Abstract
Background: Colorectal cancer represents a common malignancy and remains incurable in the metastatic stage. Identification of molecular alterations that are present in colorectal cancer has led to the introduction of targeted therapies that improve outcomes. BRAF and PIK3CA mutations are observed in a subset of colorectal cancers. Colorectal cancers bearing BRAF mutations may be treated with specific BRAF inhibitors. These drugs benefit patients with BRAF mutant colorectal cancers but responses are rather brief, and progression is the rule. In contrast, no PI3K inhibitors have proven successful yet in the disease. Thus, new treatments to supplement the currently available drugs would be welcome to further improve survival. Methods: Profiled colorectal cancer cell lines from the Cancer Cell Line Encyclopedia (CCLE) were examined for BRAF and PIK3CA mutations and were interrogated for molecular characteristics and concomitant alterations that mirror clinical sample alterations. The Genomics of Drug Sensitivity in Cancer (GDSC) project was used for determination of drug sensitivities of BRAF mutated colorectal cell lines with or without concomitant PIK3CA mutations. The Cancer Dependency Map project served as the basis for identification of molecular dependencies and vulnerabilities in these cell lines. Results: CCLE includes 84 colorectal cancer cell lines, which recapitulate the molecular landscape of colorectal cancer. Of these, 23 and 24 cell lines possess BRAF and PIK3CA mutations, respectively. Seven BRAF mutant cell lines have V600E mutations and 14 PIK3CA mutant cell lines have hotspot helical or kinase domain mutations. V600E BRAF mutant cell lines with or without hotspot PIK3CA mutations are heterogeneous in their MSI status and mimic colorectal cancer tissues in other prevalent abnormalities including APC and TP53 mutations. Essential genes for survival include CTNNB1, WRN, and pyrimidine metabolism enzyme CAD. Besides BRAF mutations, BRAF inhibitor sensitivity in colorectal cancer cell lines is conferred by SACS mutations and PRKN locus loss. Conclusions: Colorectal cancer cell lines bearing the frequent BRAF and PIK3CA mutations present many alterations of the parental cancer tissue. Described vulnerabilities represent leads for therapeutic exploration in colorectal cancers with the corresponding alterations.
1. Introduction
Colorectal cancer is the most prevalent gastrointestinal carcinoma and a major cause of cancer morbidity and mortality. An estimated 150,000 people will be diagnosed with colorectal cancer in 2022 in the United States alone and over 50,000 patients will die from the disease [1]. It represents the third leading cause of mortality from cancer in both men (after lung and prostate cancers) and women (behind lung and breast cancers). About 20% of cases are diagnosed in a metastatic stage and a significant percentage of initially stage II and stage III patients will have a metastatic relapse [2]. Metastatic colorectal cancer remains most often an incurable disease, despite progress in systemic and local therapies that have improved outcomes [3]. The elucidation of the molecular pathogenesis of colorectal cancer has resulted in introduction of targeted therapies that have improved survival of selected patients [4,5,6,7]. These include anti-EGFR monoclonal antibodies for KRAS wild type disease, combinations of anti-EGFR monoclonal antibodies with BRAF inhibitors for BRAF mutant cancers, anti-HER2 therapies for HER2 altered cancers and immune checkpoint inhibitors for microsatellite instability (MSI) high cancers. Other targeted treatments addressing small defined sub-sets of colorectal cancers include NTRK inhibitors for colorectal cancers with NTRK fusions and specific KRAS G12C inhibitors for cancers with this KRAS substitution [8,9]. Novel therapeutics based on combinations of targeted therapies are intensely investigated with the hope that several will enter the clinic in the near future [10,11].
BRAF mutations are observed in 5% to 15% of colorectal cancers and are associated with aggressive disease [12,13]. Colorectal cancers with mutations in BRAF tend to be of high grade and occur more often in the right colon [14]. The most common mutations in BRAF occur at amino-acid V600 position of the protein and substitute the normal valine at this position with glutamic acid (V600E). BRAF V600E mutations and other rarer substitutions at this codon location (V600K, V600D, V600M, and V600R) are categorized as class I BRAF mutations. These substitutions result in potent kinase activation that is independent of upstream signals from KRAS [15,16]. Mutations of BRAF in other codons, including the neighboring L597 and K601 positions lead to a protein that retains the requirement for homo-dimerization to signal downstream. These mutations that are classified as class II, as well as class III mutations, that require KRAS input for sustained signaling, are rare [14,15].
Mutations in the gene encoding for the alpha catalytic subunit of kinase PI3K, PIK3CA, are the most common colorectal cancer mutations in the PI3K/AKT/mTOR signal transduction pathway and are present in 20% to 25% of colorectal cancers [17,18,19,20]. PIK3CA point mutations are more diverse than BRAF mutations, although about half of the cases concern codons E542, E545, and Q546 of the helical domain and codon H1047 of the kinase domain. Colorectal cancers with PIK3CA mutations are more often arising in the right colon and present with a higher mutation count than cancers without PIK3CA mutations [20]. In contrast to the mutual exclusivity of mutations in oncogenes KRAS and BRAF, cancers with PIK3CA mutations have often concomitant mutations in either of these genes of the KRAS/BRAF/MEK/ERK pathway.
This investigation examines colorectal cancer cell lines bearing BRAF mutations with concomitant PIK3CA mutations and compares them to BRAF mutant cell lines without PIK3CA mutations in regard to genomic characteristics such as ploidy, MSI status, and coexisting molecular alterations. The sensitivity of these cell lines to drugs inhibiting the mutated pathways and to other inhibitors is also interrogated. The ultimate goal is to discover new therapeutic opportunities beyond the currently available BRAF inhibitors, which are currently the only approved drugs, in combination with anti-EGFR therapies, for colorectal cancers with V600E mutations.
