
Variable Phenotypic Expression of PAX2 Variants in Two Lithuanian Families with Kidney Disease


Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Compliance
2.2. Patients
2.3. Clinical Evaluation
2.4. Genetic Investigations
2.4.1. DNA Extraction
2.4.2. Next-Generation Sequencing
2.4.3. Inherited Kidney Disease Gene Panel Analysis
2.4.4. Sanger Sequencing
3. Results
3.1. Clinical Characteristics
3.2. PAX2 Variants
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CAKUT | Congenital anomalies of the kidney and urinary tract |
| CKD | Chronic kidney disease |
| eGFR | Estimated glomerular filtration rate |
| FSGS | Focal segmental glomerulosclerosis |
| NGS | Next-generation sequencing |
| OMIM | Online Mendelian Inheritance in Man |
| RCS | Renal coloboma syndrome |
References
- Torres, M.; Gómez-Pardo, E.; Dressler, G.R.; Gruss, P. Pax-2 controls multiple steps of urogenital development. Development 1995, 121, 4057–4065. [Google Scholar] [CrossRef]
- Sanyanusin, P.; Schimmenti, L.A.; McNoe, L.A.; Ward, T.A.; Pierpont, M.E.; Sullivan, M.J.; Dobyns, W.B.; Eccles, M.R. Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat. Genet. 1995, 9, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Bower, M.; Salomon, R.; Allanson, J.; Antignac, C.; Benedicenti, F.; Benetti, E.; Binenbaum, G.; Jensen, U.B.; Cochat, P.; DeCramer, S.; et al. Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database. Hum. Mutat. 2012, 33, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Muntean, C.; Chirtes, C.; Baczoni, B.; Banescu, C. PAX2 Gene Mutation in Pediatric Renal Disorders—A Narrative Review. Int. J. Mol. Sci. 2023, 24, 12737. [Google Scholar] [CrossRef]
- Chang, Y.M.; Chen, C.C.; Lee, N.C.; Sung, J.M.; Chou, Y.Y.; Chiou, Y.Y. PAX2 Mutation-Related Renal Hypodysplasia: Review of the Literature and Three Case Reports. Front. Pediatr. 2022, 9, 765929. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, P.; Wu, J.; Chang, Q. A novel PAX2 heterozygous mutation in a family with Papillorenal syndrome: A case report and review of the literature. Am. J. Ophthalmol. Case Rep. 2021, 22, 101091. [Google Scholar] [CrossRef]
