
ASC-J9 Blocks Cell Proliferation and Extracellular Matrix Production of Keloid Fibroblasts through Inhibiting STAT3 Signaling

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
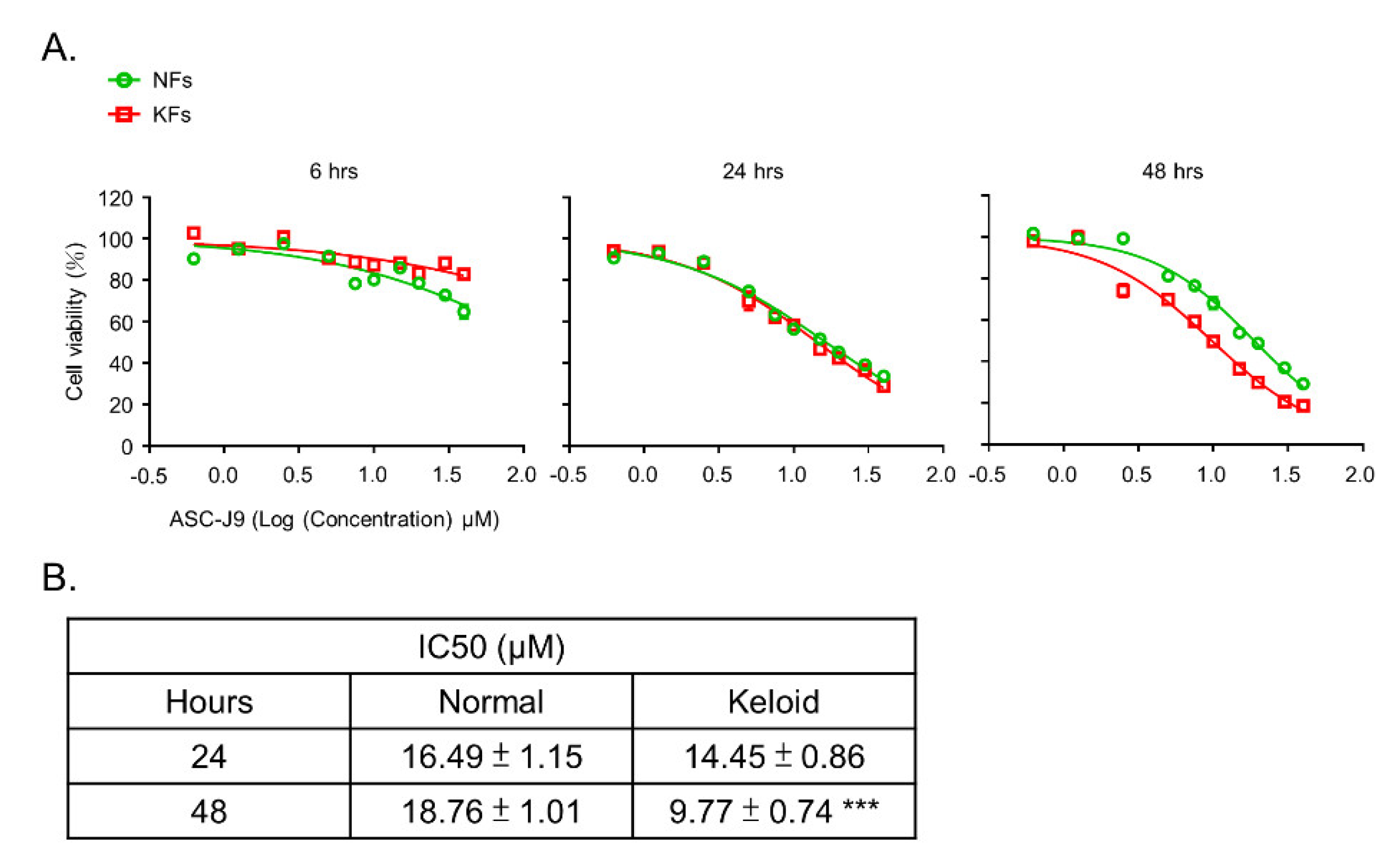
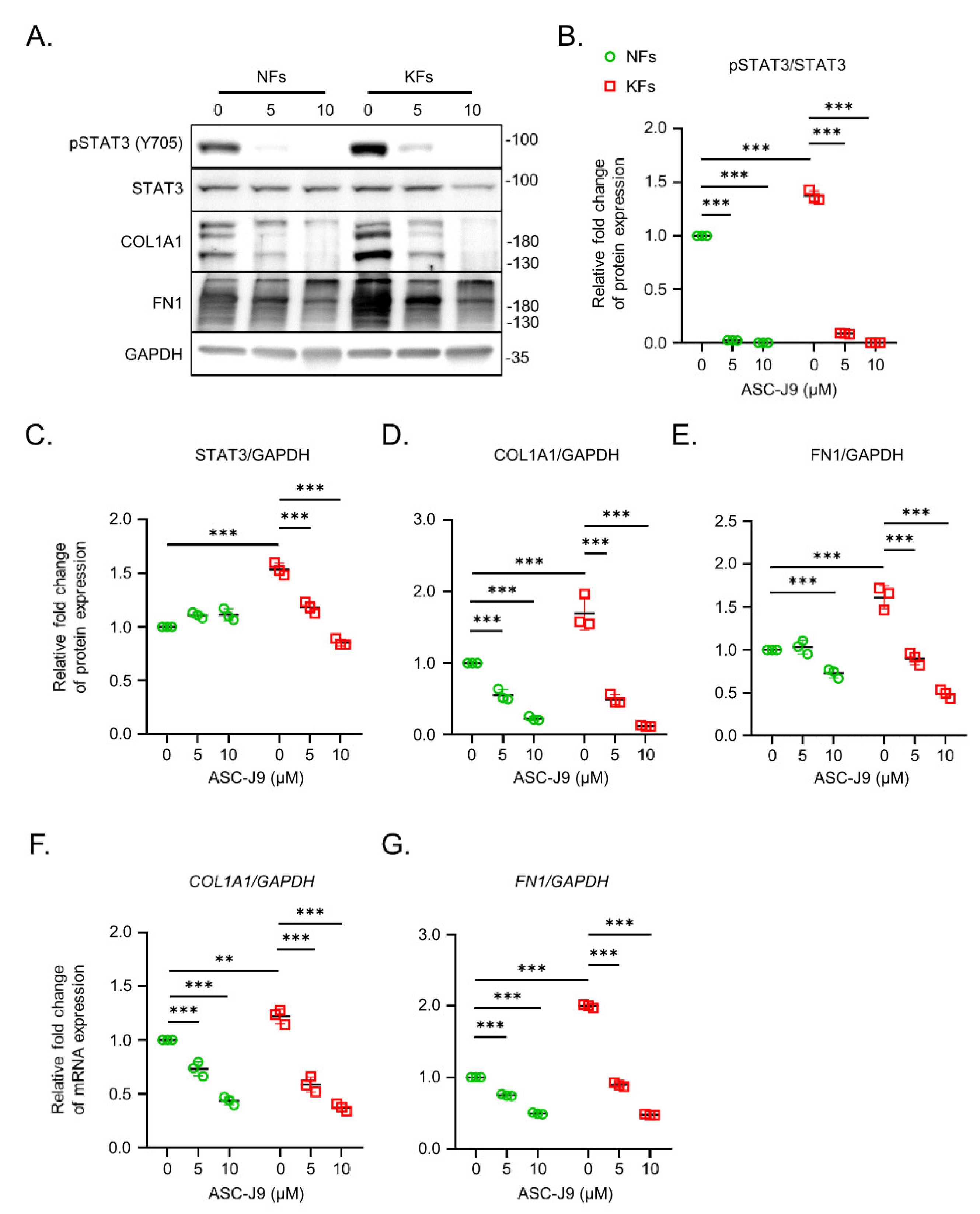
2.1. ASC-J9 Inhibits ECM Synthesis and Cell Proliferation in Keloid Fibroblasts
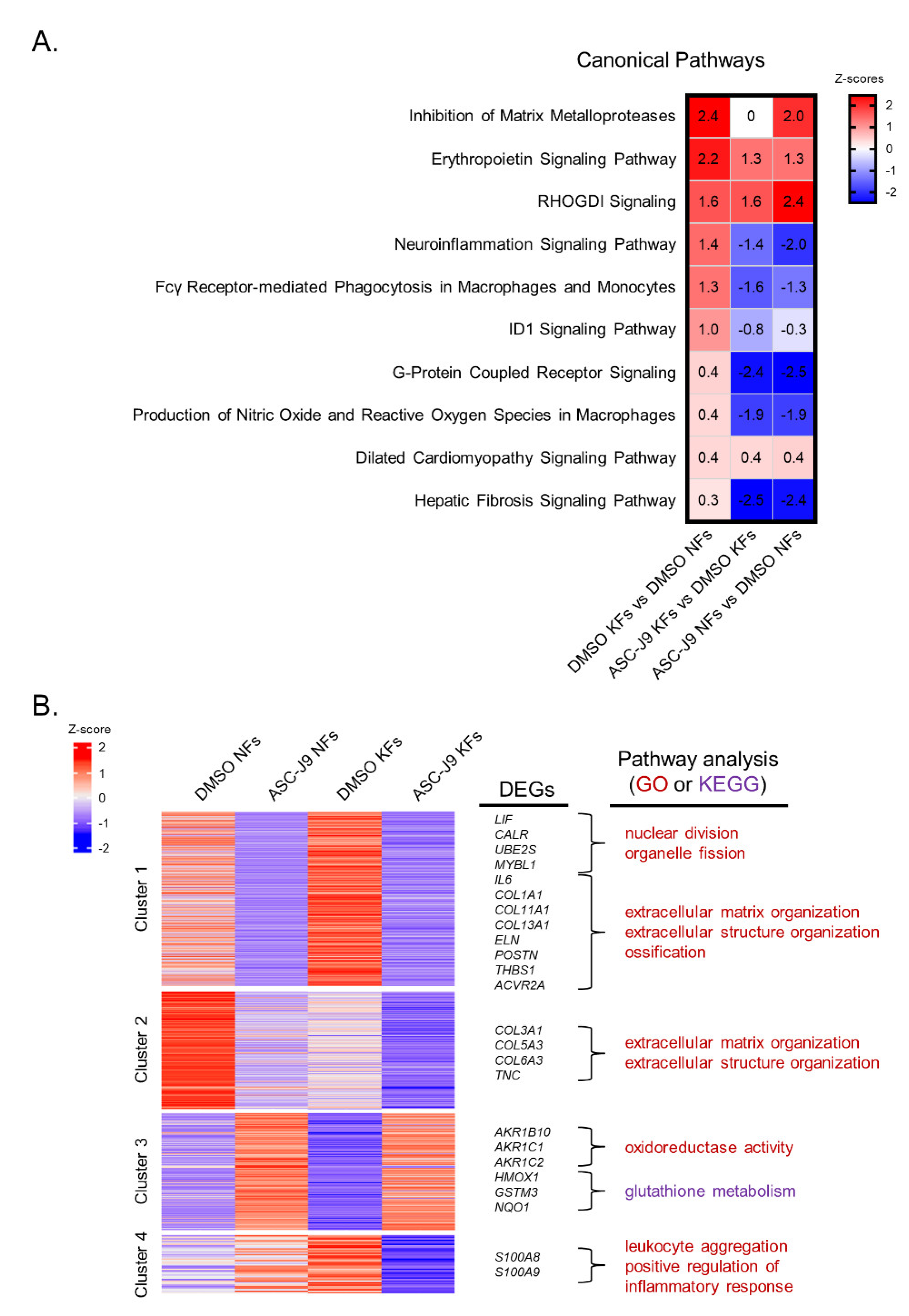
2.2. RNA-Seq Reveals the Inhibitory Effect of ASC-J9 on Inflammation, ROS Generation, and Fibrosis in Keloid Fibroblasts
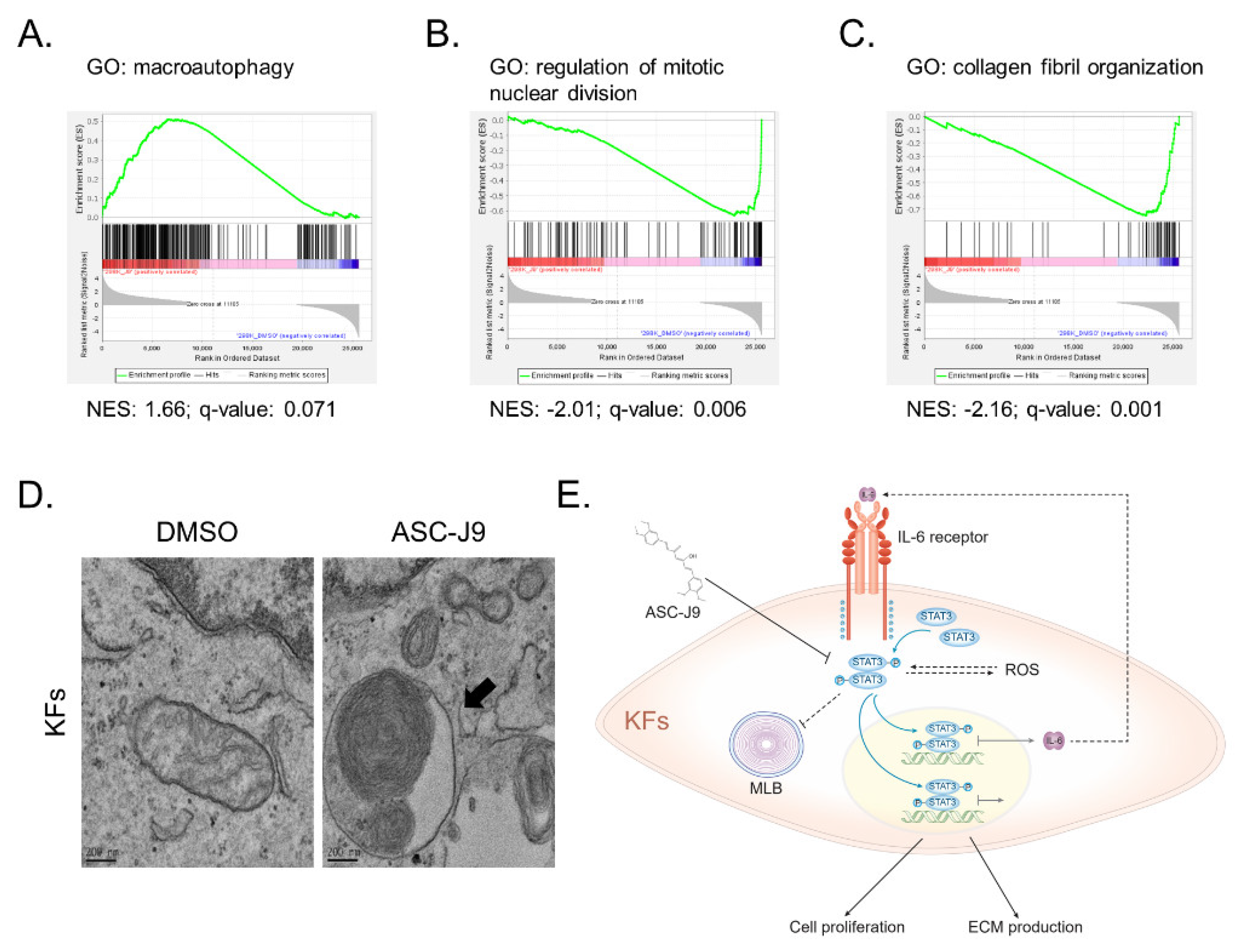
2.3. ASC-J9 Treatment Regulates Oxidative Stress Homeostasis and Inflammatory Responses
2.4. ASC-J9 Induces the Formation of Multilamellar Bodies in Keloid Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Keloid Patients
4.2. Primary Culture of Fibroblasts
4.3. Quantitative PCR (qPCR)
4.4. Western Blotting
4.5. MTT Assay
4.6. RNA Library Preparation and Sequencing
4.7. Bioinformatic Analysis
4.8. Enzyme-Linked Immunosorbent Assay (ELISA)
4.9. Transmission Electron Microscopy
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berman, B.; Maderal, A.; Raphael, B. Keloids and Hypertrophic Scars: Pathophysiology, Classification, and Treatment. Dermatol. Surg. 2017, 43 (Suppl. S1), S3–S18. [Google Scholar] [CrossRef] [PubMed]
- McGinty, S.; Siddiqui, W.J. Keloid; StatPearls: Tampa, FL, USA, 2020. [Google Scholar]
- Lee, J.Y.; Yang, C.C.; Chao, S.C.; Wong, T.W. Histopathological differential diagnosis of keloid and hypertrophic scar. Am. J. Dermatopathol. 2004, 26, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, K.; Ruan, M.; Yang, J.; Gao, Z. Gallic acid inhibits fibroblast growth and migration in keloids through the AKT/ERK signaling pathway. Acta Biochim. Biophys. Sin. 2018, 50, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Bian, W.; Cheng, L.; Zhang, L.; Jin, R.; Wang, W.; Zhang, Y. Ginsenoside