2. Methods
Cancer cell lines included in the current investigation constitute part of the Cancer Cell Line Encyclopedia (CCLE) collection [21]. The cBioportal Genomics Portal platform was used to identify colorectal cancer cell lines with BRAF mutations with or without concomitant PIK3CA mutations in CCLE [22]. cBioportal (http://www.cbioportal.org accessed on 29 July 2022) is a user-friendly, open-access platform for genomic analysis of tumors and cancer cell lines [22]. Additionally, genomic data of colorectal cancer patients from The Cancer Genome Atlas (TCGA) study cohort [17] were analyzed using cBioportal. The CCLE project employs whole-exome sequencing to discover mutations, copy number alterations, and fusions in cell lines from various types of cancer [21]. Analysis of copy number alterations in the CCLE project was performed with the GISTIC (Genomic Identification of Significant Targets in Cancer) algorithm, in which a score of 2 or above denotes putative amplification of a gene [23]. RNA expression was normalized with the RSEM algorithm and results were presented as the Log RNA sequences in Reads per Kilobase Million (RPKM) [24].
The functional assessment of mutations observed in cell lines of interest was performed with the help of OncoKB. OncoKB knowledgebase is a database of cancer-related genes and characterizes these genes as oncogenes or tumor suppressor genes [25]. On some occasions, genes are included in OncoKB as cancer associated but they are not annotated as oncogenes or tumor suppressors.
The Genomics of Drug Sensitivity in Cancer (GDSC) dataset (www.cancerrxgene.org accessed on 29 July 2022) was interrogated to obtain data on drug sensitivity of cell lines from colorectal cancer and other cancers with BRAF and PIK3CA mutations [26]. Two datasets, GDSC1 and GDSC2, are included within the GDSC project, differing in the experimental conditions used. GDSC1 experiments were performed between 2009 and 2015. These experiments used media alone in the negative control cell lines not exposed to drugs. The GDSC2 panel of experiments was performed more recently (after 2015) and employed media with vehicle (DMSO-dimethylsulfoxide) in the negative controls. Dependencies on specific genes of cell lines with BRAF and PIK3CA mutations were obtained from the Depmap portal that contains data from CRISPR arrays and RNA-interference (RNAi) arrays of included cell lines from CCLE [27,28]. CRISPR and RNAi arrays identify essential genes that are important for the survival of screened cell lines and, as a result, the knock-down of these essential genes has a significant effect in their survival and proliferation in vitro [29,30,31]. The two methodologies differ in the depth of suppression of assayed genes, with CRISPR knock out usually being stronger than the partial suppression obtained by RNA interference. As a result, the genes and dependencies discovered with the two methodologies are not completely overlapping. Data for CRISPR screening in DepMap are from project SCORE containing 323 cancer cell lines from various cancers and a library of 18,009 targeted genes [32]. Computational modelling of experiments in SCORE was initially performed with the CERES algorithm and later with the CHRONOS algorithm [33,34]. RNAi experiments were performed under the aegis of project Achilles using the DEMETER algorithm for analysis [30].
Statistical comparisons of categorical data were carried out using Fisher’s exact test or the x2 test. The Mann–Whitney U test was used to compare median values. All statistical comparisons were considered significant if p < 0.05.
All data presented in this paper are from experiments performed by the consortiums mentioned in the above methods section and are openly available in the public domain. No new laboratory experiments have been performed for this investigation.
3. Results
The colorectal cancer cohort of CCLE consisting of 84 cell lines contains 23 cell lines (27.4%) with BRAF mutations. Ten BRAF mutant cell lines contain classic V600E mutations, in three of them (OUMS23, MDST8 and HT-29) with additional non-canonical BRAF mutations (Table 1). Thirteen cell lines contain non-V600E mutations. In two of them, NCI-H508 and HT-55, mutations are oncogenic or potentially oncogenic (G596R and N581Y, respectively).

Table 1.
BRAF mutated colorectal cancer cell lines and their specific BRAF mutations and concomitant PIK3CA mutations. Data are from the Cancer Cell Line Encyclopedia (CCLE). WT: wild type.
Seven BRAF V600E mutant cell lines are wild type for PIK3CA, while three cell lines with V600E mutations (SNU-C5, RKO and HT-29) as well as cell line NCI-H508, which has a pathogenic non-V600 mutation at position G596, have concomitant pathogenic mutations in PIK3CA (Table 1). Five of the seven cell lines with V600E BRAF mutations and no PIK3CA mutations are MSS, possess a lower mutation count, are hyper-diploid and have a high Fraction of Genome Altered (FGA) (Table 2). The two V600E BRAF mutant/PIK3CA wild type colorectal cancer cell lines, LS411N and CL34, that are MSI high have consistently a high mutation count. The two cell lines with concomitant BRAF V600E and PIK3CA H1047R mutations, SNU-C5 and RKO, are MSI high, have a high mutation count, are diploid and have a low FGA (Table 2). The two other cell lines with concomitant mutations, NCI-H508 and HT-29, have non-canonical pathogenic mutations in either BRAF (NCI-H508) or in PIK3CA (HT-29) and they are both MSS, have lower mutation counts, are hyper-diploid and have a high FGA.
Table 2.
Characteristics of colorectal cancer cell lines with BRAF V600E mutations without and with concomitant PIK3CA mutations. Cell line NCI-H508 has a BRAF G596R pathogenic mutation instead of BRAF V600E mutation. Cell lines without an asterisk are without PIK3CA mutations and are presented first. Cell lines with an asterisk in the bottom lines of the table are those with concomitant PIK3CA mutations.
