
Time-Series Clustering of lncRNA-mRNA Expression during the Adipogenic Transdifferentiation of Porcine Skeletal Muscle Satellite Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation, Culture, and Identification of Porcine SMSCs
2.2. Adipogenesis Induction and Sample Collection
2.3. RNA Sequencing
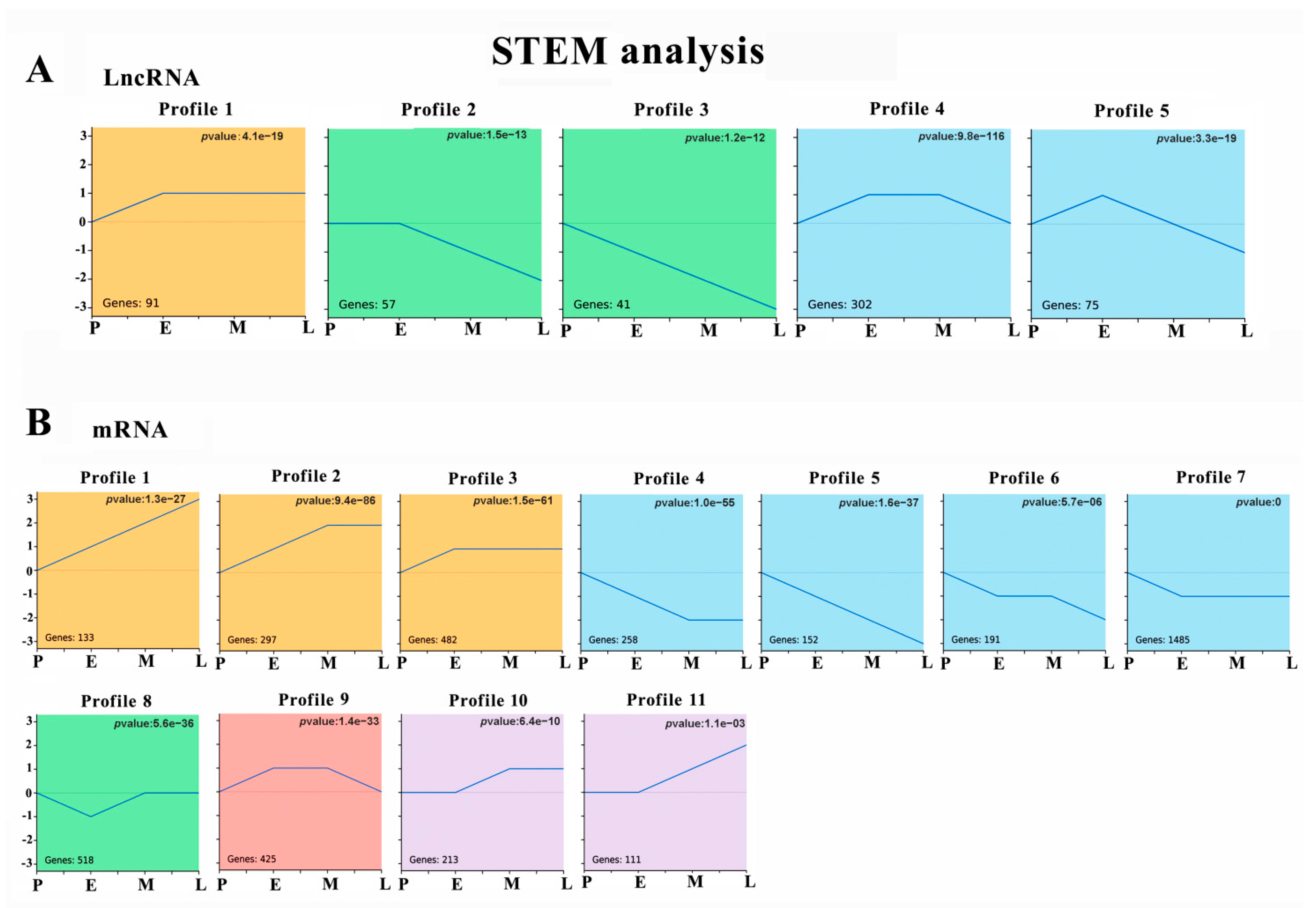
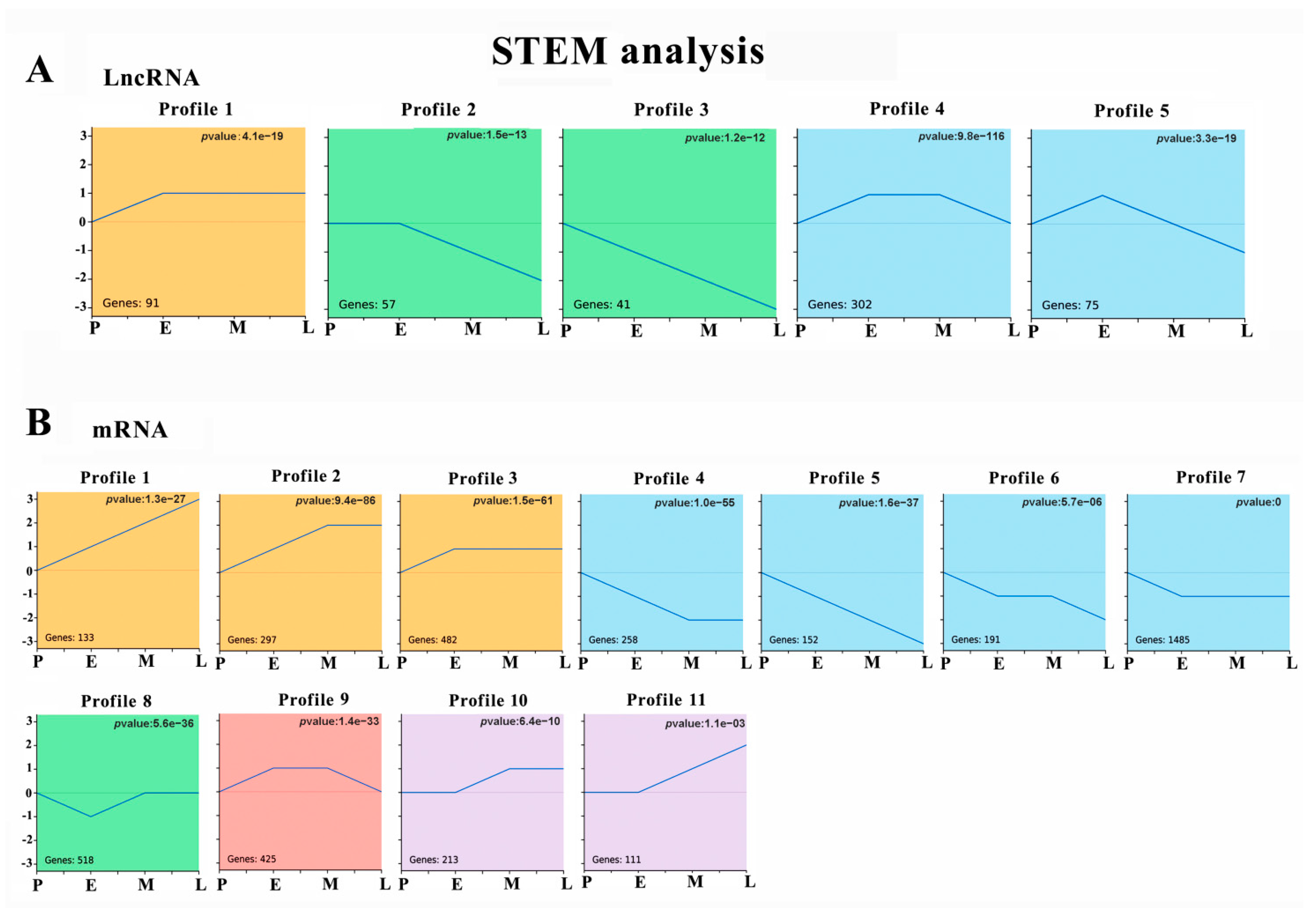
2.4. STEM Analysis and Enrichment Analysis
