
Novel Germline PHD2 Variant in a Metastatic Pheochromocytoma and Chronic Myeloid Leukemia, but in the Absence of Polycythemia

 ,
,  , , ,
, , ,  ,
,  ,
,  ,
,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
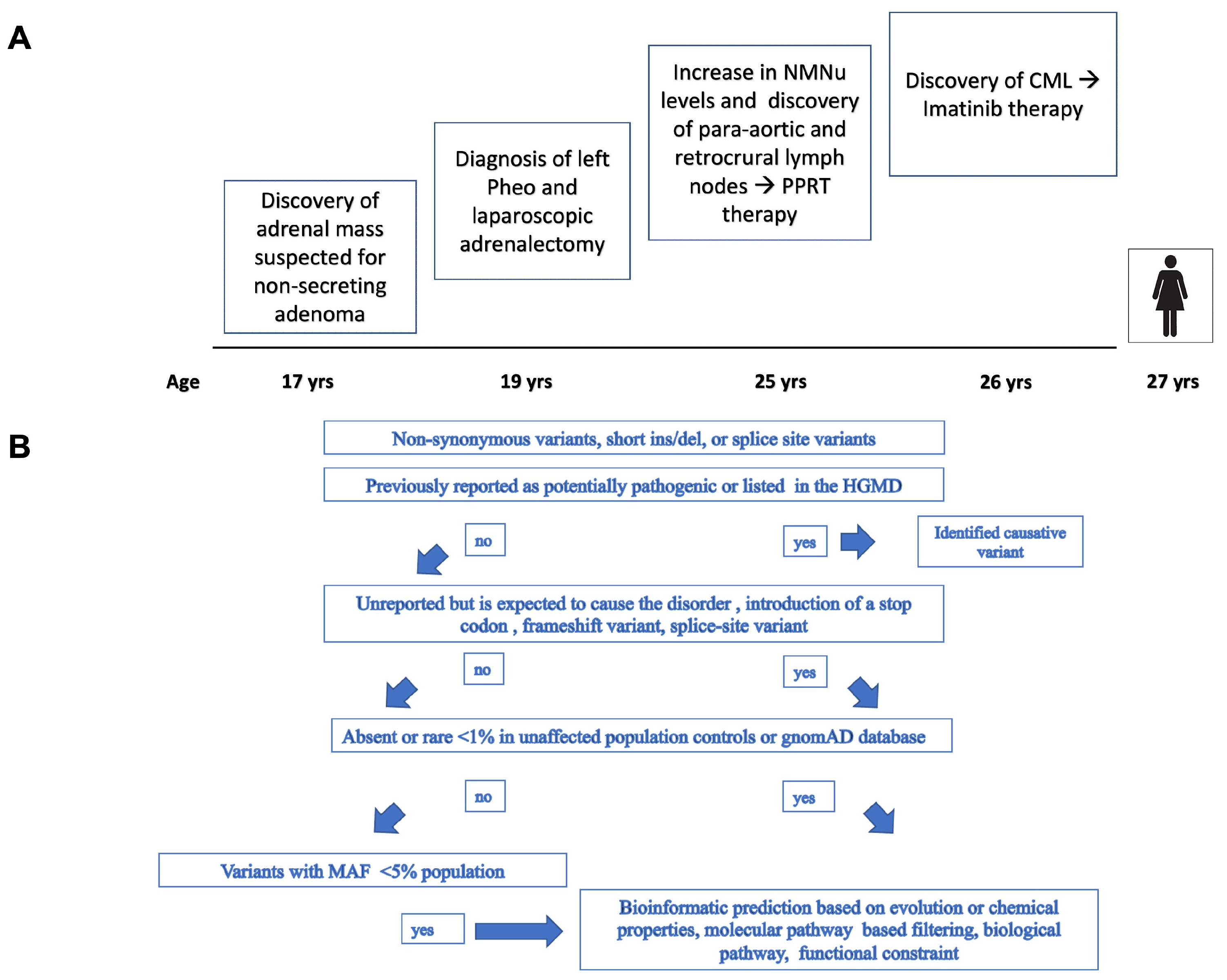
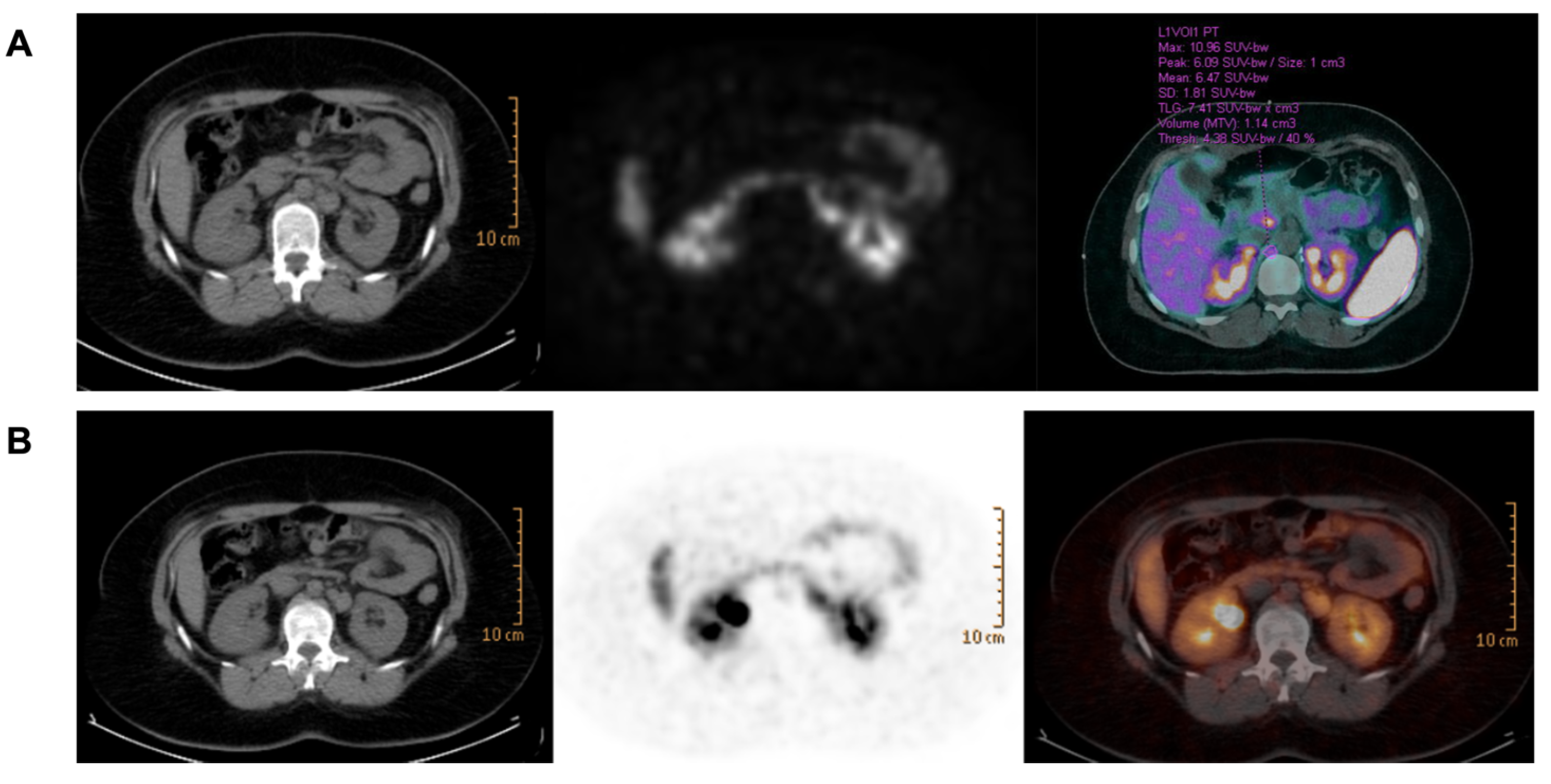
2.1. Case Presentation
2.2. Genetic Analysis
2.3. SNP-CGH Array
2.4. Whole Exome Sequencing and Bioinformatics Analysis
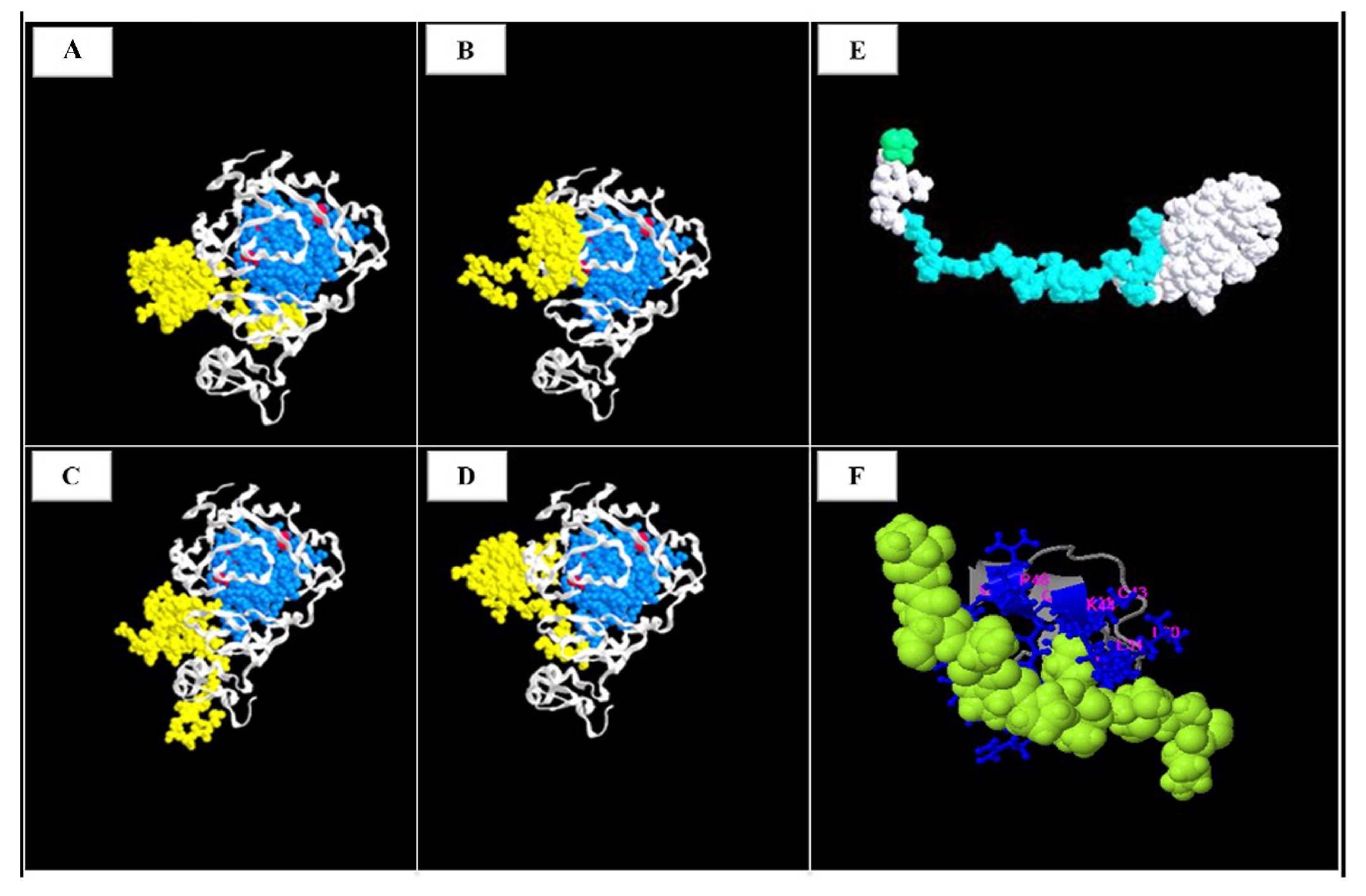
2.5. 3 D Variant Prediction
2.6. PHD2 Immunohistochemistry
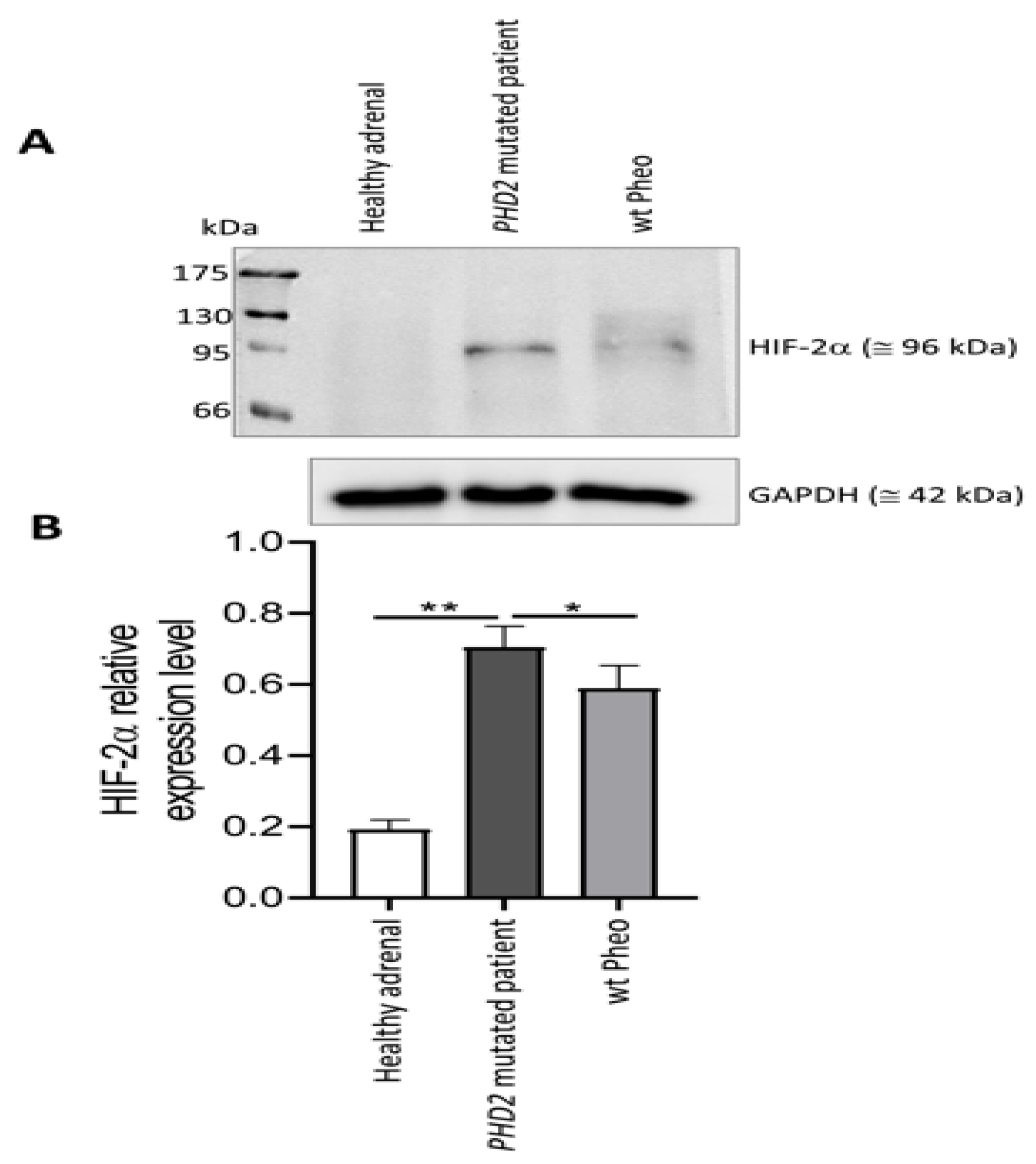
2.7. Western Blot Analysis
2.8. RNA Isolation and Quantitative Real-Time PCR
3. Results
3.1. Mutational Analysis
3.2. PHD2 and HIF Expression in the Primary Tumor
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lenders, J.W.M.; Kerstens, M.N.; Amar, L.; Prejbisz, A.; Robledo, M.; Taieb, D.; Pacak, K.; Crona, J.; Zelinka, T.; Mannelli, M.; et al. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: A position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J. Hypertens. 2020, 38, 1443–1456. [Google Scholar] [PubMed]
- Majewska, A.; Budny, B.; Ziemnicka, K.; Ruchała, M.; Wierzbicka, M. Head and Neck Paragangliomas—A Genetic Overview. Int. J. Mol. Sci. 2020, 21, 7669. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.; Murad, M.H.; Narse, M.; Pacak, K.; Young, W.F., Jr. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef]
- Rao, D.; Van Berkel, A.; Piscaer, I.; Young, W.F.; Gruber, L.; Deutschbein, T.; Fassnacht, M.; Beuschlein, F.; Spyroglou, A.; Prejbisz, A.; et al. Impact of 123I-MIBG scintigraphy on clinical decision-making in pheochromocytoma and paraganglioma. J. Clin. Endocrinol. Metab. 2019, 104, 3812–3820. [Google Scholar] [CrossRef]
- Taïeb, D.; Hicks, R.J.; Hindié, E.; Guillet, B.A.; Avram, A.; Ghedini, P.; Timmers, H.J.; Scott, A.T.; Elojeimy, S.; Rubello, D.; et al. European Association of Nuclear Medicine Practice Guideline/Society of Nuclear Medicine and Molecular Imaging Procedure Standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2112–2137. [Google Scholar]
