Development of a New Bioequivalent Omeprazole Product


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Omeprazole Pellets
2.2.1. Active Coating
2.2.2. Protective Coating
2.2.3. Enteric Coating
2.3. Dissolution Study
2.4. Stability Study
2.5. Bioequivalence Study
2.5.1. Study Products
2.5.2. Ethics Compliance and Subjects
2.5.3. Study Design and Safety
2.5.4. Analytical Method
2.5.5. Pharmacokinetic and Statistical Analysis
3. Results
3.1. Development of the Composition and the Production Technology of the Drug
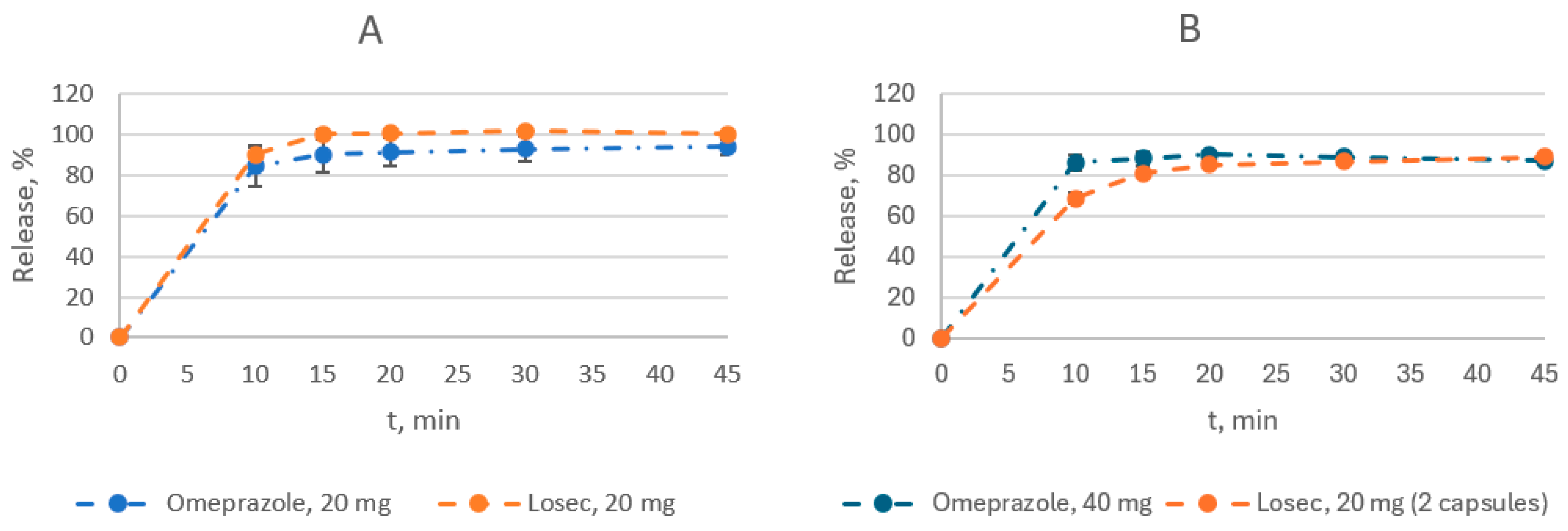
3.2. Dissolution Study
3.3. Stability Study
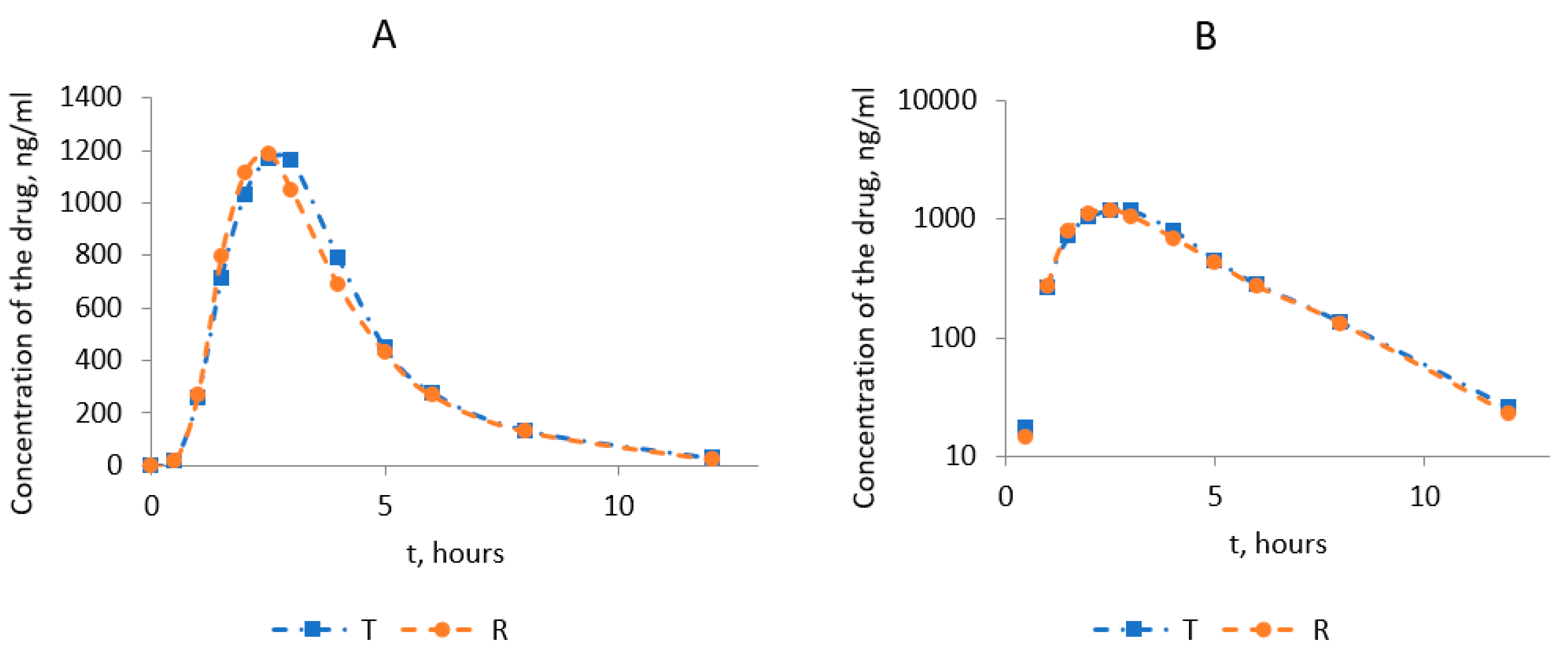
3.4. Bioequivalence Study
3.4.1. Subjects
3.4.2. Assessment of PK parameters
- lnCmax: 95.02–113.15%; μT/μR = 103.69;
- lnAUC0-t: 96.03–110.74%; μT/μR = 103.13;
- lnCmax/AUC0-t: 96.58–104.67%; μT/μR = 100.54.
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dekhnich, N.N. Proton pump inhibitors in the treatment of acid-dependent diseases. Bull. Smolensk. State Med. Acad. 2011, 1, 21–24. [Google Scholar]