- Wall, P.B.; Traboulsi, E.I. Congenital abnormalities of the optic nerve: From gene mutation to clinical expression. Curr. Neurol. Neurosci. Rep. 2013, 13, 363. [Google Scholar] [CrossRef]
- Bullich, G.; Domingo-Gallego, A.; Vargas, I.; Ruiz, P.; Lorente-Grandoso, L.; Furlano, M.; Fraga, G.; Madrid, Á.; Ariceta, G.; Borregán, M.; et al. A kidney-disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases. Kidney Int. 2018, 94, 363–371. [Google Scholar] [CrossRef]
- Rossanti, R.; Morisada, N.; Nozu, K.; Kamei, K.; Horinouchi, T.; Yamamura, T.; Minamikawa, S.; Fujimura, J.; Nagano, C.; Sakakibara, N.; et al. Clinical and genetic variability of PAX2-related disorder in the Japanese population. J. Hum. Genet. 2020, 65, 541–549. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024, 105, S117–S314. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Koressaar, T.; Lepamets, M.; Kaplinski, L.; Raime, K.; Andreson, R.; Remm, M. Primer3_masker: Integrating masking of template sequence with primer design software. Bioinformatics 2018, 34, 1937–1938. [Google Scholar] [CrossRef]
- Negrisolo, S.; Benetti, E.; Centi, S.; Della Vella, M.; Ghirardo, G.; Zanon, G.F.; Murer, L.; Artifoni, L. PAX2 gene mutations in pediatric and young adult transplant recipients: Kidney and urinary tract malformations without ocular anomalies. Clin. Genet. 2011, 80, 581–585. [Google Scholar] [CrossRef]
- Domingo-Gallego, A.; Pybus, M.; Bullich, G.; Furlano, M.; Ejarque-Vila, L.; Lorente-Grandoso, L.; Ruiz, P.; Fraga, G.; González, M.L.; Piñero-Fernández, J.A.; et al. Clinical utility of genetic testing in early-onset kidney disease: Seven genes are the main players. Nephrol. Dial. Transplant. 2021, 37, 687–696. [Google Scholar] [CrossRef]
- Hu, X.; Lin, W.; Luo, Z.; Zhong, Y.; Xiao, X.; Tang, R. Frameshift Mutation in PAX2 Related to Focal Segmental Glomerular Sclerosis: A Case Report and Literature Review. Mol. Genet. Genomic Med. 2024, 12, e70006. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhai, S.B.; Zhao, L.Y.; Zhang, Y.; Sun, B.C.; Ma, Q.S. New PAX2 heterozygous mutation in a child with chronic kidney disease: A case report and review of the literature. BMC Nephrol. 2018, 19, 245. [Google Scholar] [CrossRef]