2.5. Protein-Protein Interaction (PPI) Network Construction
2.6. Target Gene s of DE lncRNAs-mRNA
2.7. Quantitative Real-Time PCR
2.8. Statistical Analysis
3. Results
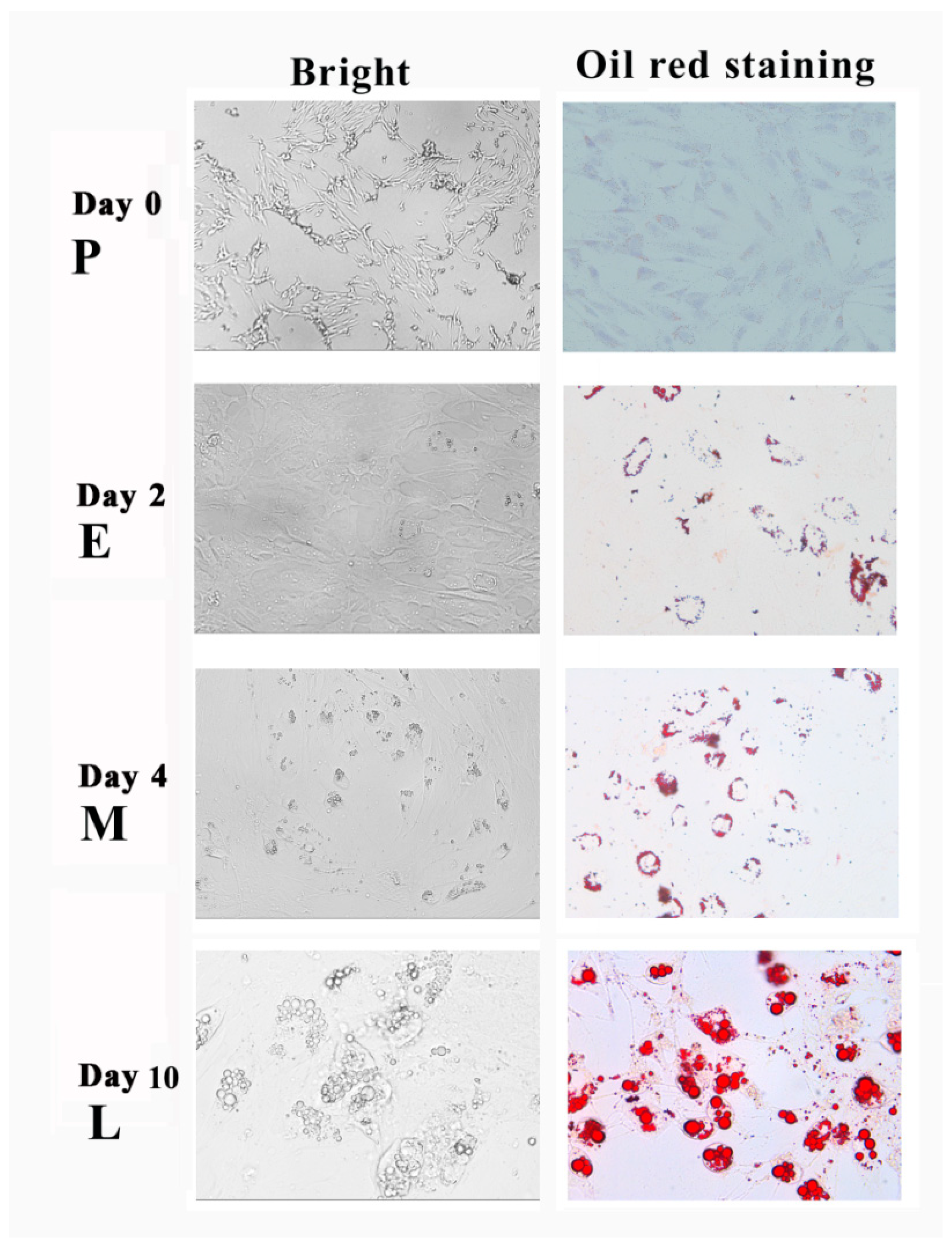
