
Case Report of Neonatal Sotos Syndrome with a New Missense Mutation in the NSD1 Gene and Literature Analysis in the Chinese Han Population

Abstract
:1. Introduction
2. Case Presentation
2.1. Clinical Data
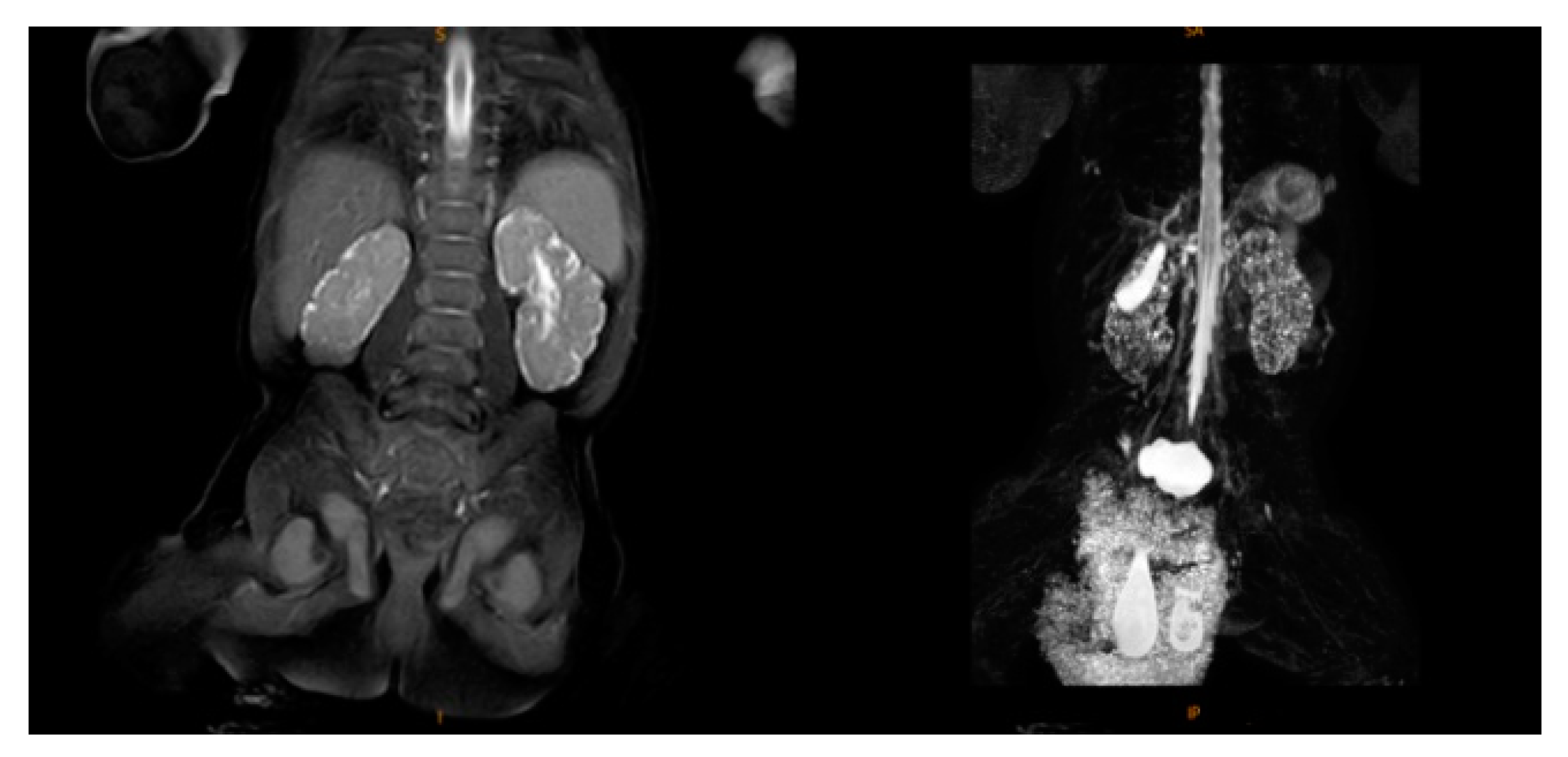
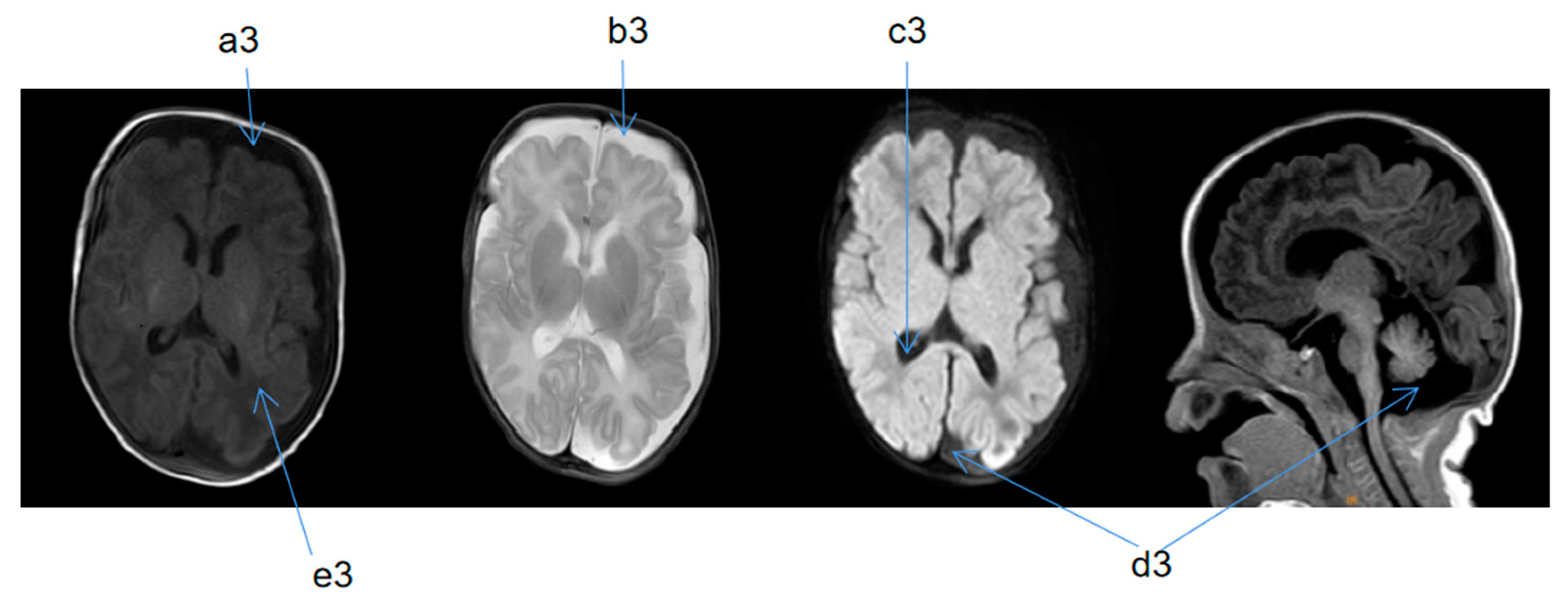
2.2. Supplementary Examination
2.3. Treatment Process and Follow-Up
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gaudreau, P.; Zizak, V.; Gallagher, T.Q. The otolaryngologic manifestations of Sotos syndrome. Int. J. Pediatr. Otorhinolaryngol. 2013, 77, 1861–1863. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.J.; Lin, Y.; Mei, M.; Fan, Z.C.; Zan, G.D. Two cases of neonatal Sotos syndrome and literature review. Chin. J. Perinat. Med. 2015, 18, 127. [Google Scholar]
- Chen, L.L. Gene mutation Sotos syndrome type 1: A case report (Chinese). J. Eugen. Genet. 2017, 25, 119. [Google Scholar]
- Xian, W.; Wan, Z.T. A case report of neonatal Sotos syndrome. J. Hubei Univ. Sci. Technol. (Med. Ed.) 2021, 35, 258–260. [Google Scholar]
- Tatton-Brown, K.; Rahman, N. Sotos syndrome. Eur. J. Hum. Genet. 2007, 15, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Douglas, J.; Coleman, K.; Baujat, G.; Cole, T.R.P.; Das, S.; Horn, D.; Hughes, H.E.; Temple, I.K.; Faravelli, F.; et al. Genotype-phenotype associations in Sotos syndrome: An analysis of 266 individuals with NSD1 aberrations. Am. J. Hum. Genet. 2005, 77, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Danis, D.O.; Bodamer, O.; Levi, J.R. The otolaryngologic manifestations of Sotos syndrome 1: A systematic review. Int. J. Pediatr. Orl. 2021, 143, 110649. [Google Scholar] [CrossRef]
- Žilina, O.; Reimand, T.; Tammur, P.; Tillmann, V.; Kurg, A.; Õunap, K. Patient with dup (5)(q35.2–q35.3) reciprocal to the common Sotos syndrome deletion and review of the literature. Eur. J. Med. Genet. 2013, 56, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.; Anand, P.; Lee, J.A.; Jones, J.R.; Kim, C.A.; Bertola, D.R.; Labonne, J.D.J.; Layman, L.C.; Wenzel, W.; Kim, H.G. Steric clash in the set domain of histone methyltransferase NSD1 as a cause of Sotos syndrome and its genetic heterogeneity in a Brazilian cohort. Genes 2016, 7, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.K.; Du, X.B.; Jiang, X.H. Sotos syndrome: A case report and literature review. J. Pract. Med. 2019, 36, 344–347. [Google Scholar]