- Lazebnik, L.B.; Lee, E.D.; Mikheeva, O.M. The use of proton pump inhibitors for the treatment of acid-related diseases. Arch. Intern. Med. 2013, 3, 56. [Google Scholar]
- Product Monograph Losec® (Omeprazole Delayed Release Capsules) 20 mg Omeprazole H+, K+-ATPase Inhibitor; AstraZeneca Canada Inc.: Mississauga, ON, Canada, 2017.
- Junggren, U.K.; Sjöstrand, S.E. Substituted Pyridylsulfinylbenzimidazoles Having Gastric Acid Secretion Properties, Pharmaceutical Preparations Containing Same, and Intermediates for Their Preparation. Patent No. EP0005129A1, 31 October 1979. [Google Scholar]
- Trends in Drug Patenting—Case Studies: The Cases: 5. Omeprazole. apps.who.int. 2 October 2010. Available online: http://apps.who.int/medicinedocs/en/d/Js4915e/2.5.html (accessed on 22 August 2019).
- Leading chemical substances dispensed in England in 2022, by number of items. Available online: https://www.statista.com/statistics/378445/prescription-cost-analysis-top-twenty-chemicals-by-items-in-england// (accessed on 25 February 2024).
- Omeprazole. Drug Usage Statistics, United States, 2013–2021. Available online: https://clincalc.com/DrugStats/Drugs/Omeprazole// (accessed on 25 February 2024).