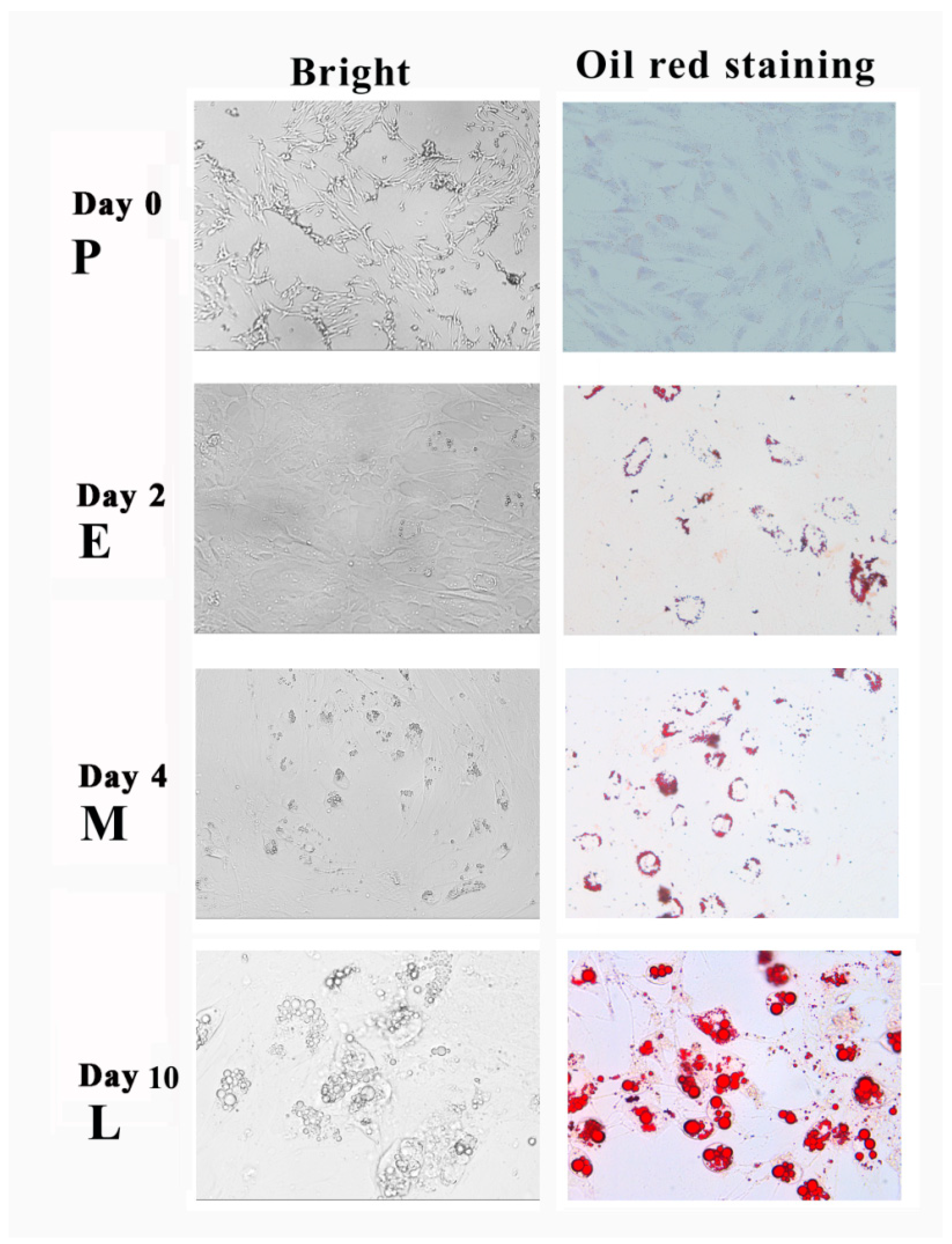
3.1. Adipogenic Transdifferentiation of SMSCs
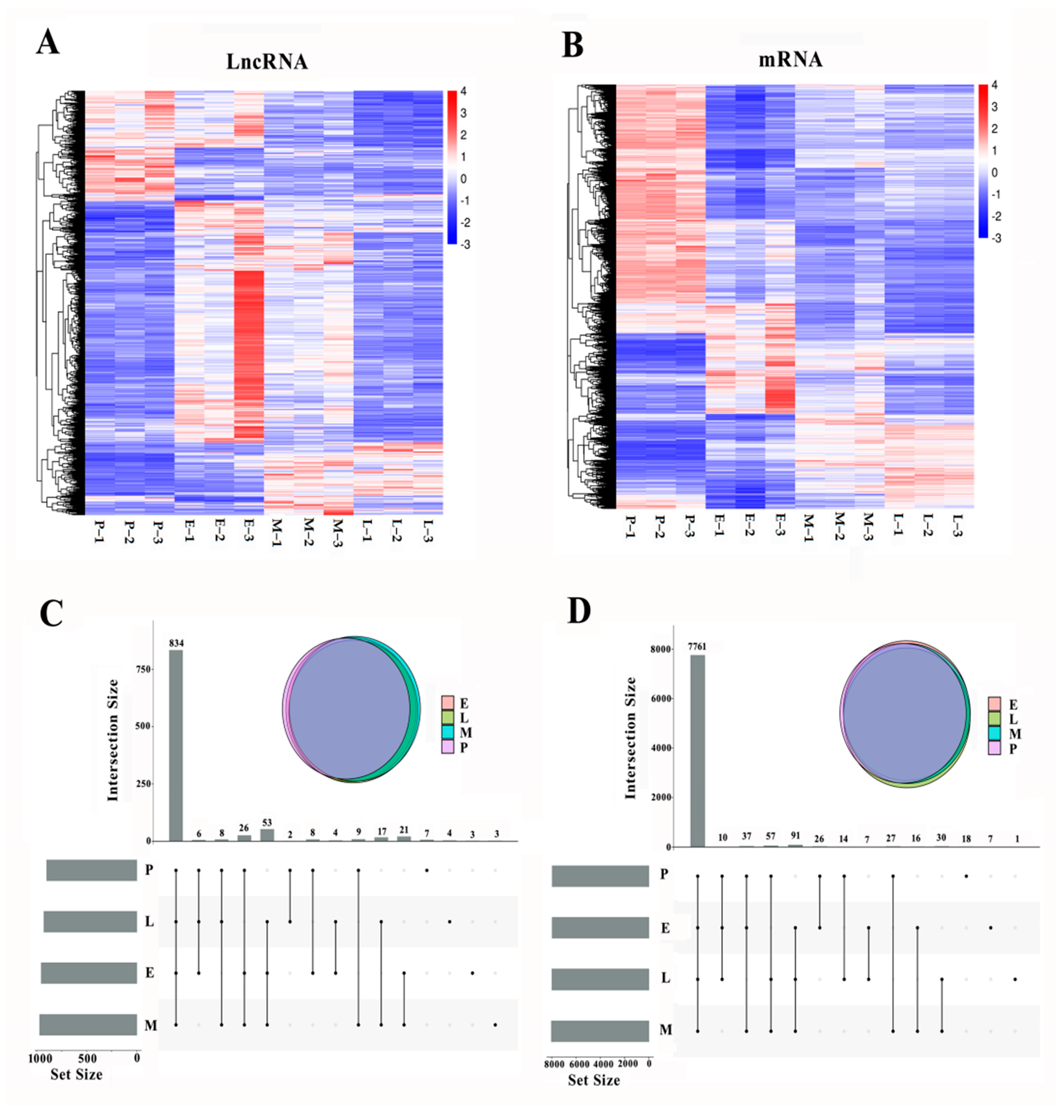
3.2. Summary of Sequencing and Gene Expression Analysis