Regarding concomitant cancer-associated mutations in V600E BRAF mutant/PIK3CA wild type colorectal cancer cell lines all seven cell lines have oncogenic mutations in APC and four have also oncogenic mutations in TP53 (Table 3). No cell lines have KRAS mutations, which tend to be mutually exclusive with BRAF mutations. Recurrent oncogenic deletions include the loci of dual specificity phosphatase DUSP22, which is present in 4 cell lines and deletions in SMAD4 and SMAD2, which are present in 3 and 2 cell lines, respectively (Table 3). Only two of the four cell lines with oncogenic mutations in both BRAF and PIK3CA have concomitant APC mutations and three of the four have also TP53 mutations (Table 3). Recurrent amplifications are observed in MYC and AGO2 that are both located at chromosome arm 8q and are present in cell lines RKO and HT-29. These cell lines and the cell line NCI-H508 also possess deletions of PRKN, encoding for ubiquitin ligase parkin, which is the only recurrent deletions in BRAF/PIK3CA double mutant colorectal cancer cell lines. HT-29 is the only double mutant cell line possessing the recurrent deletion of DUSP22, observed in cell lines with V600E BRAF mutations and wild type PIK3CA (Table 3).
Table 3.
Molecular alterations in colorectal cancer cell lines with BRAF V600E mutations without and with concomitant PIK3CA mutations. +: presence of oncogenic mutation. Cell lines with an asterisk are those with concomitant PIK3CA mutations.
Vulnerabilities of BRAF mutant cell lines with or without PIK3CA mutations were explored with interrogation of RNAi libraries for determination of preferentially essential genes and with CRISPR mediated knock out arrays (Table 4). Recurrent genes that are observed to be essential for survival in more than one BRAF mutant cell lines include CTNNB1, encoding for β-catenin, WRN, encoding for Warner syndrome ATP-dependent helicase, ALYREF which encodes for a chaperone of basal region leucine zipper (bZIP) proteins, and peptidylprolyl isomerase E (PPIE). These recurrent essential genes are in the top list of preferentially essential genes in one or more of the four cell lines with BRAF and PIK3CA mutations (Table 4). In addition, the gene encoding for CAD, an enzyme of the pyrimidine biosynthesis pathway induced by MAPK cascade, is a preferentially essential gene in two of four BRAF and PIK3CA mutant cell lines.
Table 4.
Top dependencies of BRAF V600E mutant/PIK3CA wild type and BRAF V600E mutant/PIK3CA mutant colorectal cancer cell lines, as determined by RNAi and CRISPR knock-out. RNAi experiments are from project Achilles and CRISPR experiments are from project SCORE and CHRONOS. NA: not available.
Five of the seven cell lines with BRAF mutations and without PIK3CA mutations (COLO205, MDST8, LS411N, SW1417 and CL34) have been assayed for drug sensitivities in GDSC (Table 5). Top drug sensitivities displayed by cell lines COLO205 and CL34 are to BRAF inhibitors, inhibitors of downstream MEK kinases and inhibitors of upstream receptor tyrosine kinases. LS411N cell line displays sensitivity to drugs of the pathway as well as to other kinases and the dihydrofolate reductase inhibitor pyrimethamine. In contrast, no inhibitors of BRAF or the receptor tyrosine kinase/KRAS/BRAF/MAPK pathway are among the top sensitivities of cell lines MDST8 and SW1417. Top sensitivities of these two cell lines include drugs involved in lipid metabolism and apoptosis inhibitors (Table 5). Cell lines with mutations in both BRAF and PIK3CA display sensitivities to several inhibitors of the receptor tyrosine kinase/KRAS/BRAF/MAPK pathway and PI3K/AKT cascade. Two of the four BRAF/PIK3CA double mutated cell lines, SNUC5 and RKO present additional sensitivities to the clinically used antimetabolite methotrexate, the WEE1 kinase inhibitor MK-1775, the mitotic kinases AURKA and AURKB inhibitor ZM447439 and the epigenetic modifier, BET bromodomain inhibitor JQ1. Compared with cell lines not bearing mutations in BRAF and PIK3CA, colorectal cancer cell lines with BRAF mutations with or without PIK3CA mutations show heterogeneous up-regulation in the mRNA expression of genes that are targets of the BRAF/MEK/ERK pathway. These include phosphatases DUSP5, DUSP6, AP-1 transcription factor component FOS, and apoptosis inhibitors survivin (also known as BIRC5—that is, baculoviral IAP repeat containing 5) and MCL1 (Figure 1). However, the robustness of pathway upregulation as suggested by the upregulation of these genes does not correlate with sensitivity to BRAF inhibitors. For example, cell lines SW1417 and MDST8, which display upregulation of pathway target genes, show no BRAF or other pathway inhibitors among their top inhibiting drugs (Table 5).
Table 5.
Drug sensitivities of PIK3CA wild type/BRAF V600E mutant cell lines. Data are from the Genomics of Drug Sensitivity in Cancer (GDSC).
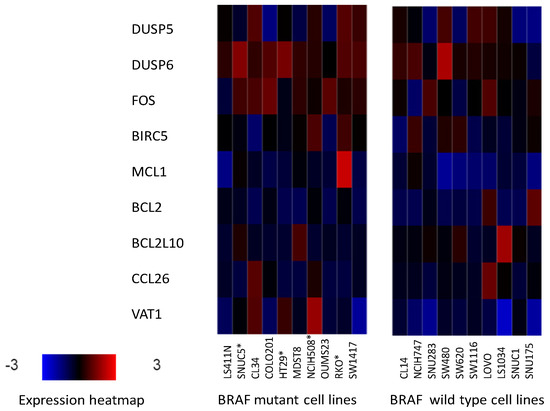
Figure 1.
mRNA expression of genes targeted by the BRAF/MEK/ERK pathway (DUSP5, DUSP6, FOS, BIRC5, and MCL1) and genes not directly targeted by the BRAF/MEK/ERK pathway (BCL2, BCL2L10, CCL26 and VAT1) as controls in representative colorectal cancer cell lines with (left panel) and without (right panel) mutations in BRAF. BRAF mutated cell lines with coexisting PIK3CA mutations are shown with an asterisk.