- Roman-Gonzalez, A.; Zhou, S.; Ayala-Ramirez, M.; Shen, C.; Waguespack, S.G.; Habra, M.A.; Karam, J.A.; Perrier Md, N.; Wood, C.G.; Jimenez, C. Impact of Surgical Resection of the Primary Tumor on Overall Survival in Patients with Metastatic Pheochromocytoma or Sympathetic Paraganglioma. Ann. Surg. 2018, 268, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.P.H.; Young, W.F.; Eng, C. Pheochromocytoma and Paraganglioma. N. Engl. J. Med. 2019, 381, 552–565. [Google Scholar] [CrossRef]
- Hescot, S.; Leboulleux, S.; Amar, L.; Vezzosi, D.; Borget, I.; Bournaud-Salinas, C.; de la Fouchardiere, C.; Libé, R.; Do Cao, C.; Niccoli, P.; et al. One-year progression-free survival of therapy-naive patients with malignant pheochromocytoma and paraganglioma. J. Clin. Endocrinol. Metab. 2013, 98, 4006–4012. [Google Scholar] [CrossRef]
- Pryma, D.A.; Chin, B.B.; Noto, R.B.; Dillon, J.S.; Perkins, S.; Solnes, L.; Kostakoglu, L.; Serafini, A.N.; Pampaloni, M.H.; Jensen, J.; et al. Efficacy and Safety of High-Specific-Activity. J. Nucl. Med. 2019, 60, 623–630. [Google Scholar] [CrossRef]
- Kolasinska-Ćwikła, A.; Pęczkowska, M.; Ćwikła, J.B.; Michałowska, I.; Pałucki, J.M.; Bodei, L.; Lewczuk-Myślicka, A.; Januszewicz, A. A Clinical Efficacy of PRRT in Patients with Advanced, Nonresectable, Paraganglioma-Pheochromocytoma, Related to SDHx Gene Mutation. J. Clin. Med. 2019, 8, 952. [Google Scholar] [CrossRef]
- Fishbein, L.; Ben-Maimon, S.; Keefe, S.; Cengel, K.; Pryma, D.A.; Loaiza-Bonilla, A.; Fraker, D.L.; Nathanson, K.L.; Cohen, D.L. Mutation carriers with malignant pheochromocytoma respond better to CVD. Endocr. -Relat. Cancer 2017, 24, L51–L55. [Google Scholar] [CrossRef] [PubMed]
- Breen, W.; Bancos, I.; Young, W.F.; Bible, K.C.; Laack, N.N.; Foote, R.L.; Hallemeier, C.L. External beam radiation therapy for advanced/unresectable malignant paraganglioma and pheochromocytoma. Adv. Radiat. Oncol. 2018, 3, 25–29. [Google Scholar] [CrossRef] [PubMed]