3.3. Temporal Gene Expression Patterns of DE lncRNAs and DE mRNAs
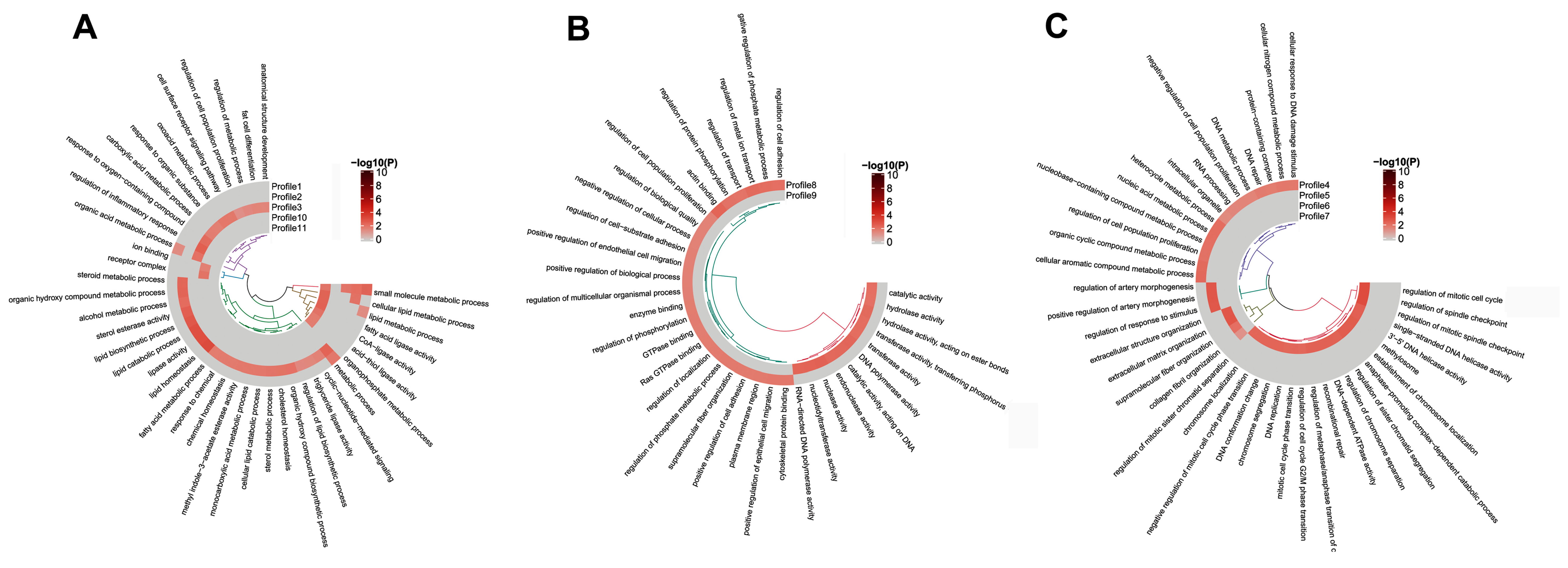
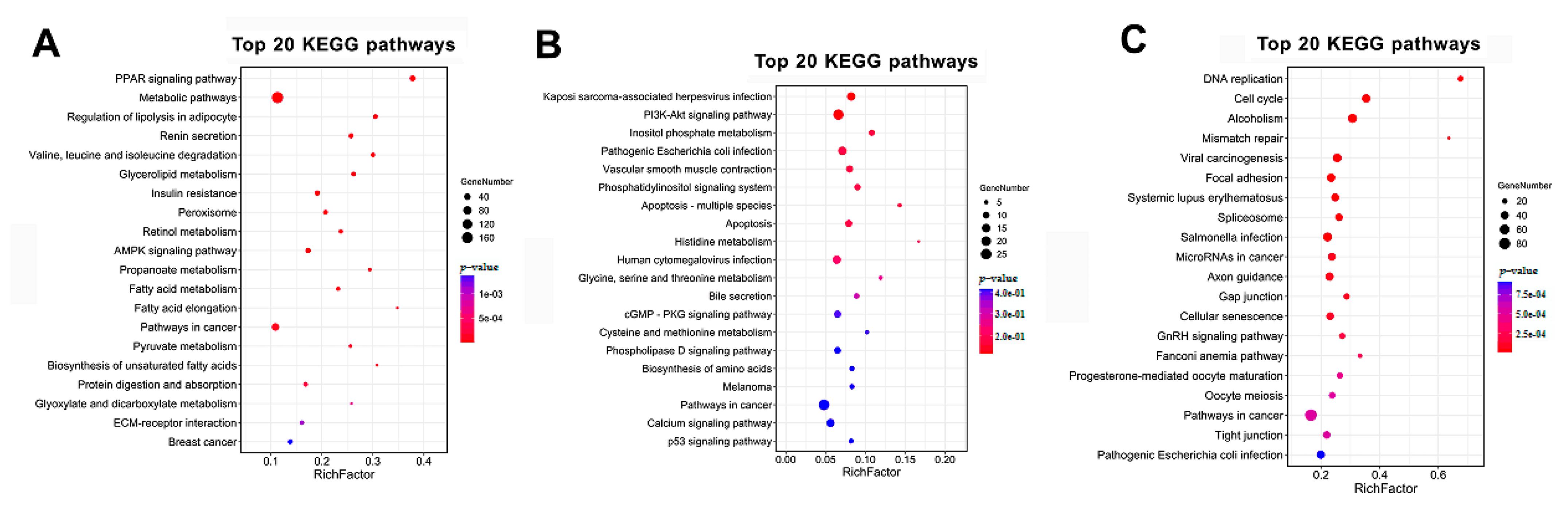
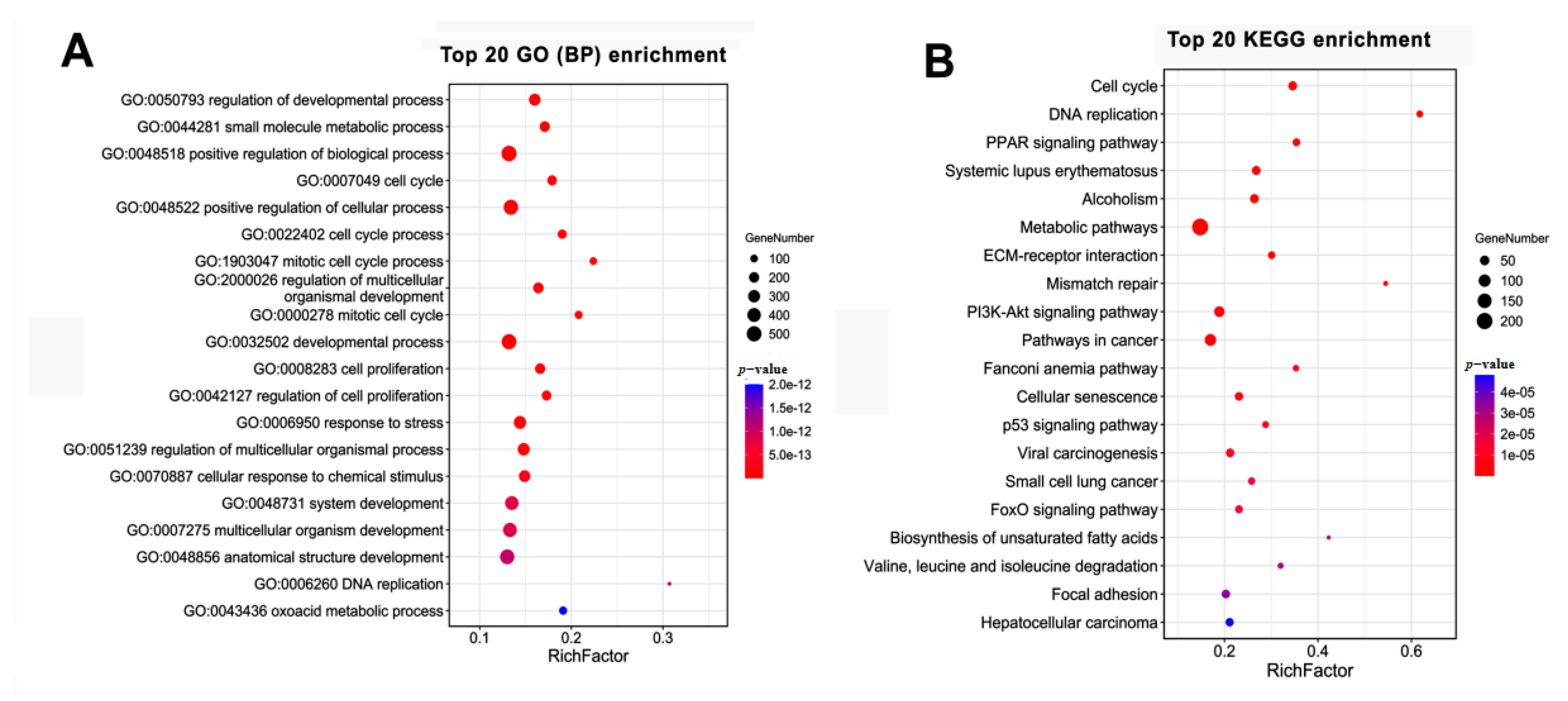
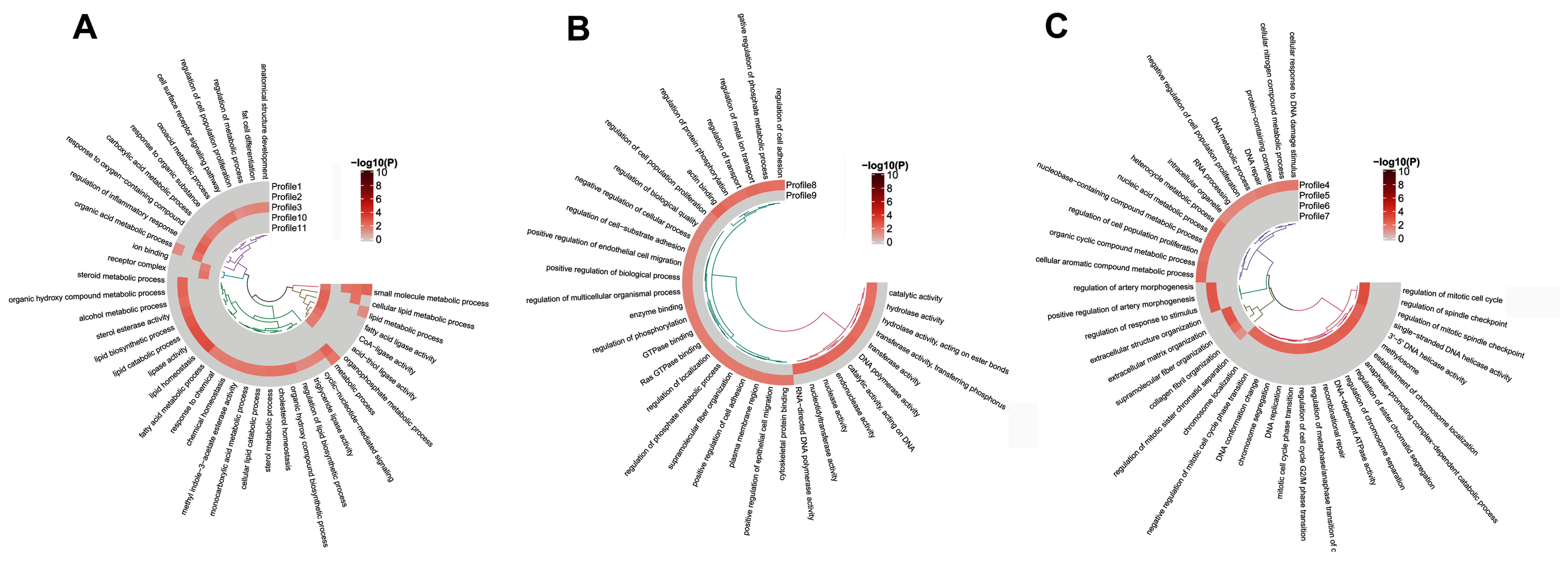