GDSC includes five specific BRAF inhibitors among the panel of assayed drugs. Recurrent molecular characteristics of the colorectal cancer cell lines panel that confer sensitivity to specific BRAF inhibitors include, as expected, BRAF mutations conferring sensitivity to 4 of the 5 inhibitors (Table 6). In addition, the presence of KRAS mutations confer resistance to 3 of the 5 BRAF inhibitors, as they tend to be mutually exclusive with BRAF mutations and segregate with BRAF wild type cell lines. Another genomic feature that is present recurrently among the abnormalities conferring BRAF inhibitor sensitivity in colorectal cancer cell lines is mutations in SACS, a gene encoding for sacsin, a chaperone protein. The most common copy number alteration that confers resistance to 3 of the 5 BRAF inhibitors is a loss at chromosome 6q26, a locus containing gene PRKN, encoding for E3 ubiquitin ligase parkin (feature cnaCOREAD24). Loss of PRKN is a feature of some BRAF mutant cell lines, as mentioned above, and it is also, rarely, encountered in BRAF mutant colorectal cancers. Thus, resistance to BRAF inhibitors associated with concomitant loss of PRKN may be of clinical significance. Interestingly, PIK3CA mutations do not feature among the molecular abnormalities conferring resistance to specific BRAF inhibitors in colorectal cancer cell lines. The only BRAF specific inhibitor that is not significantly more effective in BRAF mutant cell lines is HG6-64-1, which displays a separate private panel of mutations conferring resistance, not observed in other BRAF inhibitors. These include EGFR mutations and mutations in kinase ATM (Table 6).
Table 6.
Top molecular features with increased sensitivities to various BRAF inhibitors (statistically significant or approaching significance). Two non-specific RAF inhibitors (RAF 9304 and Sorafenib) are also shown. Data are from the Genomics of Drug Sensitivity in Cancer (GDSC).
In the pan-cancer analysis of cell lines with BRAF mutations, which is more statistically robust due to the number of cell lines assayed, pathway inhibitors (BRAF inhibitors: Dabrafenib, PLX-4720, SB59088, MEK inhibitors: selumetinib, trametinib, refametinib, PD0325901, ERK inhibitors: ulixertinib, ERK2440, ERK6604, SCH772984, VX-11e) are significantly associated with sensitivity compared to cell lines without BRAF mutations. In addition, the inhibitor of NUAK1 and NUAK2 kinases WZ4003 is statistically significantly associated with sensitivity in BRAF mutant cell lines compared with BRAF wild type cell lines (IC50 effect size: −0.34, p = 8.03 × 10−5). Specifically for colorectal cancer cell lines, BRAF mutant cell lines display also greater sensitivity to inhibitor WZ4003 compared to BRAF wild type colorectal cancer cell lines (mean IC50: 63.7 μM versus 132 μM), although, due to smaller numbers, this difference did not reach statistical significance (p = 0.08).
4. Discussion
BRAF is an oncogenic serine/threonine kinase, which is mutated in various cancers, most commonly in melanoma, thyroid carcinomas, hairy cell leukemia, lung cancers, and colorectal cancers [35]. The gene encoding for the kinase is located on the human chromosome locus 7q34. BRAF is activated by KRAS downstream of growth factor receptors and activates the Mitogen Activated Protein Kinase (MAPK)/Extracellular signal-Regulated Kinase (ERK) signaling cascade promoting cell proliferation. The importance of this pathway in cancer is highlighted by the fact that KRAS is the most frequently mutated oncogene across cancer types [36]. In parallel with the KRAS/BRAF/MAPK/ERK pathway, and also activated by growth factor receptors, the PI3K/AKT/mTOR cascade plays an important role in carcinogenesis through inhibition of apoptosis, cell growth promotion and oncogene activation [37]. PIK3CA, the gene encoding for the catalytic alpha sub-unit of kinase PI3K is often mutated in prevalent cancers such as breast cancer and colorectal adenocarcinomas. In colorectal cancer, PIK3CA is mutated in 20% to 25% of cases and is the second most commonly mutated oncogene after KRAS [17]. BRAF mutated colorectal cancers are less prevalent, representing 5% to 15% of all colorectal cancers. Most of BRAF mutations are located at amino acid position V600, substituting glutamic acid for valine that is normally at this position in the wild type protein (V600E substitution). Substitutions at position V600 render the protein independent from KRAS and result in robust kinase-mediated activation of MAPK cascade, without the physiologic input from growth factors [38]. Other less common BRAF mutations produce a protein with lower kinase activity or even a kinase-dead protein that can still activate down-stream signaling through interaction with the homologous CRAF kinase [15]. Canonical V600E BRAF mutations are mutually exclusive with KRAS mutations. In contrast, PIK3CA mutations are encountered in colorectal cancers with either KRAS or BRAF mutations with an equal or higher prevalence than in cancers with wild type KRAS and BRAF.
BRAF mutations are targeted currently in colorectal cancer in the clinic at the second line metastatic setting with a regimen that combines BRAF inhibitors and anti-EGFR monoclonal antibodies. This combination has provided superior efficacy and survival outcomes compared with chemotherapy, with a modest improvement of 3 months in Overall Survival (OS) [39]. In contrast, no therapies targeting PIK3CA mutated colorectal cancers have been approved for clinical use. Combinations of BRAF inhibitors with PI3K inhibitors have not been studied in a systematic manner in colorectal cancer, but few available retrospective data suggest that parallel inhibition of the two mutated oncogenes may provide a synergistic effect in double mutant cancers [40]. Unveiling vulnerabilities of colorectal cancers with BRAF mutations with and without concomitant PIK3CA mutations may provide new opportunities for targeted treatments.