Rg3 inhibits keloid fibroblast proliferation, angiogenesis and collagen synthesis in vitro via the TGF-β/Smad and ERK signaling pathways. Int. J. Mol. Med. 2018, 41, 1487–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipev, C.C.; Simon, M. Phenotypic differences between dermal fibroblasts from different body sites determine their responses to tension and TGFbeta1. BMC Dermatol. 2002, 2, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.K.; Lin, H.H.; Harn, H.I.; Ogawa, R.; Wang, Y.K.; Ho, Y.T.; Chen, W.R.; Lee, Y.C.; Lee, J.Y.; Shieh, S.J.; et al. Caveolin-1 Controls Hyperresponsiveness to Mechanical Stimuli and Fibrogenesis-Associated RUNX2 Activation in Keloid Fibroblasts. J. Investig. Dermatol. 2018, 138, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, R.; Tosa, M.; Dohi, T.; Akaishi, S.; Kuribayashi, S. Surgical excision and postoperative radiotherapy for keloids. Scars Burn. Health 2019, 5, 2059513119891113. [Google Scholar] [CrossRef] [Green Version]
- Chang, Z.; Wang, Y.; Zhou, X.; Long, J.E. STAT3 roles in viral infection: Antiviral or proviral? Future Virol. 2018, 13, 557–574. [Google Scholar] [CrossRef]
- Huang, G.; Yan, H.; Ye, S.; Tong, C.; Ying, Q.L. STAT3 phosphorylation at tyrosine 705 and serine 727 differentially regulates mouse ESC fates. Stem Cells 2014, 32, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Gkouveris, I.; Nikitakis, N.; Sauk, J. STAT3 signaling in cancer. J. Cancer Ther. 2015, 6, 709. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.P.; Phan, T.T.; Lim, I.J.; Cao, X. Stat3 contributes to keloid pathogenesis via promoting collagen production, cell proliferation and migration. Oncogene 2006, 25, 5416–5425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Liang, Y.C.; Wu, P.; Kulber, D.A.; Tanabe, K.; Chuong, C.M.; Widelitz, R. STAT3 signalling pathway is implicated in keloid pathogenesis by preliminary transcriptome and open chromatin analyses. Exp. Dermatol. 