
Dyspnea in Post-COVID Syndrome following Mild Acute COVID-19 Infections: Potential Causes and Consequences for a Therapeutic Approach


Abstract
1. Introduction
1.1. Hyperventilation as the Cause of Dyspnea in Post-COVID Syndrome Patients with Mild Acute COVID-19 Infections
1.2. The Potential Causes of Hyperventilation
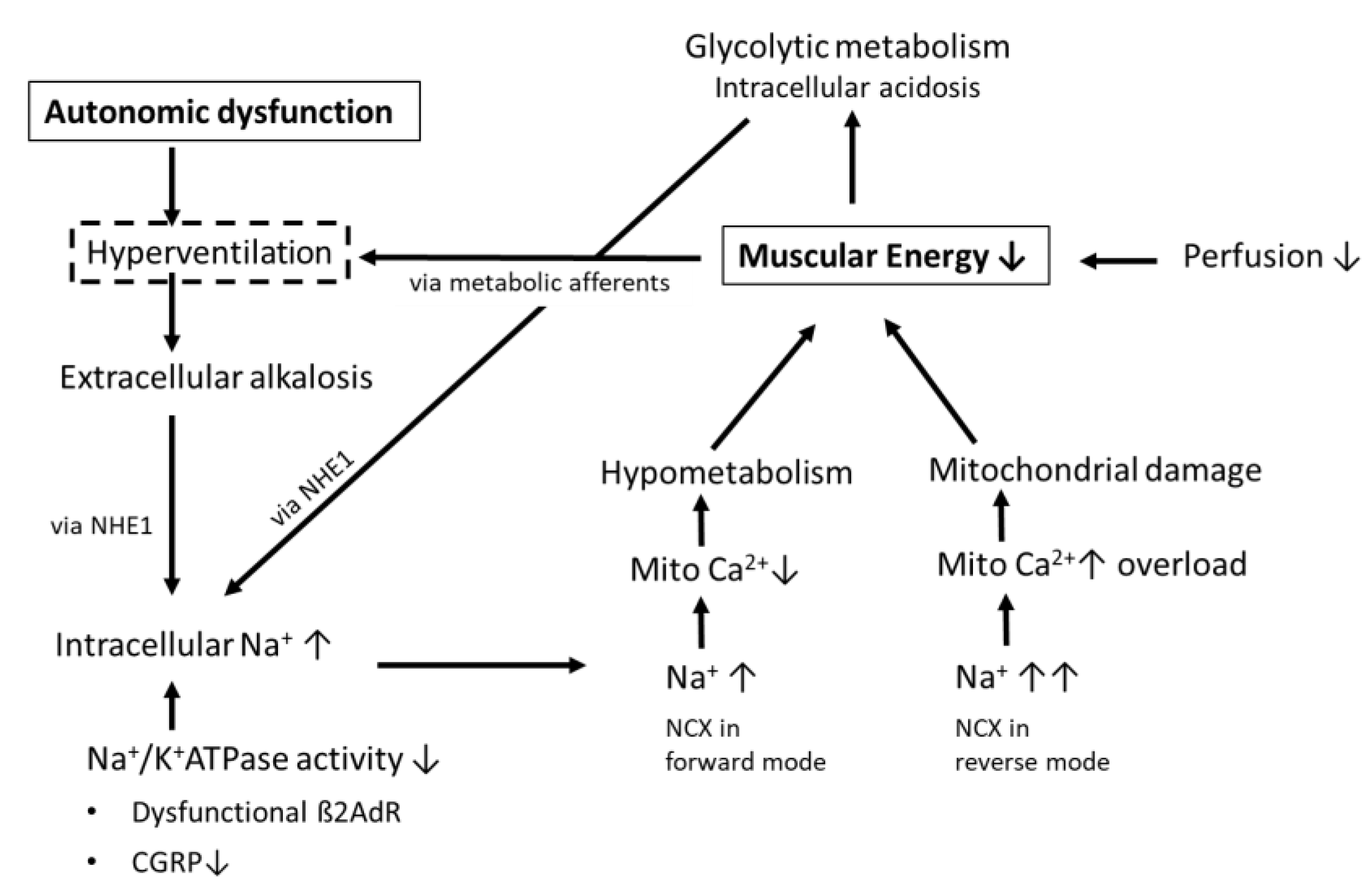
1.3. Possible Consequences of Hyperventilation and Respiratory Alkalosis on Muscular Ionic Homeostasis
1.4. Differential Mechanisms of Dyspnea following Mild and Severe COVID-19
2. Conclusions and Therapeutic Implication
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Motiejunaite, J.; Balagny, P.; Arnoult, F.; Mangin, L.; Bancal, C.; d’Ortho, M.-P.; Frija-Masson, J. Hyperventilation: A Possible Explanation for Long-Lasting Exercise Intolerance in Mild COVID-19 Survivors? Front. Physiol. 2021, 11, 614590. [Google Scholar] [CrossRef]
- Aparisi, Á.; Ybarra-Falcón, C.; García-Gómez, M.; Tobar, J.; Iglesias-Echeverría, C.; Jaurrieta-Largo, S.; Ladrón, R.; Uribarri, A.; Catalá, P.; Hinojosa, W.; et al. Exercise Ventilatory Inefficiency in Post-COVID-19 Syndrome: Insights from a Prospective Evaluation. J. Clin. Med. 2021, 10, 2591. [Google Scholar] [CrossRef]
- Mancini, D.M.; Brunjes, D.L.; Lala, A.; Trivieri, M.G.; Contreras, J.P.; Natelson, B.H. Use of Cardiopulmonary Stress Testing for Patients With Unexplained Dyspnea Post–Coronavirus Disease. JACC Heart Fail. 2021, 9, 927–937. [Google Scholar] [CrossRef]
- Freitag, H.; Szklarski, M.; Lorenz, S.; Sotzny, F.; Bauer, S.; Philippe, A.; Kedor, C.; Grabowski, P.; Lange, T.; Riemekasten, G.; et al. Autoantibodies to Vasoregulative G-Protein-Coupled Receptors Correlate with Symptom Severity, Autonomic Dysfunction and Disability in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J. Clin. Med. 2021, 10, 3675. [Google Scholar] [CrossRef]
- Jason, L.A.; Islam, M.F.; Conroy, K.; Cotler, J.; Torres, C.; Johnson, M.; Mabie, B. COVID-19 symptoms over time: Comparing long-haulers to ME/CFS. Fatigue Biomed. Health Behav. 2021, 9, 59–68. [Google Scholar] [CrossRef]
- Kedor, C.; Freitag, H.; Meyer-Arndt, L.; Wittke, K.; Zoller, T.; Steinbeis, F.; Haffke, M.; Rudolf, G.; Heidecker, B.; Volk, H.; et al. Chronic COVID-19 Syndrome and Chronic Fatigue Syndrome (ME/CFS) following the first pandemic wave in Germany—A first analysis of a prospective observational study. medRxiv 2021. [Google Scholar] [CrossRef]