The current investigation examines a panel of colorectal cancer cell lines with BRAF mutations with or without concomitant mutations in PIK3CA from the CCLE for drug sensitivities and molecular dependencies. Mutations in PIK3CA are the most frequent mutations in the receptor tyrosine kinase-initiated pathways in colorectal cancers with BRAF mutations, as the even more frequent KRAS mutations are mutually exclusive with BRAF mutations. Colorectal cancer cell line models recapitulate the presence of BRAF and PIK3CA mutations as encountered in clinical colorectal cancer samples, and also duplicate the frequent presence of MSI in these cases [41]. Mutations in tumor suppressors APC and TP53 are often present in BRAF mutant colorectal cancer cell lines, similar to clinical samples. Cell lines with BRAF mutations and wild type PIK3CA possess also deletions of signal transducers of TGFβ pathway SMAD4 and SMAD2 and of phosphatase DUSP22. The genes of these proteins are rarely deleted in clinical colorectal cancer, but they are more commonly mutated. For example, in TCGA cohort, SMAD4 mutations are observed in 16.1% of cases with BRAF mutations, SMAD2 mutations are observed in 6.5% of cases with BRAF mutations and DUSP22 mutations are encountered in 9.7% of patients with BRAF mutations [17]. The presence of mutations or deletions of these genes suggest that decreased availability and function of the resulting proteins may be essential for BRAF mutant cancers both in vitro and in vivo. The TGFβ signaling pathway and tumor suppressor SMAD4 mutations have been implicated in the serrated colon carcinogenesis pathway commonly resulting from BRAF mutations [42]. In addition, inhibitors of the TGFβ receptor TGFBR1 prevented the development of resistance to BRAF inhibitor vemurafenib in BRAF mutant melanoma cells [43]. Thus, inhibitors of the TGFβ pathway, should they become clinically available, could be candidates for combination therapies in BRAF mutated colorectal cancers. Phosphatase DUSP22 (also called JKAP- c-JUN N-terminal Kinase Associated phosphatase) is a regulator of the MAPK pathway, and as a result, it may modulate the effect of BRAF mutations in the pathway output [44]. DUSP22 showed lower mRNA expression in colorectal cancer tissues compared to adjacent normal colonic mucosa [45]. In this study that included 92 patients, patients with metastatic colorectal cancer and low expression of DUSP22 had a trend towards worse survival, although not statistically significant [45].
The analysis of molecular features associated with sensitivity or resistance to BRAF specific inhibitors reveals that, besides BRAF mutations and KRAS mutations that are associated with sensitivity and resistance to the drugs, respectively, no other abnormalities of the pathway affect sensitivity to these drugs in a consistent manner, in vitro. Unrelated molecular alterations associated with sensitization of colorectal cancer cell lines to BRAF inhibitors included mutations in SACS, encoding for chaperone protein sacsin and deletions at the locus of parkin. Sacsin is a large protein with chaperone function in the nervous system and loss of function mutations are associated with the degenerative disorder autosomal recessive spastic ataxia of Charlevoix-Saguenay [46]. Cells with sacsin loss of function have defective mitochondrial dynamics and increased oxidative stress. Mutations in SACS have not been previously linked with colorectal cancer. The protein consists of 4579 amino acids and is mutated in 12.5% of colorectal cancers of the TCGA cohort with mutations distributed equally across the length of the protein [17]. It is also mutated in 33.9% of colorectal cancers with BRAF mutations and in 19% of cancers with PIK3CA mutations. Among colorectal cancers classified as MSI high or with proofreading polymerase epsilon mutations, SACS mutations are present in 42.5% of cases, suggesting that these mutations are associated with high TMB and may be passenger [47]. Alternatively, an oncogenic role of sacsin mutations in colorectal cancer is also possible based on its function in oxidative stress and deserves to be formally confirmed or excluded.
Concomitant mutations in APC that are observed in most cell lines with BRAF mutations with or without PIK3CA mutations, as well as the fact that CTNNB1 gene, encoding for β-catenin, is a recurrent preferential essential gene in these cell lines suggest that BRAF mutated colorectal cancers remain dependent on the activity of WNT/APC/β-catenin pathway [48,49]. Two other recurrent preferentially essential genes in BRAF mutated cell lines are WRN, encoding for Werner helicase and CAD (carbamoyl-phosphate synthetase 2, aspartate transcarbamylase and dihydroorotase), encoding for a protein with trifunctional enzyme activity implicated in the de novo pyrimidine nucleotide biosynthesis. WRN helicase is involved in DNA repair and was recently identified as a vulnerability of cancer cells with MSI [27,50,51,52]. Cells with MSI are vulnerable to massive apoptosis in the absence of WRN function because of accumulation of long TA dinucleotide repeats that form secondary structures that stall DNA forks during replication [53]. Consistent with this mechanism, MSS cell lines are not dependent on WRN helicase function [52]. Indeed, the BRAF mutant colorectal cancer cell lines that show vulnerability to WRN knock-down are all MSI high, suggesting that this is the underlying molecular defect directly responsible, rather than BRAF mutations. However, given the frequent co-occurrence of the two alterations in cell lines and clinical colorectal cancers, pharmacologic inhibition of WRN helicase in these cancers can be envisioned and would be expected to spare normal cells without MSI.
The other recurrent preferentially essential gene discovered in BRAF mutated cell lines, CAD, possesses the three first enzymatic activities in the pathway of de novo pyrimidine nucleotide biosynthesis in a single polypeptide of 2225 amino acids [54]. CAD is regulated by phosphorylation by MAPK, which activates the enzyme to promote nucleotide synthesis [55]. This regulation makes CAD a target of the KRAS/BRAF/MAPK cascade in response to growth factor signaling and activates an enzymatic function that sustains nucleotide production required for cell proliferation. Moreover, in colorectal cancer, CAD is regulated by MYC and when the metabolic reprogramming observed in cancer cells as a result of MYC activation is inhibited, cell growth is blocked by shutting down CAD and other enzymes of pyrimidine biosynthesis [56]. In cancer cells with deregulated proliferation secondary to BRAF mutations, loss of CAD function would deprive them from the required de novo pyrimidine nucleotides with potential catastrophic consequences due to loss of the coordinated response to the metabolic needs derived by high cancer cell proliferation. Thus, pharmacologic CAD inhibition with novel inhibitors in development may represent a therapeutic target in BRAF mutated cells with concomitant PIK3CA mutations, given that MAPK signaling and MYC are regulated by the two oncogenes [57].