- Kumisbek, G.K.; David, V. Relevance of Application and Market Review of Proton Pump Inhibitors and Omeprazole//Natural Sciences and Medicine: Theory and Practice: Collection. Art. by Mother XL-XLI International Scientific-Practical Conf. 11–12; SibAK: Novosibirsk, Russia, 2021; pp. 42–48. [Google Scholar]
- RSE on REM “National Center for Expertise of Medicines and Medical Devices” of the Committee for Medical and Pharmaceutical Control of the Ministry of Health of the Republic of Kazakhstan Home Page. Available online: http://register.ndda.kz/category/search_prep (accessed on 7 December 2021).
- Bai, M.; Chen, S.; Xin, W.; Wu, H. Enteric-Coated Pellet Preparation of Proton Pump Inhibitor and Preparation Method Thereof Patent No. CN102119927B, 26 December 2012. CSPC Zhongqi Pharmaceutical Technology Shijiazhuang Co., Ltd.: Shijiazhuang, China.
- Chen, J.-R. Preparation of Enteric Pharmaceutical Dosage Forms for Omerprazole and Lansoprazole. Patent No. US 6,726,927 B2, 27 April 2004. Sage Pharmaceuticals, Inc.: Shreveport, LA, USA, 2012. [Google Scholar]
- Bengtsson, I.S.; Lovgren, K.I. Pharmaceutical Formulation of Omeprazole. US Patent Patent No5,690,960, 25 November 1997. Astra Aktiebolag: Sodertalje, Sweden. [Google Scholar]
- United States Pharmacopeia. USP Monographs, Omeprazole Delayed-Release Capsules; Doc ID: GUID-71E87DD7-0164-42C0-8027-A9211B069968_1_en-US; USP-NF (United States Pharmacopeia): Rockville, MD, USA, 2023. [Google Scholar] [CrossRef]
- ICH Topic Q 1 A (R2) Stability Testing of New Drug Substances and Products. Note for Guidance on Stability Testing: Stability Testing of New Drug Substances and Products (CPMP/ICH/2736/99). The ICH Official Website. Available online: https://www.ich.org/page/quality-guidelines (accessed on 23 November 2023).
- On Approval of the Rules for Conducting Stability Studies by the Manufacturer of a Medicinal Product, Establishing a Shelf Life and Re-Monitoring of Medicinal Products Order of the Minister of Health of the Republic of Kazakhstan. Dated 28 October 2020. No. KR DSM-165/2020. Registered with the Ministry of Justice of the Republic of Kazakhstan on 30 October 2020. No. 21545. Available online: https://adilet.zan.kz/rus/index/docs (accessed on 23 November 2023).
- Rules for Conducting Bioequivalence Studies of Medicinal Products within the Framework of the Eurasian Economic Union; No. 85; Council of the Eurasian Economic Commission: Moscow, Russia, 2016.
- Guidance for Industry: Bioanalytical Method Validation; US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evolution and Research (CDER), US Government Printing Office: Washington, DC, USA, 2018.
- Guideline on Bioanalytical Method Validation; EMEA/CHMP/EWP/192217/2009; European Medicines Agency, Committee for Medicinal Products for Human Use (EMEA): Amsterdam, The Netherlands, 2011.
- Mironov, N. Guidelines for Examination of Medicinal Products; Federal State Budgetary Institution “NTsESMP”: Moscow, Russia, 2014; Volume 1. [Google Scholar]
- Šalandová, J.; Franc, A.; Hofmann, J.; Dumicic, A.; Kukačková, L.; Červená, T.; Beránek, J.; Srbek, J.; Repický, A.; Vladovičová, B.; et al. The effect of the composition of a fixed dose combination on bioequivalence results. Int. J. Pharm. 2018, 546, 235–246. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Fan, L.-F.; Du, Q.; Xiang, B.; Li, C.-L.; Bai, M.; Chang, Y.-Z.; Cao, D.-Y. Design and in Vitro/in Vivo Evaluation of Multi-layer Film Coated Pellets for Omeprazole. Chem. Pharm. Bull. 2009, 57, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Joti, J.J.; Nahar, K.; Hasan, A.; Azad, M.A.; Ullah, M.A.; Islam, S.M.; Hasnat, A. Bioequivalence and Pharmacokinetic Study of Two Different Omeprazole Capsule Formulations in Healthy Bangladeshi Volunteers. Arzneimittelforschung 2009, 59, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, K.; Ghozal, M.; Behloul, S.; Adda Abbou, L.; Makhlouf, A.; Nekhoul, K.; Hadjaz, I.; Mansouri, M.B. Bioequivalence of Two Brands of Omeprazole 20 mg Gastro-Resistant Capsules in 18 Healthy Algerian Volunteers: A Pilot Study. J. Pharm. Pharmacol. 2017, 5, 877–884. [Google Scholar] [CrossRef][Green Version]
- Guideline on the Investigation of Bioequivalence; EMEA/CPMP/EWP/QWP/1401/98; European Medicines Agency, Committee for Medicinal Products for Human Use (EMEA): Amsterdam, The Netherlands, 2011.