- Mantovani, E.; Mariotto, S.; Gabbiani, D.; Dorelli, G.; Bozzetti, S.; Federico, A.; Zanzoni, S.; Girelli, D.; Crisafulli, E.; Ferrari, S.; et al. Chronic fatigue syndrome: An emerging sequela in COVID-19 survivors? J. NeuroVirology 2021, 27, 631–637. [Google Scholar] [CrossRef]
- Petracek, L.S.; Suskauer, S.J.; Vickers, R.F.; Patel, N.R.; Violand, R.L.; Swope, R.L.; Rowe, P.C. Adolescent and Young Adult ME/CFS After Confirmed or Probable COVID-19. Front. Med. 2021, 8, 668944. [Google Scholar] [CrossRef] [PubMed]
- Sudre, C.H.; Murray, B.; Varsavsky, T.; Graham, M.S.; Penfold, R.S.; Bowyer, R.C.; Pujol, J.C.; Klaser, K.; Antonelli, M.; Canas, L.S.; et al. Attributes and predictors of long COVID. Nat. Med. 2021, 27, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.L.; Weitzer, D.J. Long COVID and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)—A Systemic Review and Comparison of Clinical Presentation and Symptomatology. Medicina 2021, 57, 418. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, B.M.; van de Sande, M.I.; De Meirleir, K.L.; Klimas, N.G.; Broderick, G.; Mitchell, T.; Staines, D.; Powles, A.C.; Speight, N.; Vallings, R.; et al. Myalgic encephalomyelitis: International Consensus Criteria. J. Intern. Med. 2011, 270, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Taverne, J.; Salvator, H.; Leboulch, C.; Barizien, N.; Ballester, M.; Imhaus, E.; Chabi-Charvillat, M.-L.; Boulin, A.; Goyard, C.; Chabrol, A.; et al. High incidence of hyperventilation syndrome after COVID-19. J. Thorac. Dis. 2021, 13, 3918–3922. [Google Scholar] [CrossRef] [PubMed]
- Shevtsova, N.A.; Marchenko, V.; Bezdudnaya, T. Modulation of Respiratory System by Limb Muscle Afferents in Intact and Injured Spinal Cord. Front. Neurosci. 2019, 13, 289. [Google Scholar] [CrossRef] [PubMed]
- van Campen, C.M.C.; Rowe, P.C.; Visser, F.C. Orthostatic Symptoms and Reductions in Cerebral Blood Flow in Long-Haul COVID-19 Patients: Similarities with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Medicina 2022, 58, 28. [Google Scholar] [CrossRef] [PubMed]
- Wirth, K.; Scheibenbogen, C. A Unifying Hypothesis of the Pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): Recognitions from the finding of autoantibodies against ss2-adrenergic receptors. Autoimmun. Rev. 2020, 19, 102527. [Google Scholar] [CrossRef] [PubMed]
- Wirth, K.J.; Scheibenbogen, C. Pathophysiology of skeletal muscle disturbances in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). J. Transl. Med. 2021, 19, 162. [Google Scholar] [CrossRef] [PubMed]
- Wirth, K.J.; Scheibenbogen, C.; Paul, F. An attempt to explain the neurological symptoms of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J. Transl. Med. 2021, 19, 471. [Google Scholar] [CrossRef] [PubMed]