2019, 28, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Luo, J.; Sun, Y.; Lin, C.; Li, G.; Niu, Y.; Chang, C. ASC-J9(®) suppresses prostate cancer cell invasion via altering the sumoylation-phosphorylation of STAT3. Cancer Lett. 2018, 425, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.C.; Hu, Y.F.; Zhu, D.H.; Cheng, Q.; Gu, J.J.; Feng, Q.L.; Zhang, L.X.; Xu, Y.P.; Wang, D.; Rong, Z.; et al. Single-cell RNA-seq reveals fibroblast heterogeneity and increased mesenchymal fibroblasts in human fibrotic skin diseases. Nat. Commun. 2021, 12, 3709. [Google Scholar] [CrossRef]
- Ulrich, D.; Ulrich, F.; Unglaub, F.; Piatkowski, A.; Pallua, N. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in patients with different types of scars and keloids. J. Plast. Reconstr. Aesthetic Surg. 2010, 63, 1015–1021. [Google Scholar] [CrossRef]
- Lee, U.E.; Friedman, S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 195–206. [Google Scholar] [CrossRef]
- Wiercinska, E.; Wickert, L.; Denecke, B.; Said, H.M.; Hamzavi, J.; Gressner, A.M.; Thorikay, M.; ten Dijke, P.; Mertens, P.R.; Breitkopf, K.; et al. Id1 is a critical mediator in TGF-beta-induced transdifferentiation of rat hepatic stellate cells. Hepatology 2006, 43, 1032–1041. [Google Scholar] [CrossRef]
- Salazar, N.C.; Chen, J.; Rockman, H.A. Cardiac GPCRs: GPCR signaling in healthy and failing hearts. Biochim. Biophys. Acta 2007, 1768, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Gareri, C.; Rockman, H.A. G-Protein-Coupled Receptors in Heart Disease. Circ. Res. 2018, 123, 716–735. [Google Scholar] [CrossRef]
- Altman, N.; Krzywinski, M. Clustering. Nat. Methods 2017, 14, 545–546. [Google Scholar] [CrossRef]