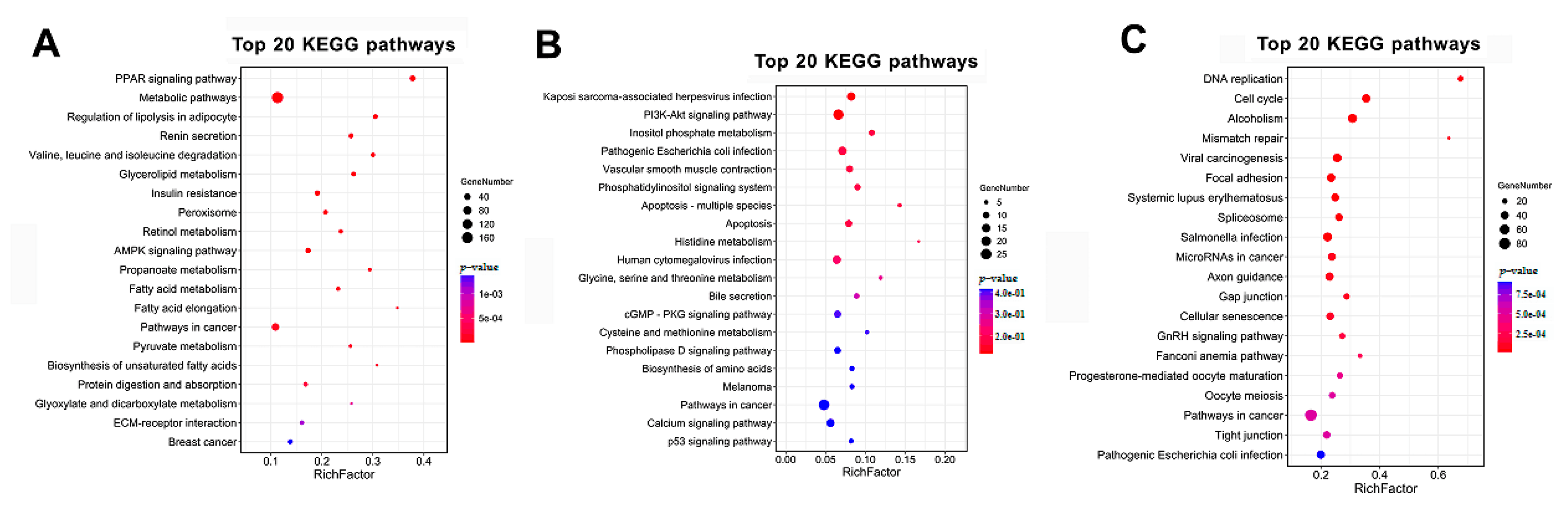
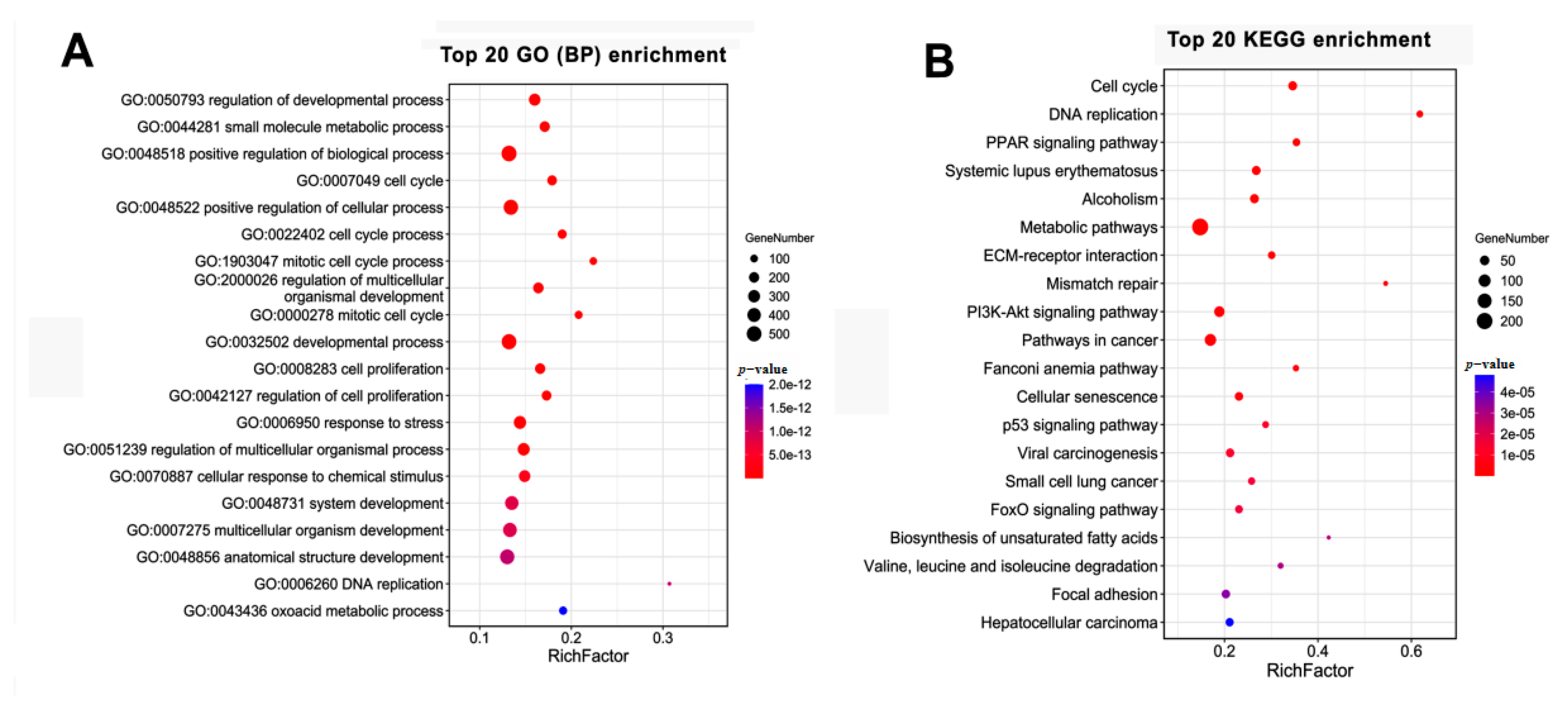
3.4. GO and KEGG Analysis of the Temporal Expressed DE mRNAs
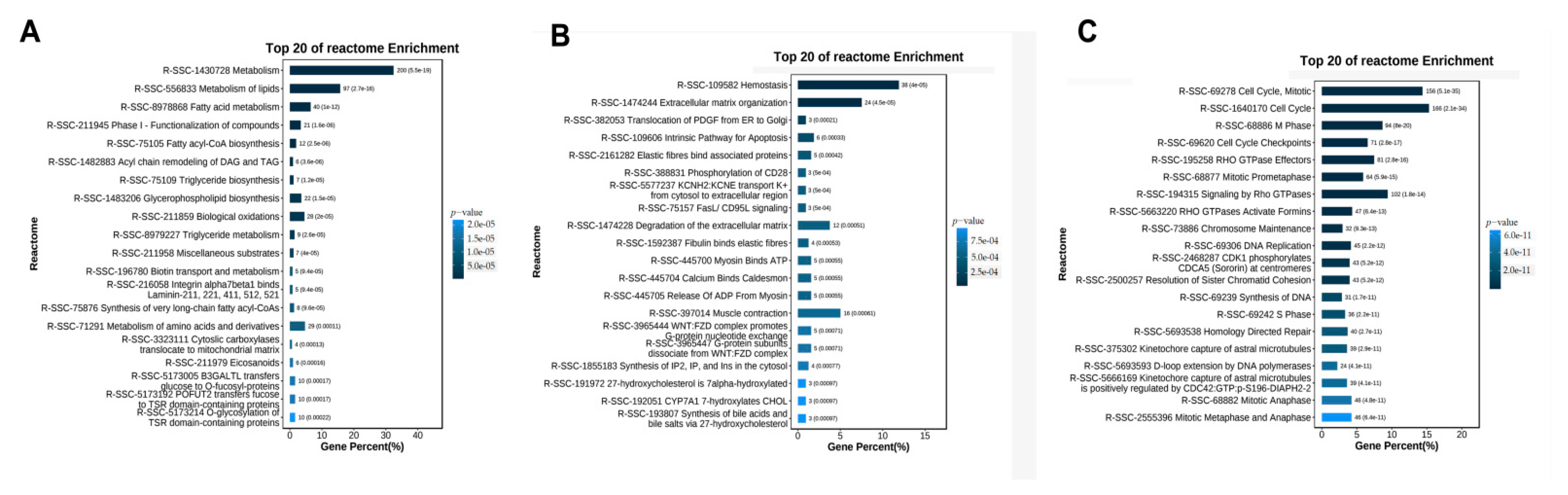