- O’Kane, G.M.; Ezzat, S.; Joshua, A.; Bourdeau, I.; Leibowitz-Amit, R.; Olney, H.J.; Krzyzanowska, M.; Reuther, D.; Chin, S.; Wang, L.; et al. A phase 2 trial of sunitinib in patients with progressive paraganglioma or pheochromocytoma: The SNIPP trial. Br. J. Cancer 2019, 120, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Tong, A.; Li, M.; Cui, Y.; Ma, X.; Wang, H.; Li, Y. Temozolomide Is a Potential Therapeutic Tool for Patients with Metastatic Pheochromocytoma/Paraganglioma-Case Report and Review of the Literature. Front. Endocrinol. 2020, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, C.; Subbiah, V.; Stephen, B.; Ma, J.; Milton, D.; Xu, M.; Zarifa, A.; Akhmedzhanov, F.O.; Tsimberidou, A.; Habra, M.A.; et al. Phase II Clinical Trial of Pembrolizumab in Patients with Progressive Metastatic Pheochromocytomas and Paragangliomas. Cancers 2020, 12, 2307. [Google Scholar] [CrossRef]
- Buffet, A.; Burnichon, N.; Favier, J.; Gimenez-Roqueplo, A.-P. An overview of 20 years of genetic studies in pheochromocytoma and paraganglioma. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101416. [Google Scholar]
- Casey, R.; Neumann, H.P.H.; Maher, E.R. Genetic stratification of inherited and sporadic phaeochromocytoma and paraganglioma: Implications for precision medicine. Hum. Mol. Genet. 2020, 29, R128–R137. [Google Scholar] [CrossRef]
- Pacak, K.; Wimalawansa, S.J. Pheochromocytoma and paraganglioma. Endocr. Pract. 2015, 21, 406–412. [Google Scholar] [CrossRef]
- Jochmanova, I.; Pacak, K. Genomic Landscape of Pheochromocytoma and Paraganglioma. Trends Cancer 2017, 4, 6–9. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Pacak, K.; Huynh, T.-T.; Qin, N.; Bratslavsky, G.; Linehan, W.M.; Mannelli, M.; Friberg, P.; Grebe, S.K.; Timmers, H.J.; et al. Catecholamine metabolomic and secretory phenotypes in phaeochromocytoma. Endocr. -Relat. Cancer 2010, 18, 97–111. [Google Scholar] [CrossRef]
- Favier, J.; Gimenez-Roqueplo, A.P. Genetics of paragangliomas and pheochromocytomas. Med. Sci. 2012, 28, 625–632. [Google Scholar]
