharides
3.6. Condition of HPLC
4. Conclusions
Supplementary Files
Acknowledgments
References
- Sokolova, R.V.; Ermakova, S.P.; Awada, S.M.; Zvyagintseva, T.N.; Kanaan, H.M. Composition, structureal characteristics, and antitumor properties of polysaccharides from the brown algal Dictyopteris polypodioides and Sargassum sp. Chem. Nat. Compd. 2011, 47, 329–334. [Google Scholar] [CrossRef]
- Synytsya, A.; Kim, W.-J.; Kim, S.-M.; Pohl, R.; Synytsya, A.; Kvasnička, F.; Čopíková, J.; Park, Y.I. Structure and antitumour activity of fucoidan isolated from sporophyll of Korean brown seaweed Undaria pinnatifida. Carbohydr. Polym. 2010, 81, 41–48. [Google Scholar] [CrossRef]
- Rhee, K.; Lee, K. Protective effects of fucoidan against γ-radiation-induced damage of blood cells. Arch.Pharm. Res. 2011, 34, 645–651. [Google Scholar] [CrossRef]
- Koyanagi, S.; Tanigawa, N.; Nakagawa, H.; Soeda, S.; Shimeno, H. Oversulfation of fucoidan enhances its anti-angiogenic and antitumor activity. Biochem. Pharmacol. 2003, 65, 173–179. [Google Scholar]
- Caipang, C.M.A.; Lazado, C.C.; Berg, I.; Brinchmann, M.F.; Kiron, V. Influence of alginic acid and fucoidan on the immune responses of head kidney leukocytes in cod. Fish Physiol. Biochem. 2010, 37, 603–612. [Google Scholar]
- Ermakova, S.; Sokolova, R.; Kim, S.-M.; Um, B.-H.; Isakov, V.; Zvyagintseva, T. Fucoidans from Brown Seaweeds Sargassum hornery, Eclonia cava, Costaria costata: Structural characteristics and anticancer activity. Appl. Biochem. Biotechnol. 2011, 164, 841–850. [Google Scholar] [CrossRef]
- Bilan, M.I.; Grachev, A.A.; Ustuzhanina, N.E.; Shashkov, A.S.; Nifantiev, N.E.; Usov, A.I. Structure of a fucoidan from the brown seaweed Fucus evanescens C.Ag. Carbohydr. Res. 2002, 337, 719–730. [Google Scholar] [CrossRef]
- Croci, D.O.; Cumashi, A.; Ushakova, N.A.; Preobrazhenskaya, M.E.; Piccoli, A.; Totani, L.; Ustyuzhanina, N.E.; Bilan, M.I.; Usov, A.I.; Grachev, A.A.; et al. Fucans, but not Fucomannoglucuronans, determine the biological activities of sulfated polysaccharides from Laminaria saccharina brown seaweed. PLoS ONE 2011, 6, e17283. [Google Scholar]
- Duarte, M.E.R.; Cardoso, M.A.; Noseda, M.D.; Cerezo, A.S. Structural studies on fucoidans from the brown seaweed Sargassum stenophyllum. Carbohydr. Res. 2001, 333, 281–293. [Google Scholar] [CrossRef]
- Mestechkina, N.M.; Shcherbukhin, V.D. Sulfated polysaccharides and their anticoagulant activity: A review. Appl. Biochem. Microbiol. 2010, 46, 267–273. [Google Scholar] [CrossRef]
- Jiao, G.; Yu, G.; Zhang, J.; Ewart, H.S. Chemical structures and bioactivities of sulfated polysaccharides from marine algae. Mar. Drugs 2011, 9, 196–223. [Google Scholar] [CrossRef]
- Bilan, M.I.; Grachev, A.A.; Shashkov, A.S.; Kelly, M.; Sanderson, C.J.; Nifantiev, N.E.; Usov, A.I. Further studies on the composition and structure of a fucoidan preparation from the brown alga Saccharina latissima. Carbohydr. Res. 2010, 345, 2038–2047. [Google Scholar] [CrossRef]
- Anastyuk, S.D.; Shevchenko, N.M.; Nazarenko, E.L.; Dmitrenok, P.S.; Zvyagintseva, T.N. Structural analysis of a fucoidan from the brown alga Fucus evanescens by MALDI-TOF and tandem ESI mass spectrometry. Carbohydr. Res. 2009, 344, 779–787. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Q.; Zhang, Z.; Li, Z. Antioxidant activity of sulfated polysaccharide fractions extracted from Laminaria japonica. Int. J. Biologic. Macromol. 2008, 42, 127–132. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Q.; Zhang, Z.; Zhang, H.; Niu, X. Structural studies on a novel fucogalactan sulfate extracted from the brown seaweed Laminaria japonica. Int. J. Biologic. Macromolec. 2010, 47, 126–131. [Google Scholar] [CrossRef]