- Mejia-Renteria, H.; Travieso, A.; Sagir, A.; Martínez-Gómez, E.; Carrascosa-Granada, A.; Toya, T.; Núñez-Gil, I.J.; Estrada, V.; Lerman, A.; Escaned, J. In-vivo evidence of systemic endothelial vascular dysfunction in COVID-19. Int. J. Cardiol. 2021, 345, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Charfeddine, S.; Ibn Hadj Amor, H.; Jdidi, J.; Torjmen, S.; Kraiem, S.; Hammami, R.; Bahloul, A.; Kallel, N.; Moussa, N.; Touil, I.; et al. Long COVID 19 Syndrome: Is It Related to Microcirculation and Endothelial Dysfunction? Insights From TUN-EndCOV Study. Front. Cardiovasc. Med. 2021, 8, 1702. [Google Scholar]
- Sang, C.J.; Burkett, A.; Heindl, B.; Litovsky, S.H.; Prabhu, S.D.; Benson, P.V.; Rajapreyar, I. Cardiac pathology in COVID-19: A single center autopsy experience. Cardiovasc. Pathol. 2021, 54, 107370. [Google Scholar] [CrossRef] [PubMed]
- Kubánková, M.; Hohberger, B.; Hoffmanns, J.; Fürst, J.; Herrmann, M.; Guck, J.; Kräter, M. Physical phenotype of blood cells is altered in COVID-19. Biophys. J. 2021, 120, 2838–2847. [Google Scholar] [CrossRef] [PubMed]
- Avkiran, M.; Gross, G.; Karmazyn, M.; Klein, H.; Murphy, E.; Ytrehus, K. Na+/H+ exchange in ischemia, reperfusion and preconditioning. Cardiovasc. Res. 2001, 50, 162–166. [Google Scholar] [CrossRef]
- Karmazyn, M.; Sawyer, M.; Fliegel, L. The Na(+)/H(+) exchanger: A target for cardiac therapeutic intervention. Curr. Drug. Targets. Cardiovasc. Haematol. Disord. 2005, 5, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Esfandyarpour, R.; Kashi, A.; Nemat-Gorgani, M.; Wilhelmy, J.; Davis, R.W. A nanoelectronics-blood-based diagnostic biomarker for myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Proc. Natl. Acad. Sci. USA 2019, 116, 10250–10257. [Google Scholar] [CrossRef] [PubMed]
- Krump, E.; Nikitas, K.; Grinstein, S. Induction of tyrosine phosphorylation and Na+/H+ exchanger activation during shrinkage of human neutrophils. J. Biol. Chem. 1997, 272, 17303–17311. [Google Scholar] [CrossRef] [PubMed]
- Barizien, N.; Le Guen, M.; Russel, S.; Touche, P.; Huang, F.; Vallée, A. Clinical characterization of dysautonomia in long COVID-19 patients. Sci. Rep. 2021, 11, 14042. [Google Scholar] [CrossRef] [PubMed]
- Innes, J.A.; De Cort, S.C.; Evans, P.J.; Guz, A. Central command influences cardiorespiratory response to dynamic exercise in humans with unilateral weakness. J. Physiol. 1992, 448, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Zakharycheva, T.; Makhovskaya, T.; Shirokova, A.; Shikina, I. Autonomic Dysregulation Syndrome in COVID-19 Convalescents: Possible Causes and Approaches to Its Correction; Springer Nature: Cham, Switzerland, 2022. [Google Scholar]