- Dahia, P.L.M. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Cancer 2014, 14, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Berends, A.M.A.; Eisenhofer, G.; Fishbein, L.; Horst-Schrivers, A.N.A.V.; Kema, I.P.; Links, T.P.; Lenders, J.W.M.; Kerstens, M.N. Intricacies of the Molecular Machinery of Catecholamine Biosynthesis and Secretion by Chromaffin Cells of the Normal Adrenal Medulla and in Pheochromocytoma and Paraganglioma. Cancers 2019, 11, 1121. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, S.; Maggi, M.; Rapizzi, E. Pheochromocytoma/paraganglioma preclinical models: Which to use and why? Endocr. Connect. 2020, 9, R251–R260. [Google Scholar] [CrossRef] [PubMed]
- The NGS in PPGL (NGSnPPGL) Study Group; Toledo, R.; Burnichon, N.; Cascon, A.; Benn, D.E.; Bayley, J.-P.; Welander, J.; Tops, C.M.; Firth, H.; Dwight, T.; et al. Consensus Statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat. Rev. Endocrinol. 2017, 13, 233–247. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Activation of the HIF pathway in cancer. Curr. Opin. Genet. Dev. 2001, 11, 293–299. [Google Scholar] [CrossRef]
- Lau, K.W.; Tian, Y.-M.; Raval, R.R.; Ratcliffe, P.; Pugh, C.W. Target gene selectivity of hypoxia-inducible factor-α in renal cancer cells is conveyed by post-DNA-binding mechanisms. Br. J. Cancer 2007, 96, 1284–1292. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Hrašćan, R.; Pecina-Slaus, N.; Martić, T.N.; Čolić, J.F.; Gall-Trošelj, K.; Pavelić, K.; Karapandža, N. Analysis of Selected Genes in Neuroendocrine Tumours: Insulinomas and Phaeochromocytomas. J. Neuroendocr. 2008, 20, 1015–1022. [Google Scholar] [CrossRef]
- Crona, J.; Verdugo, A.D.; Maharjan, R.; Stålberg, P.; Granberg, D.; Hellman, P.; Björklund, P. Somatic mutations in H-RAS in sporadic pheochromocytoma and paraganglioma identified by exome sequencing. J. Clin. Endocrinol. Metab. 2013, 98, E1266–E1271. [Google Scholar] [CrossRef]