- Volk, S.W.; Wang, Y.; Mauldin, E.A.; Liechty, K.W.; Adams, S.L. Diminished type III collagen promotes myofibroblast differentiation and increases scar deposition in cutaneous wound healing. Cells Tissues Organs 2011, 194, 25–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribitsch, I.; Bileck, A.; Aldoshin, A.D.; Kandula, M.M.; Mayer, R.L.; Egerbacher, M.; Gabner, S.; Auer, U.; Gultekin, S.; Huber, J.; et al. Molecular Mechanisms of Fetal Tendon Regeneration Versus Adult Fibrous Repair. Int. J. Mol. Sci. 2021, 22, 5619. [Google Scholar] [CrossRef] [PubMed]
- Theocharidis, G.; Drymoussi, Z.; Kao, A.P.; Barber, A.H.; Lee, D.A.; Braun, K.M.; Connelly, J.T. Type VI Collagen Regulates Dermal Matrix Assembly and Fibroblast Motility. J. Investig. Dermatol. 2016, 136, 74–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Joon Lee, H.; Jakovcevski, I.; Shah, R.; Bhagat, N.; Loers, G.; Liu, H.Y.; Meiners, S.; Taschenberger, G.; Kügler, S.; et al. The extracellular matrix glycoprotein tenascin-C is beneficial for spinal cord regeneration. Mol. Ther. 2010, 18, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanyam, M.; Koteswari, A.A.; Kumar, R.S.; Monickaraj, S.F.; Maheswari, J.U.; Mohan, V. Curcumin-induced inhibition of cellular reactive oxygen species generation: Novel therapeutic implications. J. Biosci. 2003, 28, 715–721. [Google Scholar] [CrossRef] [Green Version]
- Barzegar, A.; Moosavi-Movahedi, A.A. Intracellular ROS protection efficiency and free radical-scavenging activity of curcumin. PLoS ONE 2011, 6, e26012. [Google Scholar] [CrossRef] [Green Version]
- Larasati, Y.A.; Yoneda-Kato, N.; Nakamae, I.; Yokoyama, T.; Meiyanto, E.; Kato, J.Y. Curcumin targets multiple enzymes involved in the ROS metabolic pathway to suppress tumor cell growth. Sci. Rep. 2018, 8, 2039. [Google Scholar] [CrossRef]