- Zheng, H.X.; Yao, C.; Yin, L.P.; Li, X.; Ding, Y. Clinical characteristics and gene variation analysis of children with Sotos syndrome. Lab. Med. 2021, 36, 130–134. [Google Scholar]
- Hou, Y.H.; Hong, J. A new missense mutation of NSD1 gene in Sotos syndrome and literature review. J. Precis. Med. 2021, 36, 59–62. [Google Scholar]
- Nagai, T.; Matsumoto, N.; Kurotaki, N.; Harada, N.; Niikawa, N.; Ogata, T.; Imaizumi, K.; Kurosawa, K.; Kondoh, T.; Ohashi, H.; et al. Sotos syndrome and haploinsufficiency of NSD1: Clinical features of intragenic mutations and submicroscopic deletions. J. Med. Genet. 2003, 40, 285–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagali, C.; Kok, F.; Nicola, P.; Kim, C.; Bertola, D.; Albano, L.; Koiffmann, C.P. MLPA analysis in 30 Sotos syndrome patients revealed one total NSD1 deletion and two partial deletions not previously reported. Eur. J. Med. Genet. 2009, 52, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Fortin, O.; Vincelette, C.; Khan, A.Q.; Berrahmoune, S.; Dassi, C.; Karimi, M.; Scheffer, I.E.; Lu, J.; Davis, K.; Myers, K.A. Seizures in Sotos syndrome: Phenotyping in 49 patients. Epilepsia Open 2021, 6, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Machida, M.; Katoh, H.; Miyake, A.; Taira, K.; Ohashi, H. The Association of Scoliosis and NSD1 gene deletion in Sotos syndrome patients. Spine (Phila Pa 1976) 2021, 46, 162. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Gene | Chromosome Position | Transcript Number | Exon/Intron | Nucleotide Change | Amino Acid Change | Heterozygous/ Homozygous | Related Diseases | Inheritance Mode | Variation Classification | Source of Variation |
|---|---|---|---|---|---|---|---|---|---|---|
| NSD1 | chr5:176687023 | NM_02245 5.4 | Exon14 | c.5000C>A | p.A1667E | Heterozygous | Sotos syndrome type 1 | AD | Likely pathogenic | De novo mutation |
| Sample Name | Sample Number | Amount of Detection Data (bp) | Average Sequencing Depth | Target Area Coverage | 10× above Coverage Interval Proportion | 20× above Coverage Interval Proportion |
|---|---|---|---|---|---|---|
| Child | WES21120171 | 11703902400 | 152.34 | 99.92% | 99.78% | 99.47% |