- Toledo, R.A.; Qin, Y.; Srikantan, S.; Morales, N.P.; Li, Q.; Deng, Y.; Kim, S.-W.; Pereira, M.A.A.; Toledo, S.P.A.; Su, X.; et al. In vivo and in vitro oncogenic effects of HIF2A mutations in pheochromocytomas and paragangliomas. Endocr.-Relat. Cancer 2013, 20, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Percy, M.J.; Furlow, P.W.; Beer, P.A.; Lappin, T.R.J.; McMullin, M.F.; Lee, F.S. A novel erythrocytosis-associated PHD2 mutation suggests the location of a HIF binding groove. Blood 2007, 110, 2193–2196. [Google Scholar] [CrossRef] [PubMed]
- Taïeb, D.; Yang, C.; Delenne, B.; Zhuang, Z.; Barlier, A.; Sebag, F.; Pacak, K. First report of bilateral pheochromocytoma in the clinical spectrum of HIF2A-related polycythemia-paraganglioma syndrome. J. Clin. Endocrinol. Metab. 2013, 98, E908–E913. [Google Scholar] [CrossRef] [PubMed]
- Ladroue, C.; Carcenac, R.; Leporrier, M.; Gad, S.; Le Hello, C.; Galateau-Salle, F.; Feunteun, J.; Pouysségur, J.; Richard, S.; Gardie, B. PHD2 mutation and congenital erythrocytosis with paraganglioma. N. Engl. J. Med. 2008, 359, 2685–2692. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhuang, Z.; Fliedner, S.M.; Shankavaram, U.; Sun, M.G.; Bullova, P.; Zhu, R.; Elkahloun, A.G.; Kourlas, P.J.; Merino, M.; et al. Germ-line PHD1 and PHD2 mutations detected in patients with pheochromocytoma/paraganglioma-polycythemia. J. Mol. Med. 2015, 93, 93–104. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef]
- Vakser, I.A. Evaluation of GRAMM low-resolution docking methodology on the hemagglutinin-antibody complex. Proteins 1997, 29 (Suppl. 1), 226–230. [Google Scholar] [CrossRef]
- Vakser, I.A.; Matar, O.G.; Lam, C.F. A systematic study of low-resolution recognition in protein—protein complexes. Proc. Natl. Acad. Sci. USA 1999, 96, 8477–8482. [Google Scholar] [CrossRef]
- Rapizzi, E.; Ercolino, T.; Canu, L.; Giach’e, V.; Francalanci, M.; Pratesi, C.; Valeri, A.; Mannelli, M. Mitochondrial function and content in pheochromocytoma/paraganglioma of succinate dehydrogenase mutation carriers. Endocr. -Relat. Cancer 2012, 19, 261–269. [Google Scholar] [CrossRef]