3.5. REACTOME Pathways Analysis of the Temporal Expressed DE mRNAs
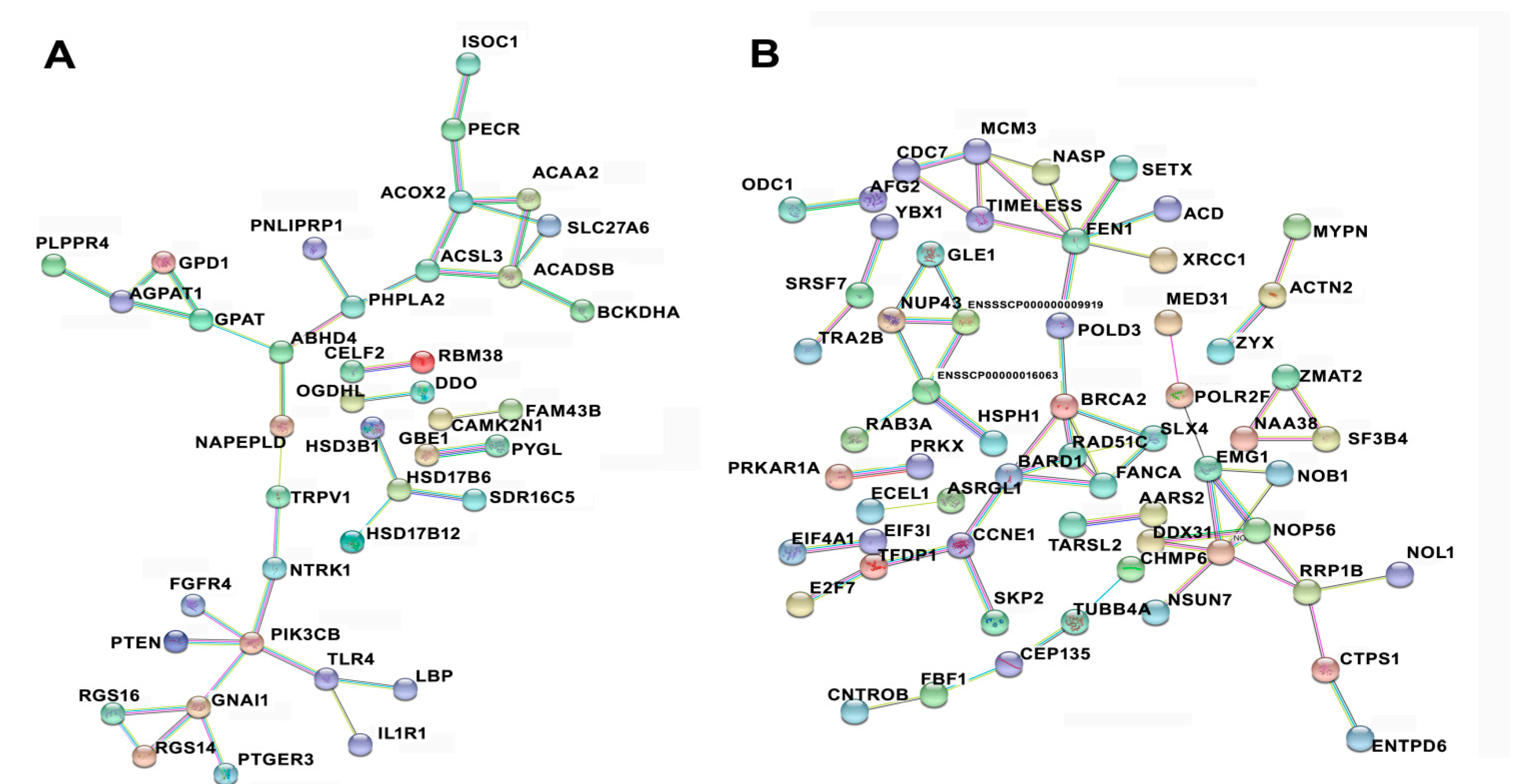
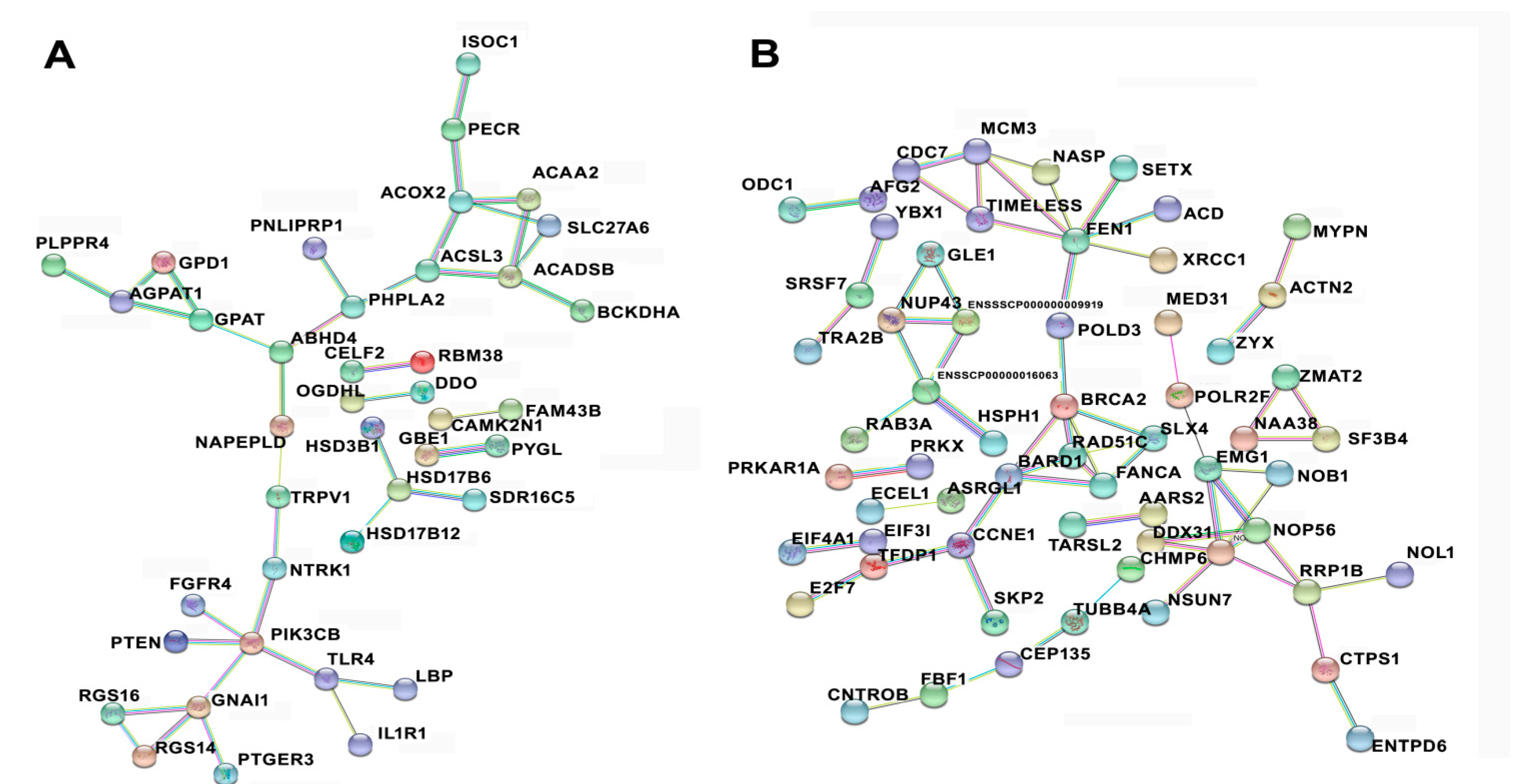
3.6. Protein–Protein Interaction among Temporal Expressed DE mRNAs