- Yoon, S.; Woo, S.U.; Kang, J.H.; Kim, K.; Kwon, M.H.; Park, S.; Shin, H.J.; Gwak, H.S.; Chwae, Y.J. STAT3 transcriptional factor activated by reactive oxygen species induces IL6 in starvation-induced autophagy of cancer cells. Autophagy 2010, 6, 1125–1138. [Google Scholar] [CrossRef] [Green Version]
- Seiwert, N.; Wecklein, S.; Demuth, P.; Hasselwander, S.; Kemper, T.A.; Schwerdtle, T.; Brunner, T.; Fahrer, J. Heme oxygenase 1 protects human colonocytes against ROS formation, oxidative DNA damage and cytotoxicity induced by heme iron, but not inorganic iron. Cell Death Dis. 2020, 11, 787. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hariri, M.; Millane, G.; Guimond, M.P.; Guay, G.; Dennis, J.W.; Nabi, I.R. Biogenesis of multilamellar bodies via autophagy. Mol. Biol. Cell 2000, 11, 255–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.I.; Na, H.J.; Ding, Y.; Wang, Z.; Lee, S.J.; Choi, M.E. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-β1. J. Biol. Chem. 2012, 287, 11677–11688. [Google Scholar] [CrossRef] [Green Version]
- Kawano, S.; Torisu, T.; Esaki, M.; Torisu, K.; Matsuno, Y.; Kitazono, T. Autophagy promotes degradation of internalized collagen and regulates distribution of focal adhesions to suppress cell adhesion. Biol. Open 2017, 6, 1644–1653. [Google Scholar] [CrossRef] [Green Version]
- Pathania, A.S.; Guru, S.K.; Kumar, S.; Kumar, A.; Ahmad, M.; Bhushan, S.; Sharma, P.R.; Mahajan, P.; Shah, B.A.; Sharma, S.; et al. Interplay between cell cycle and autophagy induced by boswellic acid analog. Sci. Rep. 2016, 6, 33146. [Google Scholar] [CrossRef] [Green Version]
- Calderon, M.; Lawrence, W.T.; Banes, A.J. Increased proliferation in keloid fibroblasts wounded in vitro. J. Surg. Res. 1996, 61, 343–347. [Google Scholar] [CrossRef]