- Rapizzi, E.; Fucci, R.; Giannoni, E.; Canu, L.; Richter, S.; Cirri, P.; Mannelli, M. Role of microenvironment on neuroblastoma SK-N-AS SDHB-silenced cell metabolism and function. Endocr.-Relat. Cancer 2015, 22, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Ruggiero, C.; Cantini, G.; Canu, L.; Baroni, G.; Armignacco, R.; Jouinot, A.; Santi, R.; Ercolino, T.; Ragazzon, B.; et al. Fascin-1 Is a Novel Prognostic Biomarker Associated with Tumor Invasiveness in Adrenocortical Carcinoma. J. Clin. Endocrinol. Metab. 2018, 104, 1712–1724. [Google Scholar] [CrossRef]
- Cantini, G.; Fei, L.; Canu, L.; Lazzeri, E.; Sottili, M.; Francalanci, M.; Angelotti, M.L.; De Filpo, G.; Ercolino, T.; Gelmini, S.; et al. Stimulated Expression of CXCL12 in Adrenocortical Carcinoma by the PPARgamma Ligand Rosiglitazone Impairs Cancer Progression. J. Pers. Med. 2021, 11, 1097. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Peng, J.; Baxter, S.; Peng, Z. AutoPVS1: An automatic classification tool for PVS1 interpretation of null variants. Hum. Mutat. 2020, 41, 1488–1498. [Google Scholar] [CrossRef] [PubMed]
- Welander, J.; Andreasson, A.; Juhlin, C.C.; Wiseman, R.W.; Bäckdahl, M.; Höög, A.; Larsson, C.; Gimm, O.; Söderkvist, P. Rare germline mutations identified by targeted next-generation sequencing of susceptibility genes in pheochromocytoma and paraganglioma. J. Clin. Endocrinol. Metab. 2014, 99, E1352–E1360. [Google Scholar] [CrossRef]
- -Sheikh, M.; Moradkhani, K.; Lopez, M.; Wajcman, H.; Préhu, C. Disturbance in the HIF-1α pathway associated with erythrocytosis: Further evidences brought by frameshift and nonsense mutations in the prolyl hydroxylase domain protein 2 (PHD2) gene. Blood Cells Mol. Dis. 2008, 40, 160–165. [Google Scholar] [CrossRef]
- Wilson, R.; Syed, N.; Shah, P. Erythrocytosis due to PHD2 Mutations: A Review of Clinical Presentation, Diagnosis, and Genetics. Case Rep. Hematol. 2016, 2016, 6373706. [Google Scholar]
- Jochmanová, I.; Zelinka, T.; Widimský, J.; Pacak, K. HIF signaling pathway in pheochromocytoma and other neuroendocrine tumors. Physiol. Res. 2014, 63, S251–S262. [Google Scholar] [CrossRef]