- Vivante, A.; Chacham, O.S.; Shril, S.; Schreiber, R.; Mane, S.M.; Pode-Shakked, B.; Soliman, N.A.; Koneth, I.; Schiffer, M.; Anikster, Y.; et al. Dominant PAX2 mutations may cause steroid-resistant nephrotic syndrome and FSGS in children. Pediatr. Nephrol. 2019, 34, 1607–1613. [Google Scholar] [CrossRef]
- Ohtsubo, H.; Morisada, N.; Kaito, H.; Nagatani, K.; Nakanishi, K.; Iijima, K. Alport-like glomerular basement membrane changes with renal-coloboma syndrome. Pediatr. Nephrol. 2012, 27, 1189–1192. [Google Scholar] [CrossRef]
- Yamada, Y.; Yokoyama, H.; Kinoshita, R.; Kitamoto, K.; Kawaba, Y.; Okada, S.; Horie, T.; Nagano, C.; Nozu, K.; Namba, N. Familial focal segmental glomerulosclerosis with Alport-like glomerular basement changes caused by paired box protein 2 gene variant. CEN Case Rep. 2024, 13, 204–208. [Google Scholar] [CrossRef]
- Favor, J.; Sandulache, R.; Neuhäuser-Klaus, A.; Pretsch, W.; Chatterjee, B.; Senft, E.; Wurst, W.; Blanquet, V.; Grimes, P.; Spörle, R.; et al. The mouse Pax21Neu mutation is identical to a human PAX2 mutation in a family with renal-coloboma syndrome and results in developmental defects of the brain, ear, eye, and kidney. Proc. Natl. Acad. Sci. USA 1996, 93, 13870–13875. [Google Scholar] [CrossRef]
- Yang, X.; Li, Y.; Fang, Y.; Shi, H.; Xiang, T.; Liu, J.; Liu, J.; Tang, X.; Fang, X.; Chen, J.; et al. Phenotypic spectrum and genetics of PAX2-related disorder in the Chinese cohort. BMC Med. Genom. 2021, 14, 250. [Google Scholar] [CrossRef] [PubMed]
- Madariaga, L.; Morinière, V.; Jeanpierre, C.; Bouvier, R.; Loget, P.; Martinovic, J.; Dechelotte, P.; Leporrier, N.; Thauvin-Robinet, C.; Jensen, U.B.; et al. Severe prenatal renal anomalies associated with mutations in HNF1B or PAX2 genes. Clin. J. Am. Soc. Nephrol. 2013, 8, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.Y.; Shi, Y.Q.; Zhong, C.; Yang, Q.; Zhang, G.; Yang, H.; Wu, D.; Chen, Y.; Li, Q.; Wang, M. Detection of De Novo PAX2 Variants and Phenotypes in Chinese Population: A Single-Center Study. Front. Genet. 2022, 13, 799562. [Google Scholar] [CrossRef]