- Abrams, R.M.C.; Simpson, D.M.; Navis, A.; Jette, N.; Zhou, L.; Shin, S.C. Small fiber neuropathy associated with SARS-CoV-2 infection. Muscle Nerve 2021, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Tal, I.; Kozlovsky, T.; Brisker, D.; Giladi, M.; Khananshvili, D. Kinetic and equilibrium properties of regulatory Ca(2+)-binding domains in sodium-calcium exchangers 2 and 3. Cell Calcium 2016, 59, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Naschitz, J.E.; Mussafia-Priselac, R.; Kovalev, Y.; Zaigraykin, N.; Elias, N.; Rosner, I.; Slobodin, G. Patterns of Hypocapnia on Tilt in Patients with Fibromyalgia, Chronic Fatigue Syndrome, Nonspecific Dizziness, and Neurally Mediated Syncope. Am. J. Med. Sci. 2006, 331, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Sophie, B.; Alan, K.O.; Jemina, F.; Florian, L.; Sylvain, C.; Aline, S.; Thomas, S.; Marina, S.P.; Bruno, H.; Dan, A.; et al. Virtual reality exercise to help COVID patients with refractory breathlessness. medRxiv 2021. [Google Scholar] [CrossRef]
- Laursen, J.C.; Hansen, C.S.; Bordino, M.; Vistisen, D.; Zobel, E.H.; Winther, S.A.; Groop, P.H.; Frimodt-Møller, M.; Bernardi, L.; Rossing, P. Hyperoxia improves autonomic function in individuals with long-duration type 1 diabetes and macroalbuminuria. Diabet. Med. 2020, 37, 1561–1568. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wirth, K.J.; Scheibenbogen, C. Dyspnea in Post-COVID Syndrome following Mild Acute COVID-19 Infections: Potential Causes and Consequences for a Therapeutic Approach. Medicina 2022, 58, 419. https://doi.org/10.3390/medicina58030419
Wirth KJ, Scheibenbogen C. Dyspnea in Post-COVID Syndrome following Mild Acute COVID-19 Infections: Potential Causes and Consequences for a Therapeutic Approach. Medicina. 2022; 58(3):419. https://doi.org/10.3390/medicina58030419
Chicago/Turabian StyleWirth, Klaus J., and Carmen Scheibenbogen. 2022. "Dyspnea in Post-COVID Syndrome following Mild Acute COVID-19 Infections: Potential Causes and Consequences for a Therapeutic Approach" Medicina 58, no. 3: 419. https://doi.org/10.3390/medicina58030419
APA StyleWirth, K. J., & Scheibenbogen, C. (2022). Dyspnea in Post-COVID Syndrome following Mild Acute COVID-19 Infections: Potential Causes and Consequences for a Therapeutic Approach. Medicina, 58(3), 419. https://doi.org/10.3390/medicina58030419