- Gardie, B.; Percy, M.J.; Hoogewijs, D.; Chowdhury, R.; Bento, C.; McMullin, M.F.; Schofield, C.J.; Lee, F.S.; Richard, S.; Arsenault, P.R.; et al. The role of PHD2 mutations in the pathogenesis of erythrocytosis. Hypoxia 2014, 2, 71–90. [Google Scholar] [CrossRef]
- Rijken, J.; Niemeijer, N.; Jonker, M.; Eijkelenkamp, K.; Jansen, J.; van Berkel, A.; Timmers, H.; Kunst, H.; Bisschop, P.; Kerstens, M.; et al. The penetrance of paraganglioma and pheochromocytoma in SDHB germline mutation carriers. Clin. Genet. 2018, 93, 60–66. [Google Scholar] [CrossRef]
- Ladroue, C.; Hoogewijs, D.; Gad, S.; Carcenac, R.; Storti, F.; Barrois, M.; Gimenez-Roqueplo, A.-P.; Leporrier, M.; Casadevall, N.; Hermine, O.; et al. Distinct deregulation of the hypoxia inducible factor by PHD2 mutants identified in germline DNA of patients with polycythemia. Haematologica 2011, 97, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Won, H.-S.; Lee, Y.-M.; Choi, J.-W.; Oh, T.-I.; Jang, J.-H.; Choi, D.-K.; Lim, B.-O.; Kim, Y.J.; Park, J.-W.; et al. Oxidative Dimerization of PHD2 is Responsible for its Inactivation and Contributes to Metabolic Reprogramming via HIF-1α Activation. Sci. Rep. 2016, 6, 18928. [Google Scholar] [CrossRef] [PubMed]
- Mayerhofer, M.; Valent, P.; Sperr, W.R.; Griffin, J.D.; Sillaber, C. BCR/ABL induces expression of vascular endothelial growth factor and its transcriptional activator, hypoxia inducible factor-1α, through a pathway involving phosphoinositide 3-kinase and the mammalian target of rapamycin. Blood 2002, 100, 3767–3775. [Google Scholar] [CrossRef] [PubMed]
- Parmar, K.; Mauch, P.; Vergilio, J.-A.; Sackstein, R.; Down, J.D. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc. Natl. Acad. Sci. USA 2007, 104, 5431–5436. [Google Scholar] [CrossRef]
- Zhang, H.; Li, H.; Xi, H.S.; Li, S. HIF1α is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012, 119, 2595–2607. [Google Scholar] [CrossRef]
- Giuntoli, S.; Rovida, E.; Barbetti, V.; Cipolleschi, M.G.; Olivotto, M.; Dello Sbarba, P. Hypoxia suppresses BCR/Abl and selects imatinib-insensitive progenitors within clonal CML populations. Leukemia 2006, 20, 1291–1293. [Google Scholar] [CrossRef]