- Stevenson, M.; Pagnamenta, A.T.; Reichart, S.; Phlpott, C.; Lines, K.E.; OxClinWGS; Gorvin, C.M.; Lhotta, K.; Taylor, J.C.; Thakker, R.V. Whole genome sequence analysis identifies a PAX2 mutation to establish a correct diagnosis for a syndromic form of hyperuricemia. Am. J. Med. Genet. A 2020, 182, 2521–2528. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wu, Y.; Wang, C.; Zhang, W. Inhibition of PAX2 Gene Expression by siRNA (Polyethylenimine) in Experimental Model of Obstructive Nephropathy. Ren. Fail. 2012, 34, 1288–1296. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, H.; Gao, M.; Lv, Y.; Song, W.; Duan, C.; Liu, Y. An induced pluripotent stem cell line (SDQLCHi062-A) from a patient carrying a mutation in the PAX2 gene. Stem Cell Res. 2023, 73, 103260. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Patient 1 | Patient 2 | Patient 3 | Reference Range | ||||
|---|---|---|---|---|---|---|---|
| Age, Years | 27 | 30 | 6 | 10 | 36 | 39 | |
| Urea, mmol/L | 13.5 | 17.2 | 12.38 | 13.7 | 20 | 20.4 | 2.5–7.5 (adults); 1.7–8.3 (children) |
| Creatinine, μmol/L | 174 | 228 | 79 | 110 | 233 | 354 | 64–104 (adults); 25–42 (children) |
| eGFR, mL/min/1.73 m2 | 46 | 32 | 55.7 | 47 | 30 | 18 | >90 |
| Uric acid, μmol/L | 398 | 338 | 371 | 476 | 683 | 379 | 208–428 (adults); 205–420 (children) |
| Yang et al. [21] | Madariaga et al. [22] | Family A Patient 1 | Bower et al. [3] | Family B Patient 2 | Family B Patient 3 | |
|---|---|---|---|---|---|---|
| Gender | F | M | M | Not specified | F | M |
| Age at first presentation | 2.8 years | Prenatal | 11 years | 5.5 years | After birth | 20 years |
| Phenotype category | CAKUT | CAKUT | CAKUT | N/A | Nephrosis | Nephrosis |
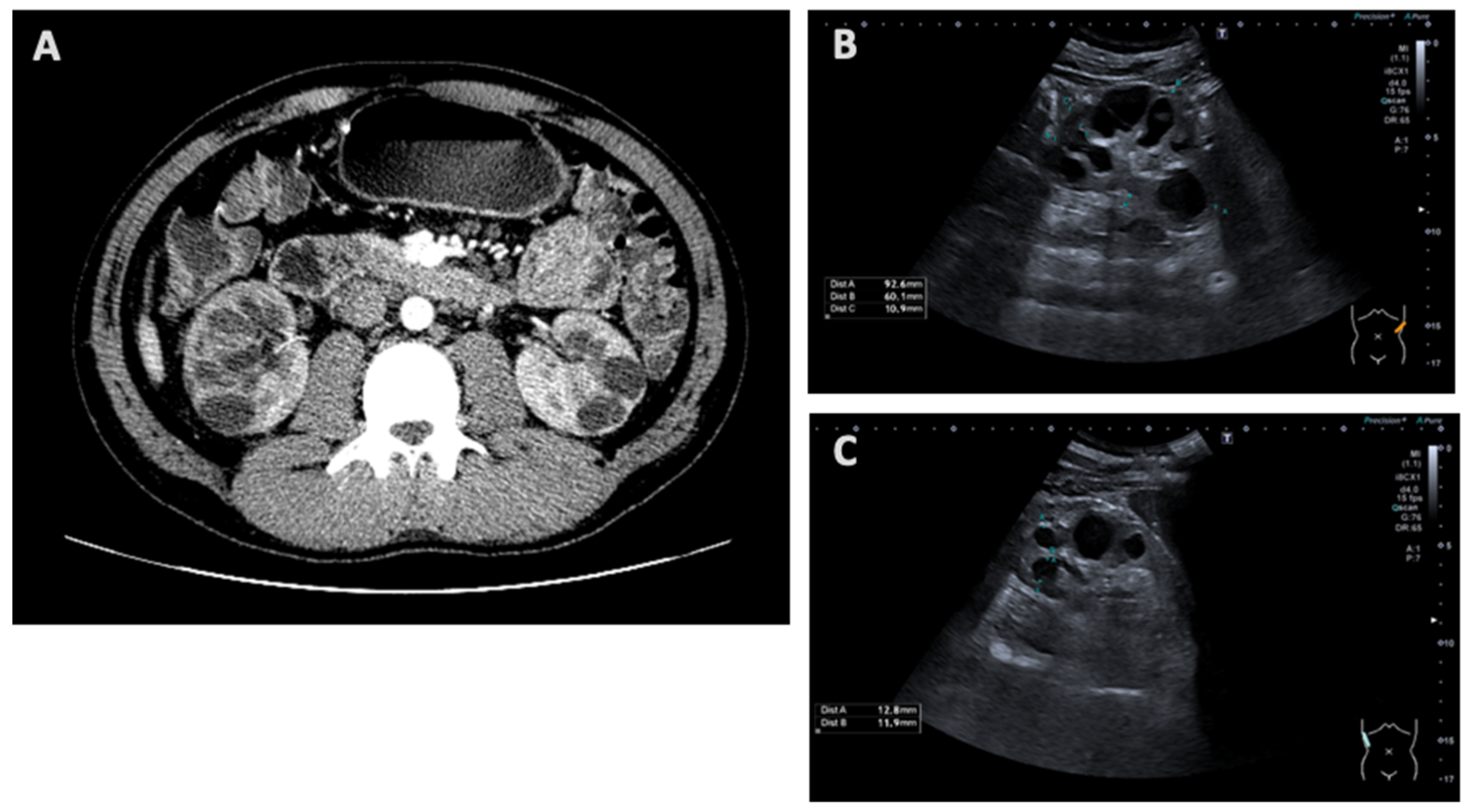
| Kidney imaging | N/A | Bilateral hyperechogenic kidneys | Multicystic dysplastic kidney/polycystic kidney | N/A | Signs of chronic kidney disease, microcalcifications and simple cysts | Increased parenchymal echogenicity |
| Kidney histology | N/A | Bilateral hypoplasia with cortical tubular microcysts | N/A | N/A | N/A | Focal segmental glomerulosclerosis |