- Syed, F.; Ahmadi, E.; Iqbal, S.A.; Singh, S.; McGrouther, D.A.; Bayat, A. Fibroblasts from the growing margin of keloid scars produce higher levels of collagen I and III compared with intralesional and extralesional sites: Clinical implications for lesional site-directed therapy. Br. J. Dermatol. 2011, 164, 83–96. [Google Scholar] [CrossRef]
- Zhou, Y.; Sun, Y.; Hou, W.; Ma, L.; Tao, Y.; Li, D.; Xu, C.; Bao, J.; Fan, W. The JAK2/STAT3 pathway inhibitor, AG490, suppresses the abnormal behavior of keloid fibroblasts in vitro. Int. J. Mol. Med. 2020, 46, 191–200. [Google Scholar] [CrossRef]
- Jiao, H.; Zhang, T.; Fan, J.; Xiao, R. The Superficial Dermis May Initiate Keloid Formation: Histological Analysis of the Keloid Dermis at Different Depths. Front. Physiol. 2017, 8, 885. [Google Scholar] [CrossRef]
- Ashcroft, K.J.; Syed, F.; Bayat, A. Site-specific keloid fibroblasts alter the behaviour of normal skin and normal scar fibroblasts through paracrine signalling. PLoS ONE 2013, 8, e75600. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Stark, G.R. Roles of unphosphorylated STATs in signaling. Cell Res. 2008, 18, 443–451. [Google Scholar] [CrossRef] [PubMed]
- De Felice, B.; Garbi, C.; Santoriello, M.; Santillo, A.; Wilson, R.R. Differential apoptosis markers in human keloids and hypertrophic scars fibroblasts. Mol. Cell. Biochem. 2009, 327, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.Y.; Wu, C.H.; Hong, C.H.; Chang, K.L.; Lee, C.H. GLUT-1 Enhances Glycolysis, Oxidative Stress, and Fibroblast Proliferation in Keloid. Life 2021, 11, 505. [Google Scholar] [CrossRef]
- Sampson, N.; Koziel, R.; Zenzmaier, C.; Bubendorf, L.; Plas, E.; Jansen-Dürr, P.; Berger, P. ROS signaling by NOX4 drives fibroblast-to-myofibroblast differentiation in the diseased prostatic stroma. Mol. Endocrinol. 2011, 25, 503–515. [Google Scholar] [CrossRef] [Green Version]