| Father | WES21120172 | 12428848500 | 162.82 | 99.93% | 99.80% | 99.50% |
| Mother | WES21120173 | 12989484300 | 170.16 | 99.80% | 99.68% | 99.41% |
| This Study | Sun, B.J. [2] | Chen, L.L. [3] | Xian, W. [4] | ||
|---|---|---|---|---|---|
| Case | 1 | 2 | 3 | 4 | 5 |
| Gender | Male | Male | Male | Male | Male |
| Age | 2 days | 6 h | 3 days | 9 days | 9 days |
| Premature delivery | + | - | - | + | - |
| Time of overgrowth appearance | Intrauterine | At birth | 3 months | At birth | At birth |
| Special facial features | + | + | − | − | + |
| Hypoglycemia | + | + | - | - | + |
| Dystonia | + | − | − | − | + |
| Cranial MRI abnormalities | + | + | + | + | + |
| Abnormal hearing | + | − | − | + | − |
| Abnormal EEG | + | − | − | + | − |
| Congenital heart defect | + | − | − | + | − |
| Urinary system problems | + | + | + | − | − |
| Developmental retardation | + | + | + | + | + |
| NSD1 mutation | chr5 176687023 | 5q35.2–5q35.1.97 MB missing in zone 3 | The long arm of chromosome 5 | Yes, loci ambiguous | 5q35,2q35.3 region missing |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, H.-Y.; Li, H.-F.; Xu, J.-L.; Hui, W.; Ruan, W.-C.; Lv, C.-C.; Xu, R.-A.; Qiang, S. Case Report of Neonatal Sotos Syndrome with a New Missense Mutation in the NSD1 Gene and Literature Analysis in the Chinese Han Population. Medicina 2022, 58, 968. https://doi.org/10.3390/medicina58070968
Jin H-Y, Li H-F, Xu J-L, Hui W, Ruan W-C, Lv C-C, Xu R-A, Qiang S. Case Report of Neonatal Sotos Syndrome with a New Missense Mutation in the NSD1 Gene and Literature Analysis in the Chinese Han Population. Medicina. 2022; 58(7):968. https://doi.org/10.3390/medicina58070968
Chicago/Turabian StyleJin, Hui-Ying, Hai-Feng Li, Jia-Lu Xu, Wang Hui, Wen-Cong Ruan, Cheng-Cheng Lv, Ren-Ai Xu, and Shu Qiang. 2022. "Case Report of Neonatal Sotos Syndrome with a New Missense Mutation in the NSD1 Gene and Literature Analysis in the Chinese Han Population" Medicina 58, no. 7: 968. https://doi.org/10.3390/medicina58070968
APA StyleJin, H.-Y., Li, H.-F., Xu, J.-L., Hui, W., Ruan, W.-C., Lv, C.-C., Xu, R.-A., & Qiang, S. (2022). Case Report of Neonatal Sotos Syndrome with a New Missense Mutation in the NSD1 Gene and Literature Analysis in the Chinese Han Population. Medicina, 58(7), 968. https://doi.org/10.3390/medicina58070968