A final interesting finding of the current investigation with potential future therapeutic implications is the identification of a NUAK family kinase (NUAK) inhibitor as one of the top hits in the pan-cancer BRAF mutant cell line screening. NUAK1 and NUAK2 are AMPK (AMP-activated Protein Kinase) related kinases with diverse functions in cancer cells [58]. NUAK1 promotes motility, invasion, and metastases of cancer cells [59,60]. NUAK1 shows higher expression in advanced stage colorectal cancers and in biopsies from liver metastatic sites, compared to primary tumors [61]. An important role of the kinase has been described in cancer cells with oncogene MYC overexpression, related to protection from oxidative stress resulting from MYC activity [62]. Mechanistically, NUAK1 contributes to mitochondrial plasticity and adaptation which is critical for cells bearing induction of oxidative respiratory chain component proteins effectuated by MYC [63]. Only 2 colorectal cancer cell lines with BRAF mutations RKO and HT-29 show MYC amplifications and both are more sensitive to the NUAK inhibitor WZ4003 than the mean sensitivity of the BRAF mutant group of colorectal cancer cell lines. Although these observations are based on a small number of cell lines, they suggest that BRAF mutant colorectal cancers with concomitant aberrations increasing oxidative stress could be candidates for combination therapies with NUAK kinases inhibitors.
A limitation of the current study is that relies exclusively in in silico publicly available data and no further experimental confirmation was performed. In addition, in the drug sensitivity analysis based on GDSC, cell lines are exposed to the assayed drugs as monotherapies and no data exist to inform combination therapies. Combinations of targeted anti-neoplastic drug therapies are increasingly recognized as being necessary for improvement of response in cancers which accumulate molecular alterations over time for their survival. Another limitation of the current study is that the cell line data do not definitely allow differentiation of a direct dependency on BRAF or PIK3CA mutations versus indirect effects related to other vulnerabilities such as MSI commonly co-occurring in these cell lines as the example of WRN helicase dependency illustrates. Moreover, it is expected that additional vulnerabilities that are not revealed with the approach used here exist in BRAF mutant colorectal cancers. For example, RANBP2, a binding protein of RAN (RAS related nuclear protein), a small GTPase of the RAS family, has been proposed as essential for survival of BRAF V600E mutant colorectal cancer cells and cells with a similar genomic signature [64].
In conclusion, targeted therapies of colorectal cancers that possess BRAF mutations with or without PIK3CA mutations could be developed based on the global molecular environment of these cancers and based on vulnerabilities uncovered in in vitro models. It is reassuring for the validity of the vulnerabilities discovered from cell lines models, that some of them, such as, for example, the synthetic lethality of MSI and WRN helicase, had previously been reported in pertinent systems. Leads discussed here need to be confirmed in in vivo studies followed by human trials in the population of interest.
Funding
This research received no external funding.
Institutional Review Board Statement
This research does not involve human subjects or animals and IRB approval was not required or obtained.
Informed Consent Statement
Not applicable.
Data Availability Statement
There are no data available beyond data included in the manuscript.
Conflicts of Interest
The author declares no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef]
- Riedesser, J.E.; Ebert, M.P.; Betge, J. Precision medicine for metastatic colorectal cancer in clinical practice. Ther. Adv. Med. Oncol. 2022, 14, 17588359211072703. [Google Scholar] [CrossRef] [PubMed]
- Rankin, A.; Klempner, S.J.; Erlich, R.; Sun, J.X.; Grothey, A.; Fakih, M.; George, T.J., Jr.; Lee, J.; Ross, J.S.; Stephens, P.J.; et al. Broad Detection of Alterations Predicted to Confer Lack of Benefit from EGFR Antibodies or Sensitivity to Targeted Therapy in Advanced Colorectal Cancer. Oncologist 2016, 21, 1306–1314. [Google Scholar] [CrossRef]
- Tabernero, J.; Grothey, A.; Van Cutsem, E.; Yaeger, R.; Wasan, H.; Yoshino, T.; Desai, J.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib Plus Cetuximab as a New Standard of Care for Previously Treated BRAF V600E-Mutant Metastatic Colorectal Cancer: Updated Survival Results and Subgroup Analyses from the BEACON Study. J. Clin. Oncol. 2021, 39, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Siena, S.; Di Bartolomeo, M.; Raghav, K.; Masuishi, T.; Loupakis, F.; Kawakami, H.; Yamaguchi, K.; Nishina, T.; Fakih, M.; Elez, E.; et al. Trastuzumab deruxtecan (DS-8201) in patients with HER2-expressing metastatic colorectal cancer (DESTINY-CRC01): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2021, 22, 779–789. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. KEYNOTE-177 Investigators. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Ratti, M.; Grizzi, G.; Passalacqua, R.; Lampis, A.; Cereatti, F.; Grassia, R.; Hahne, J.C. NTRK fusions in colorectal cancer: Clinical meaning and future perspective. Expert Opin. Ther. Targets 2021, 25, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Wang, C.; Fakih, M. Targeting KRASG12C-Mutated Advanced Colorectal Cancer: Research and Clinical Developments. Onco. Targets Ther. 2022, 15, 747–756. [Google Scholar] [CrossRef]