- Lima, C.F.; Pereira-Wilson, C.; Rattan, S.I. Curcumin induces heme oxygenase-1 in normal human skin fibroblasts through redox signaling: Relevance for anti-aging intervention. Mol. Nutr. Food Res. 2011, 55, 430–442. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Oh, J.M.; Choi, H.; Kim, S.W.; Kim, S.W.; Kim, B.G.; Cho, J.H.; Lee, J.; Lee, D.C. Activation of the Nrf2/HO-1 pathway by curcumin inhibits oxidative stress in human nasal fibroblasts exposed to urban particulate matter. BMC Complement. Med. Ther. 2020, 20, 101. [Google Scholar] [CrossRef]
- Ryter, S.W.; Choi, A.M. Heme oxygenase-1: Redox regulation of a stress protein in lung and cell culture models. Antioxid. Redox Signal. 2005, 7, 80–91. [Google Scholar] [CrossRef]
- Poss, K.D.; Tonegawa, S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10925–10930. [Google Scholar] [CrossRef] [Green Version]
- Hedblom, A.; Hejazi, S.M.; Canesin, G.; Choudhury, R.; Hanafy, K.A.; Csizmadia, E.; Persson, J.L.; Wegiel, B. Heme detoxification by heme oxygenase-1 reinstates proliferative and immune balances upon genotoxic tissue injury. Cell Death Dis. 2019, 10, 72. [Google Scholar] [CrossRef]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczepanek, K.; Lesnefsky, E.J.; Larner, A.C. Multi-tasking: Nuclear transcription factors with novel roles in the mitochondria. Trends Cell Biol. 2012, 22, 429–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, H.; McCauley, R.L.; Zhang, W. Elevated interleukin-6 expression in keloid fibroblasts. J. Surg. Res. 2000, 89, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Ghazizadeh, M.; Tosa, M.; Shimizu, H.; Hyakusoku, H.; Kawanami, O. Functional implications of the IL-6 signaling pathway in keloid pathogenesis. J. Investig. Dermatol. 