| Clinical diagnosis of kidney disease | Bilateral renal hypodysplasia | N/A | Bilateral multicystic kidney dysplasia | N/A | Bilateral renal hypoplasia | Focal segmental glomerulosclerosis |
| Renal function | CKD stage 5 at 6 years old | N/A | CKD stage 3 at 27 years old | N/A | CKD stage 3 at 6 years old | CKD stage 3 at 36 years old |
| Ocular findings | Intermittent strabismus | Focal retinal dysplasia | Bilateral optic nerve dysplasia | Bilateral optic nerve coloboma; enlarged optic nerve with pits; photophobia | Normal examination | Optic disc pits |
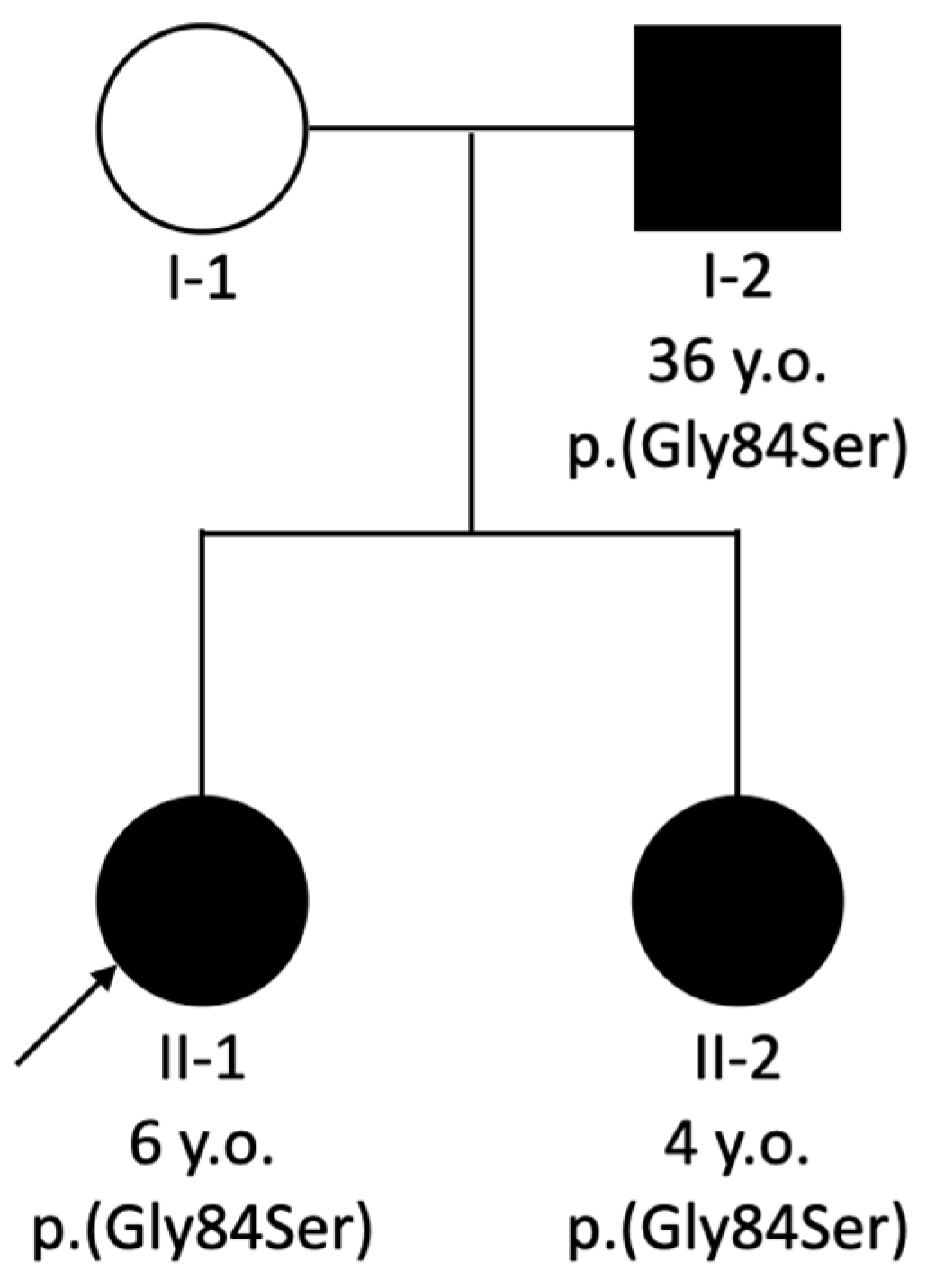
| PAX2 gene variant | c.685C>T, p.Arg229* | c.754C>T, p.Arg252* | c.685C>T, p.Arg229* | c.250G>A, p.(Gly84Ser) | c.250G>A, p.(Gly84Ser) | c.250G>A, p.(Gly84Ser) |
| Type of mutation | Nonsense | Nonsense | Nonsense | Missense | Missense | Missense |
| Zygosity | Het | Het | Het | Het | Het | Het |
| Segregation | N/A | Affected father | N/A | Affected father and siblings | Affected father and sibling | Affected daughters |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Lithuanian University of Health Sciences. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brazdziunaite, D.; Mazur, G.; Miglinas, M.; Utkus, A. Variable Phenotypic Expression of PAX2 Variants in Two Lithuanian Families with Kidney Disease. Medicina 2025, 61, 597. https://doi.org/10.3390/medicina61040597
Brazdziunaite D, Mazur G, Miglinas M, Utkus A. Variable Phenotypic Expression of PAX2 Variants in Two Lithuanian Families with Kidney Disease. Medicina. 2025; 61(4):597. https://doi.org/10.3390/medicina61040597
Chicago/Turabian StyleBrazdziunaite, Deimante, Gabija Mazur, Marius Miglinas, and Algirdas Utkus. 2025. "Variable Phenotypic Expression of PAX2 Variants in Two Lithuanian Families with Kidney Disease" Medicina 61, no. 4: 597. https://doi.org/10.3390/medicina61040597
APA StyleBrazdziunaite, D., Mazur, G., Miglinas, M., & Utkus, A. (2025). Variable Phenotypic Expression of PAX2 Variants in Two Lithuanian Families with Kidney Disease. Medicina, 61(4), 597. https://doi.org/10.3390/medicina61040597