- Giuntoli, S.; Tanturli, M.; Di Gesualdo, F.; Barbetti, V.; Rovida, E.; Dello Sbarba, P. Glucose availability in hypoxia regulates the selection of chronic myeloid leukemia progenitor subsets with different resistance to imatinib-mesylate. Haematologica 2011, 96, 204–212. [Google Scholar] [CrossRef]
- Zhao, F.; Mancuso, A.; Bui, T.V.; Tong, X.; Gruber, J.J.; Swider, C.R.; Sanchez, P.V.; Lum, J.J.; Sayed, N.; Melo, J.V.; et al. Imatinib resistance associated with BCR-ABL upregulation is dependent on HIF-1α-induced metabolic reprograming. Oncogene 2010, 29, 2962–2972. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Provenzano, A.; Chetta, M.; De Filpo, G.; Cantini, G.; La Barbera, A.; Nesi, G.; Santi, R.; Martinelli, S.; Rapizzi, E.; Luconi, M.; et al. Novel Germline PHD2 Variant in a Metastatic Pheochromocytoma and Chronic Myeloid Leukemia, but in the Absence of Polycythemia. Medicina 2022, 58, 1113. https://doi.org/10.3390/medicina58081113
Provenzano A, Chetta M, De Filpo G, Cantini G, La Barbera A, Nesi G, Santi R, Martinelli S, Rapizzi E, Luconi M, et al. Novel Germline PHD2 Variant in a Metastatic Pheochromocytoma and Chronic Myeloid Leukemia, but in the Absence of Polycythemia. Medicina. 2022; 58(8):1113. https://doi.org/10.3390/medicina58081113
Chicago/Turabian StyleProvenzano, Aldesia, Massimiliano Chetta, Giuseppina De Filpo, Giulia Cantini, Andrea La Barbera, Gabriella Nesi, Raffaella Santi, Serena Martinelli, Elena Rapizzi, Michaela Luconi, and et al. 2022. "Novel Germline PHD2 Variant in a Metastatic Pheochromocytoma and Chronic Myeloid Leukemia, but in the Absence of Polycythemia" Medicina 58, no. 8: 1113. https://doi.org/10.3390/medicina58081113
APA StyleProvenzano, A., Chetta, M., De Filpo, G., Cantini, G., La Barbera, A., Nesi, G., Santi, R., Martinelli, S., Rapizzi, E., Luconi, M., Maggi, M., Mannelli, M., Ercolino, T., & Canu, L. (2022). Novel Germline PHD2 Variant in a Metastatic Pheochromocytoma and Chronic Myeloid Leukemia, but in the Absence of Polycythemia. Medicina, 58(8), 1113. https://doi.org/10.3390/medicina58081113