2007, 127, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Qin, B.; Zhou, Z.; He, J.; Yan, C.; Ding, S. IL-6 inhibits starvation-induced autophagy via the STAT3/Bcl-2 signaling pathway. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.Y.; Yi, C.G.; Li, X.; Ma, B.; Li, Z.J.; Chen, X.L.; Guo, S.Z.; Gao, W.Y. Troglitazone suppresses transforming growth factor-beta1-induced collagen type I expression in keloid fibroblasts. Br. J. Dermatol. 2009, 160, 762–770. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, Y.-K.; Wu, C.-H.; Lin, Y.-C.; Huang, Y.-L.; Hung, K.-S.; Pai, T.-P.; Liu, Y.-T.; Chen, T.-C.; Chan, H.; Hsu, C.-K. ASC-J9 Blocks Cell Proliferation and Extracellular Matrix Production of Keloid Fibroblasts through Inhibiting STAT3 Signaling. Int. J. Mol. Sci. 2022, 23, 5549. https://doi.org/10.3390/ijms23105549
Hong Y-K, Wu C-H, Lin Y-C, Huang Y-L, Hung K-S, Pai T-P, Liu Y-T, Chen T-C, Chan H, Hsu C-K. ASC-J9 Blocks Cell Proliferation and Extracellular Matrix Production of Keloid Fibroblasts through Inhibiting STAT3 Signaling. International Journal of Molecular Sciences. 2022; 23(10):5549. https://doi.org/10.3390/ijms23105549
Chicago/Turabian StyleHong, Yi-Kai, Chen-Han Wu, Yu-Chen Lin, Yu-Lun Huang, Kuo-Shu Hung, Tsung-Pin Pai, Yen-Ting Liu, Tzu-Chi Chen, Hardy Chan, and Chao-Kai Hsu. 2022. "ASC-J9 Blocks Cell Proliferation and Extracellular Matrix Production of Keloid Fibroblasts through Inhibiting STAT3 Signaling" International Journal of Molecular Sciences 23, no. 10: 5549. https://doi.org/10.3390/ijms23105549
APA StyleHong, Y.-K., Wu, C.-H., Lin, Y.-C., Huang, Y.-L., Hung, K.-S., Pai, T.-P., Liu, Y.-T., Chen, T.-C., Chan, H., & Hsu, C.-K. (2022). ASC-J9 Blocks Cell Proliferation and Extracellular Matrix Production of Keloid Fibroblasts through Inhibiting STAT3 Signaling. International Journal of Molecular Sciences, 23(10), 5549. https://doi.org/10.3390/ijms23105549