- Rosati, G.; Aprile, G.; Colombo, A.; Cordio, S.; Giampaglia, M.; Cappetta, A.; Porretto, C.M.; De Stefano, A.; Bilancia, D.; Avallone, A. Colorectal Cancer Heterogeneity and the Impact on Precision Medicine and Therapy Efficacy. Biomedicines 2022, 10, 1035. [Google Scholar] [CrossRef]
- Ochsenreither, S.; Fiedler, W.M.; Conte, G.D.; Macchini, M.; Matos, I.; Habel, B.; Ahrens-Fath, I.; Raspagliesi, F.; Lorusso, D.; Keilholz, U.; et al. Safety and preliminary activity results of the GATTO study, a phase Ib study combining the anti-TA-MUC1 antibody gatipotuzumab with the anti-EGFR tomuzotuximab in patients with refractory solid tumors. ESMO Open 2022, 7, 100447. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Fakih, M.; Tabernero, J. Management of BRAF-mutant metastatic colorectal cancer: A review of treatment options and evidence-based guidelines. Ann. Oncol. 2021, 32, 959–967. [Google Scholar] [CrossRef]
- Kayhanian, H.; Goode, E.; Sclafani, F.; Ang, J.E.; Gerlinger, M.; Gonzalez de Castro, D.; Shepherd, S.; Peckitt, C.; Rao, S.; Watkins, D.; et al. Treatment and Survival Outcome of BRAF-Mutated Metastatic Colorectal Cancer: A Retrospective Matched Case-Control Study. Clin. Color. Cancer 2018, 17, e69. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.C.; Renfro, L.A.; Al-Shamsi, H.O.; Schrock, A.B.; Rankin, A.; Zhang, B.Y.; Kasi, P.M.; Voss, J.S.; Leal, A.D.; Sun, J.; et al. Non-V600 BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J. Clin. Oncol. 2017, 35, 2624–2630. [Google Scholar] [CrossRef] [PubMed]
- Sahin, I.H.; Klostergaard, J. BRAF Mutations as Actionable Targets: A Paradigm Shift in the Management of Colorectal Cancer and Novel Avenues. JCO Oncol. Pract. 2021, 17, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell. 2018, 33, 125–136.e3. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A. The Landscape of PIK3CA Mutations in Colorectal Cancer. Clin. Color. Cancer 2021, 20, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, PO.17.00011. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Boehm, J.S.; Garnett, M.J.; Adams, D.J.; Francies, H.E.; Golub, T.R.; Hahn, W.C.; Iorio, F.; McFarland, J.M.; Parts, L.; Vazquez, F. Cancer research needs a better map. Nature 2021, 589, 514–516. [Google Scholar] [CrossRef]
- van der Meer, D.; Barthorpe, S.; Yang, W.; Lightfoot, H.; Hall, C.; Gilbert, J.; Francies, H.E.; Garnett, M.J. Cell Model Passports-a hub for clinical, genetic and functional datasets of preclinical cancer models. Nucleic Acids Res. 2019, 47, D923–D929. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef]
- Marcotte, R.; Sayad, A.; Brown, K.R.; Sanchez-Garcia, F.; Reimand, J.; Haider, M.; Virtanen, C.; Bradner, J.E.; Bader, G.D.; Mills, G.B.; et al. Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell 2016, 164, 293–309. [Google Scholar] [CrossRef] [PubMed]
- Dwane, L.; Behan, F.M.; Gonçalves, E.; Lightfoot, H.; Yang, W.; van der Meer, D.; Shepherd, R.; Pignatelli, M.; Iorio, F.; Garnett, M.J. Project Score database: A resource for investigating cancer cell dependencies and prioritizing therapeutic targets. Nucleic Acids Res. 2021, 49, D1365–D1372. [Google Scholar] [CrossRef] [PubMed]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef]
- Dempster, J.M.; Boyle, I.; Vazquez, F.; Root, D.E.; Boehm, J.S.; Hahn, W.C.; Tsherniak, A.; McFarland, J.M. Chronos: A cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome Biol. 2021, 22, 343. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Sullivan, R.J.; Yaeger, R. Molecular Pathways and Mechanisms of BRAF in Cancer Therapy. Clin Cancer Res. 2022, OF1–OF11. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed]
- Solit, D.B.; Garraway, L.A.; Pratilas, C.A.; Sawai, A.; Getz, G.; Basso, A.; Ye, Q.; Lobo, J.M.; She, Y.; Osman, I.; et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006, 439, 358–362. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef]
- van Geel, R.M.J.M.; Tabernero, J.; Elez, E.; Bendell, J.C.; Spreafico, A.; Schuler, M.; Yoshino, T.; Delord, J.P.; Yamada, Y.; Lolkema, M.P.; et al. A Phase Ib Dose-Escalation Study of Encorafenib and Cetuximab with or without Alpelisib in Metastatic BRAF-Mutant Colorectal Cancer. Cancer Discov. 2017, 7, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A. The genomic environment of BRAF mutated and BRAF/PIK3CA double mutated colorectal cancers. J. Clin. Med. 2022, 11, 5132. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.; Kothari, O.A.; Haro, K.S.; Panda, A.; Bandari, M.M.; Carrick, J.N.; Hur, J.J.; Zhang, L.; Chan, C.S.; Xing, J.; et al. SMAD4 is critical in suppression of BRAF-V600E serrated tumorigenesis. Oncogene 2021, 40, 6034–6048. [Google Scholar] [CrossRef] [PubMed]
- Spender, L.C.; Ferguson, G.J.; Liu, S.; Cui, C.; Girotti, M.R.; Sibbet, G.; Higgs, E.B.; Shuttleworth, M.K.; Hamilton, T.; Lorigan, P.; et al. Mutational activation of BRAF confers sensitivity to transforming growth factor beta inhibitors in human cancer cells. Oncotarget 2016, 7, 81995–82012. [Google Scholar] [CrossRef]