- Chow, S.C.; Liu, J.P. Design and Analysis of Bioavailability and Bioequivalence Studies; Marcel Dekker Inc.: New York, NY, USA, 1992. [Google Scholar]
- Hauschke, D.; Kieser, M.; Diletti, E.; Burke, M. Sample size determination for proving equivalence based on the ratio of two means for normally distributed data. Stat. Med. 1999, 18, 93–105. [Google Scholar] [CrossRef]
- Berger, R.L.; Hsu, J.C. Bioequivalence Trials, Intersection-Union Tests and Confidence Sets. Stat. Sci. 1996, 11, 283–319. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Period of Study/Procedure | Screening | Study Periods | Final Examination | ||
|---|---|---|---|---|---|
| I | Washout Period | II | |||
| Thermometry (temperature should be between 36.0° and 36.6°) | Before screening | Before period I | Seven days | Before period II | |
| Duration of period | up to 10 days | 13 h | 13 h | ||
| Informed consent | X | ||||
| Inclusion criteria | X | X | X | ||
| Non-inclusion criteria | X | X | X | ||
| Demographic and anthropometric data | X | ||||
| Medical history | X | ||||
| Physical examination, assessment of vital signs (blood pressure, heart rate, body temperature) | X | X | X | X | |
| Laboratory examination (clinical blood test, biochemical blood test, general urine test) | X | X | |||
| Laboratory examination (clinical blood test, biochemical blood test, general urine test) | X | ||||
| Urine pregnancy test | X | X | X | ||
| Urine analysis for psychotropic and narcotic substances, psychoactive drugs | X | X | X | ||
| Alcohol test (breathalyzer/test strips) | X | X | X | ||
| Hospitalization | X | X | |||
| Electrocardiogram | X | X | |||
| Randomization | X | ||||
| Administration of T or R drug | X | X | |||
| Blood sampling for PC | X | X | |||
| Assessment of A.E.s/SAEs | X | X | X | X | X |
| Component | Function | Composition 1 (%) | Composition 2 (%) | Composition 3 (%) | Composition 4 (%) |
|---|---|---|---|---|---|
| Microcrystalline cellulose (MCC) pellets | Neutral cores | 60.45 | 64.8 | 62.75 | 62.44 |
| Omeprazole | API | 10.91 | 9.09 | 8.81 | 8.76 |
| Lactose monohydrate | Filler | 9.28 | 7.73 | 7.48 | 7.44 |
| Sodium lauryl sulfate | Solubilizer | 0.33 | 0.27 | 0.26 | 0.26 |
| Sodium pyrophosphate dodecahydrate | Buffer agent | 1.42 | 1.18 | 1.14 | 1.14 |
| Hydrohypropylmethyl- cellulose | Film-former/ Binder | 7.97 | 8.55 | 8.27 | 8.67 |
| Hydrohypropylcellulose | Film-former/ Binder | 2.18 | 0.91 | 0.88 | 0.44 |
| Eudragit L30-D55 | Enteric coating agent | 5.76 | 6.29 | 8.79 | 8.75 |
| PlasAcryl | Anti-tacking /Plasticizer | - | 1.07 | 1.5 | 1.49 |
| Polyethylene glycol | 1.6 | - | - | - | |
| Titanium dioxide | Colorant | - | - | - | 0.44 |
| Talc | Lubricant | 0.1 | 0.11 | 0,12 | 0,18 |
| Purified water 1 | Solvent | + | + | + | + |
| Total capsule content weight, % | 100 | 100 | 100 | 100 | |
| Weight of capsule content: 20 mg | 218.0 | 220.0 | 227.27 | 228.4 | |
| Weight of capsule content: 40 mg | 436.0 | 440.0 | 454.54 | 456.8 | |
| Capsule shell—body and cap composition, %: | |||||