- Sakai, T.; Kimura, H.; Kojima, K.; Shimanaka, K.; Ikai, K.; Kato, I. Marine bacterial sulfated fucoglucuronomannan (SFGM) lyase digests brown algal SFGM into trisaccharides. Mar. Biotechnol. 2003, 5, 70–78. [Google Scholar] [CrossRef]
- Li, B.; Wei, X.-J.; Sun, J.-L.; Xu, S.-Y. Structural investigation of a fucoidan containing a fucose-free core from the brown seaweed, Hizikia fusiforme. Carbohydr. Res. 2006, 341, 1135–1146. [Google Scholar] [CrossRef]
- Tissot, B.; Salpin, J.; Martinez, M.; Gaigeot, M.; Daniel, R. Differentiation of the fucoidan sulfated l-fucose isomers constituents by CE-ESIMS and molecular modeling. Carbohydr. Res. 2006, 341, 598–609. [Google Scholar] [CrossRef]
- Yu, G.; Zhao, X.; Yang, B.; Ren, S.; Guan, H.; Zhang, Y.; Lawson, A.M.; Chai, W. Sequence determination of sulfated carrageenan-derived oligosaccharides by high-sensitivity negative-ion electrospray tandem mass spectrometry. Anal. Chem. 2006, 78, 8499–8505. [Google Scholar]
- Daniel, R.; Chevolot, L.; Carrascal, M.; Tissot, B.; Mourão, P.A.S.; Abian, J. Electrospray ionization mass spectrometry of oligosaccharides derived from fucoidan of Ascophyllum nodosum. Carbohydr. Res. 2007, 342, 826–834. [Google Scholar] [CrossRef]
- Anastyuk, S.D.; Shevchenko, N.M.; Nazarenko, E.L.; Imbs, T.I.; Gorbach, V.I.; Dmitrenok, P.S.; Zvyagintseva, T. Structural analysis of a highly sulfated fucan from the brown alga Laminaria cichorioides by tandem MALDI and ESI mass spectrometry. Carbohydr. Res. 2010, 345, 2206–2212. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, X.; Lv, Y.; Liu, Y.; Lang, Y.; Wu, J.; Liu, X.; Li, M.; Yu, G. Analysis of structural heterogeneity of fucoidan from Hizikia fusiforme by ES-CID-MS/MS. Carbohydr. Polym. 2012, 90, 602–607. [Google Scholar] [CrossRef]
- Saad, O.M.; Leary, J.A. Delineating mechanisms of dissociation for isomeric heparin disaccharides using isotope labeling and ion trap tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2004, 15, 1274–1286. [Google Scholar] [CrossRef]
- Zhang, J.J.; Zhang, Q.B.; Wang, J.; Shi, X.L.; Zhang, Z.S. Analysis of the monosaccharide composition of fucoidan by precolumn derivation HPLC. Chin. J. Oceanol. Limnol. 2009, 27, 578–582. [Google Scholar] [CrossRef]
- Bitter, T.; Muir, H.M. A modified uronic acid carbazole reaction. Anal. Biochem. 1962, 4, 330–334. [Google Scholar] [CrossRef]
- Samples Availability: Available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jin, W.; Wang, J.; Ren, S.; Song, N.; Zhang, Q. Structural Analysis of a Heteropolysaccharide from Saccharina japonica by Electrospray Mass Spectrometry in Tandem with Collision-Induced Dissociation Tandem Mass Spectrometry (ESI-CID-MS/MS). Mar. Drugs 2012, 10, 2138-2152. https://doi.org/10.3390/md10102138
Jin W, Wang J, Ren S, Song N, Zhang Q. Structural Analysis of a Heteropolysaccharide from Saccharina japonica by Electrospray Mass Spectrometry in Tandem with Collision-Induced Dissociation Tandem Mass Spectrometry (ESI-CID-MS/MS). Marine Drugs. 2012; 10(10):2138-2152. https://doi.org/10.3390/md10102138
Chicago/Turabian StyleJin, Weihua, Jing Wang, Sumei Ren, Ni Song, and Quanbin Zhang. 2012. "Structural Analysis of a Heteropolysaccharide from Saccharina japonica by Electrospray Mass Spectrometry in Tandem with Collision-Induced Dissociation Tandem Mass Spectrometry (ESI-CID-MS/MS)" Marine Drugs 10, no. 10: 2138-2152. https://doi.org/10.3390/md10102138
APA StyleJin, W., Wang, J., Ren, S., Song, N., & Zhang, Q. (2012). Structural Analysis of a Heteropolysaccharide from Saccharina japonica by Electrospray Mass Spectrometry in Tandem with Collision-Induced Dissociation Tandem Mass Spectrometry (ESI-CID-MS/MS). Marine Drugs, 10(10), 2138-2152. https://doi.org/10.3390/md10102138