- Chen, A.J.; Zhou, G.; Juan, T.; Colicos, S.M.; Cannon, J.P.; Cabriera-Hansen, M.; Meyer, C.F.; Jurecic, R.; Copeland, N.G.; Gilbert, D.J.; et al. The dual specificity JKAP specifically activates the c-Jun N-terminal kinase pathway. J. Biol. Chem. 2002, 277, 36592–36601. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Li, Z.; Gan, M.; Zhang, H.; Yin, X.; Tang, S.; Wan, L.; Tian, Y.; Zhang, S.; Zhu, Y.; et al. Decreased expression of dual specificity phosphatase 22 in colorectal cancer and its potential prognostic relevance for stage IV CRC patients. Tumour Biol. 2015, 36, 8531–8535. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, T.Y.; Romano, L.E.; Duncan, E.J.; Nethisinghe, S.; Abeti, R.; Michael, G.J.; Giunti, P.; Vermeer, S.; Chapple, J.P. A reduction in Drp1-mediated fission compromises mitochondrial health in autosomal recessive spastic ataxia of Charlevoix Saguenay. Hum. Mol. Genet. 2016, 25, 3232–3244. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. Polymerase epsilon mutations and concomitant β2-microglobulin mutations in cancer. Gene 2018, 647, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Riemer, P.; Sreekumar, A.; Reinke, S.; Rad, R.; Schäfer, R.; Sers, C.; Bläker, H.; Herrmann, B.G.; Morkel, M. Transgenic expression of oncogenic BRAF induces loss of stem cells in the mouse intestine, which is antagonized by β-catenin activity. Oncogene 2015, 34, 3164–3175. [Google Scholar] [CrossRef]
- Morkel, M.; Riemer, P. Cell hierarchies in colorectal cancer: Focus on APC and BRAF. Oncoscience 2015, 2, 675–676. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.M.; Shibue, T.; McFarland, J.M.; Gaeta, B.; Ghandi, M.; Dumont, N.; Gonzalez, A.; McPartlan, J.S.; Li, T.; Zhang, Y.; et al. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 2019, 568, 551–556. [Google Scholar] [CrossRef]
- Lieb, S.; Blaha-Ostermann, S.; Kamper, E.; Rippka, J.; Schwarz, C.; Ehrenhöfer-Wölfer, K.; Schlattl, A.; Wernitznig, A.; Lipp, J.J.; Nagasaka, K.; et al. Werner syndrome helicase is a selective vulnerability of microsatellite instability-high tumor cells. eLife 2019, 8, e43333. [Google Scholar] [CrossRef] [PubMed]
- Kategaya, L.; Perumal, S.K.; Hager, J.H.; Belmont, L.D. Werner Syndrome Helicase Is Required for the Survival of Cancer Cells with Microsatellite Instability. iScience 2019, 13, 488–497. [Google Scholar] [CrossRef]
- van Wietmarschen, N.; Sridharan, S.; Nathan, W.J.; Tubbs, A.; Chan, E.M.; Callen, E.; Wu, W.; Belinky, F.; Tripathi, V.; Wong, N.; et al. Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 2020, 586, 292–298. [Google Scholar] [CrossRef]
- Del Caño-Ochoa, F.; Ramón-Maiques, S. Deciphering CAD: Structure and function of a mega-enzymatic pyrimidine factory in health and disease. Protein Sci. 2021, 30, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Graves, L.M.; Guy, H.I.; Kozlowski, P.; Huang, M.; Lazarowski, E.; Pope, R.M.; Collins, M.A.; Dahlstrand, E.N.; Earp, H.S., 3rd; Evans, D.R. Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature 2000, 403, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Yachida, S.; Sugimoto, M.; Oshima, M.; Nakagawa, T.; Akamoto, S.; Tabata, S.; Saitoh, K.; Kato, K.; Sato, S.; et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc. Natl. Acad. Sci. USA 2017, 114, E7697–E7706. [Google Scholar] [CrossRef]
- Lei, Z.; Wang, B.; Lu, Z.; Wang, N.; Tan, H.; Zheng, J.; Jia, Z. New regulatory mechanism-based inhibitors of aspartate transcarbamoylase for potential anticancer drug development. FEBS J. 2020, 287, 3579–3599. [Google Scholar] [CrossRef]
- Port, J.; Muthalagu, N.; Raja, M.; Ceteci, F.; Monteverde, T.; Kruspig, B.; Hedley, A.; Kalna, G.; Lilla, S.; Neilson, L.; et al. Colorectal Tumors Require NUAK1 for Protection from Oxidative Stress. Cancer Discov. 2018, 8, 632–647. [Google Scholar] [CrossRef] [PubMed]
- Kusakai, G.; Suzuki, A.; Ogura, T.; Kaminishi, M.; Esumi, H. Strong association of ARK5 with tumor invasion and metastasis. J. Exp. Clin. Cancer Res. 2004, 23, 263–268. [Google Scholar]
- Zagórska, A.; Deak, M.; Campbell, D.G.; Banerjee, S.; Hirano, M.; Aizawa, S.; Prescott, A.R.; Alessi, D.R. New roles for the LKB1-NUAK pathway in controlling myosin phosphatase complexes and cell adhesion. Sci. Signal. 2010, 3, ra25. [Google Scholar] [CrossRef]
- Kusakai, G.; Suzuki, A.; Ogura, T.; Miyamoto, S.; Ochiai, A.; Kaminishi, M.; Esumi, H. ARK5 expression in colorectal cancer and its implications for tumor progression. Am. J. Pathol. 2004, 164, 987–995. [Google Scholar] [CrossRef]
- Liu, L.; Ulbrich, J.; Müller, J.; Wüstefeld, T.; Aeberhard, L.; Kress, T.R.; Muthalagu, N.; Rycak, L.; Rudalska, R.; Moll, R.; et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature 2012, 483, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell. Biol. 2005, 25, 6225–6234. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, L.; Gambino, V.; Raaijmakers, J.; Schlicker, A.; Fumagalli, A.; Russo, M.; Villanueva, A.; Beerling, E.; Bartolini, A.; Mollevi, D.G.; et al. A Vulnerability of a Subset of Colon Cancers with Potential Clinical Utility. Cell 2016, 165, 317–330. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).