| Titanium dioxide | Colorant | 2.0 | 2.0 | 2.0 | 2.0 |
| Gelatin | Base | up to 100 | up to 100 | up to 100 | up to 100 |
| Group | Parameter | T1/2, h | tmax, h | Cmax, ng/mL | AUC0-t, ng/mL·h | AUC0-∞ ng/mL·h | (AUC0-t/AUC0-∞), % | Cmax/AUC0-t, h−1 | Cmax/AUC0-∞, h−1 | kel |
|---|---|---|---|---|---|---|---|---|---|---|
| Test | Arithmetic mean | 1.83 | 2.54 | 1320.578 | 4520.85 | 4635.99 | 97.347 | 0.2936 | 0.2856 | 0.3819 |
| Geometric mean | 1.82 | 2.50 | 1299.250 | 4448.57 | 4571.08 | 97.320 | 0.2921 | 0.2842 | 0.3799 | |
| Median | 1.84 | 2.50 | 1304.045 | 4416.58 | 4496.93 | 98.164 | 0.2878 | 0.2803 | 0.3776 | |
| Minimum | 1.42 | 2.00 | 945.489 | 2880.87 | 3044.90 | 89.859 | 0.2415 | 0.2390 | 0.3341 | |
| Maximum | 2.07 | 3.00 | 1984.091 | 6454.10 | 6571.64 | 98.969 | 0.3503 | 0.3374 | 0.4897 | |
| Standard deviation | 0.18 | 0.44 | 249.011 | 840.65 | 814.21 | 2.295 | 0.0311 | 0.0292 | 0.0415 | |
| CV, % | 9.8 | 17.3 | 18.9 | 18.6 | 17.6 | 2.4 | 10.6 | 10.2 | 10.9 | |
| Reference | Arithmetic mean | 1.82 | 2.44 | 1273.837 | 4370.70 | 4502.17 | 96.841 | 0.2914 | 0.2821 | 0.3844 |
| Geometric mean | 1.81 | 2.40 | 1253.036 | 4313.69 | 4456.15 | 96.803 | 0.2905 | 0.2812 | 0.3824 | |
| Median | 1.85 | 2.50 | 1284.560 | 4491.26 | 4568.94 | 98.192 | 0.2932 | 0.2878 | 0.3751 | |
| Minimum | 1.44 | 2.00 | 782.761 | 2773.41 | 2988.49 | 89.755 | 0.2546 | 0.2509 | 0.3145 | |
| Maximum | 2.20 | 3.00 | 1789.898 | 5566.99 | 5650.57 | 98.532 | 0.3403 | 0.3244 | 0.4810 | |
| Standard deviation | 0.18 | 0.43 | 233.311 | 695.23 | 640.08 | 2.742 | 0.0233 | 0.0226 | 0.0418 | |
| CV, % | 9.9 | 17.4 | 18.3 | 15.9 | 14.2 | 2.8 | 8.0 | 8.0 | 10.9 |
| Drug | Geometric Mean Values of Pharmacokinetic Parameters | AUC0-t/ AUC0-∞, % | Criteria for the Observation Period Sufficiency: AUC0-t > 80% × AUC0-∞ | |
|---|---|---|---|---|
| AUC0-t, ng/mL·h | AUC0-∞, ng/mL·h | |||
| Test | 4448.57 | 4571.08 | 97.3 | Completed |
| Reference | 4313.69 | 4456.15 | 96.8 | Completed |
| PK Parameter | MSEW (σ2) | CVintra | CVintra (%) | Significance Level | Group Sizes | Power |
|---|---|---|---|---|---|---|
| ln(Cmax) | 0.03105 | 0.1776 | 17.76 | α = 5% | (12, 12) | 99.9 |
| ln(AUC0→t) | 0.02067 | 0.1445 | 14.45 | 100.0 | ||
| ln(Cmax/AUC0→t) | 0.00659 | 0.0813 | 8.13 | 100.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumisbek, G.; Vetchý, D.; Kadyrbay, A. Development of a New Bioequivalent Omeprazole Product. Medicina 2024, 60, 427. https://doi.org/10.3390/medicina60030427
Kumisbek G, Vetchý D, Kadyrbay A. Development of a New Bioequivalent Omeprazole Product. Medicina. 2024; 60(3):427. https://doi.org/10.3390/medicina60030427
Chicago/Turabian StyleKumisbek, Gulzina, David Vetchý, and Arshyn Kadyrbay. 2024. "Development of a New Bioequivalent Omeprazole Product" Medicina 60, no. 3: 427. https://doi.org/10.3390/medicina60030427
APA StyleKumisbek, G., Vetchý, D., & Kadyrbay, A. (2024). Development of a New Bioequivalent Omeprazole Product. Medicina, 60(3), 427. https://doi.org/10.3390/medicina60030427