Related to Lipid Metabolism and Cell Cycle
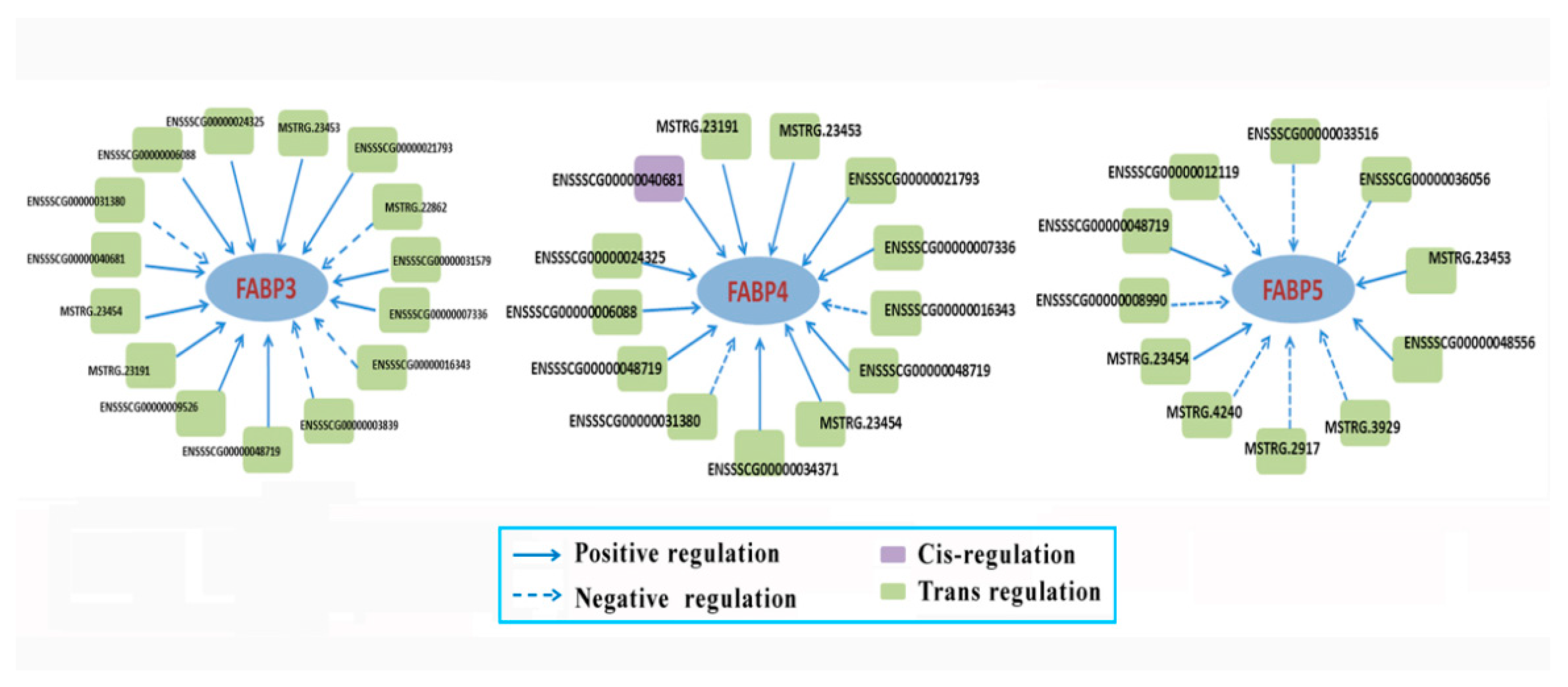
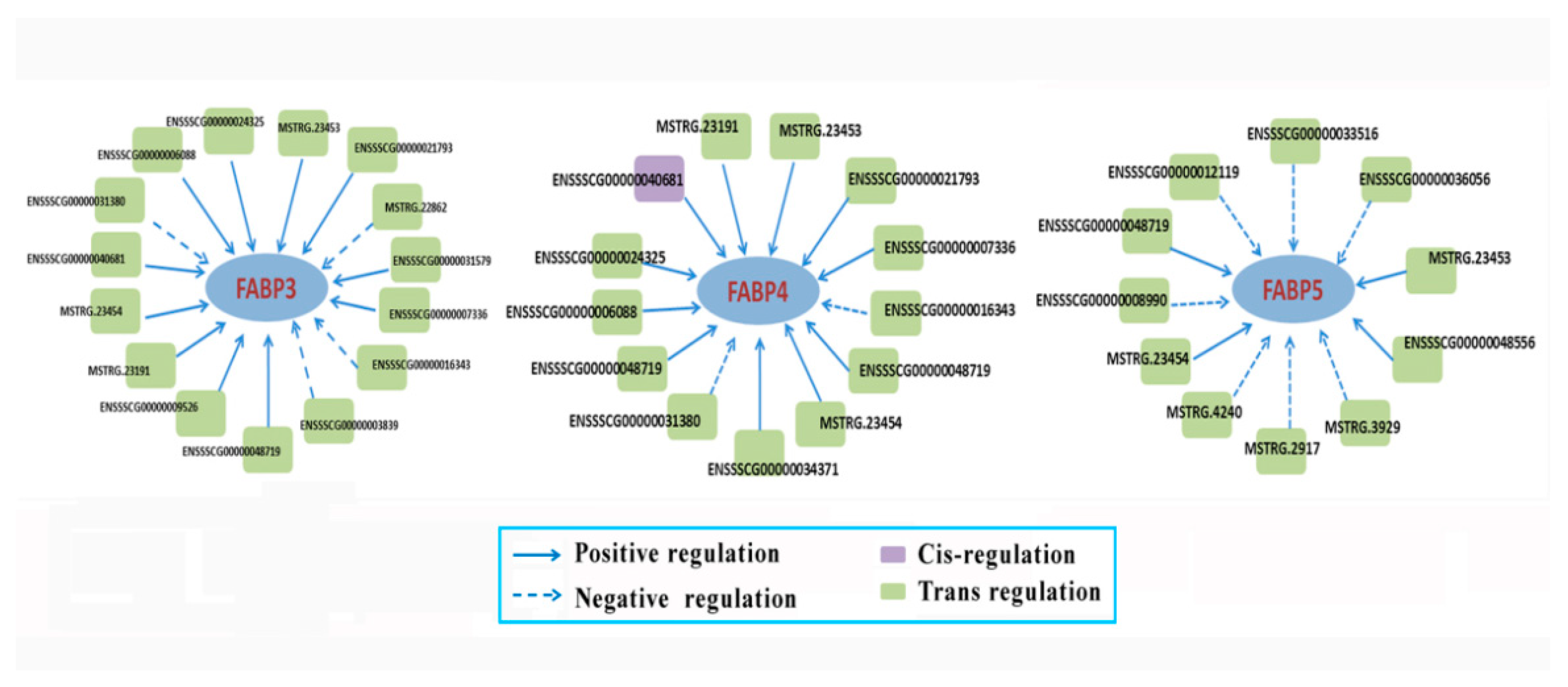
3.7. The Targeted mRNAs of DE lncRNAs
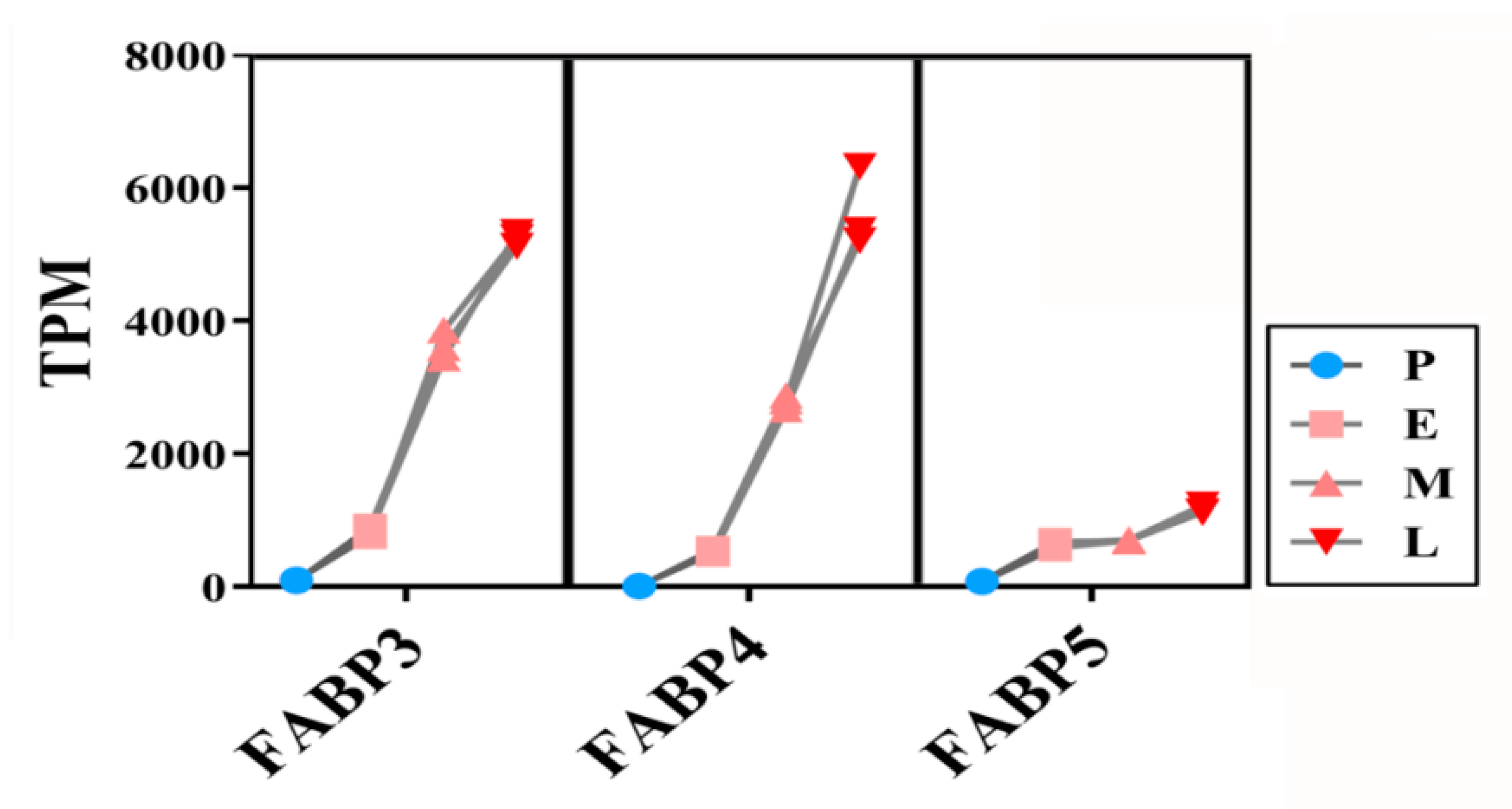
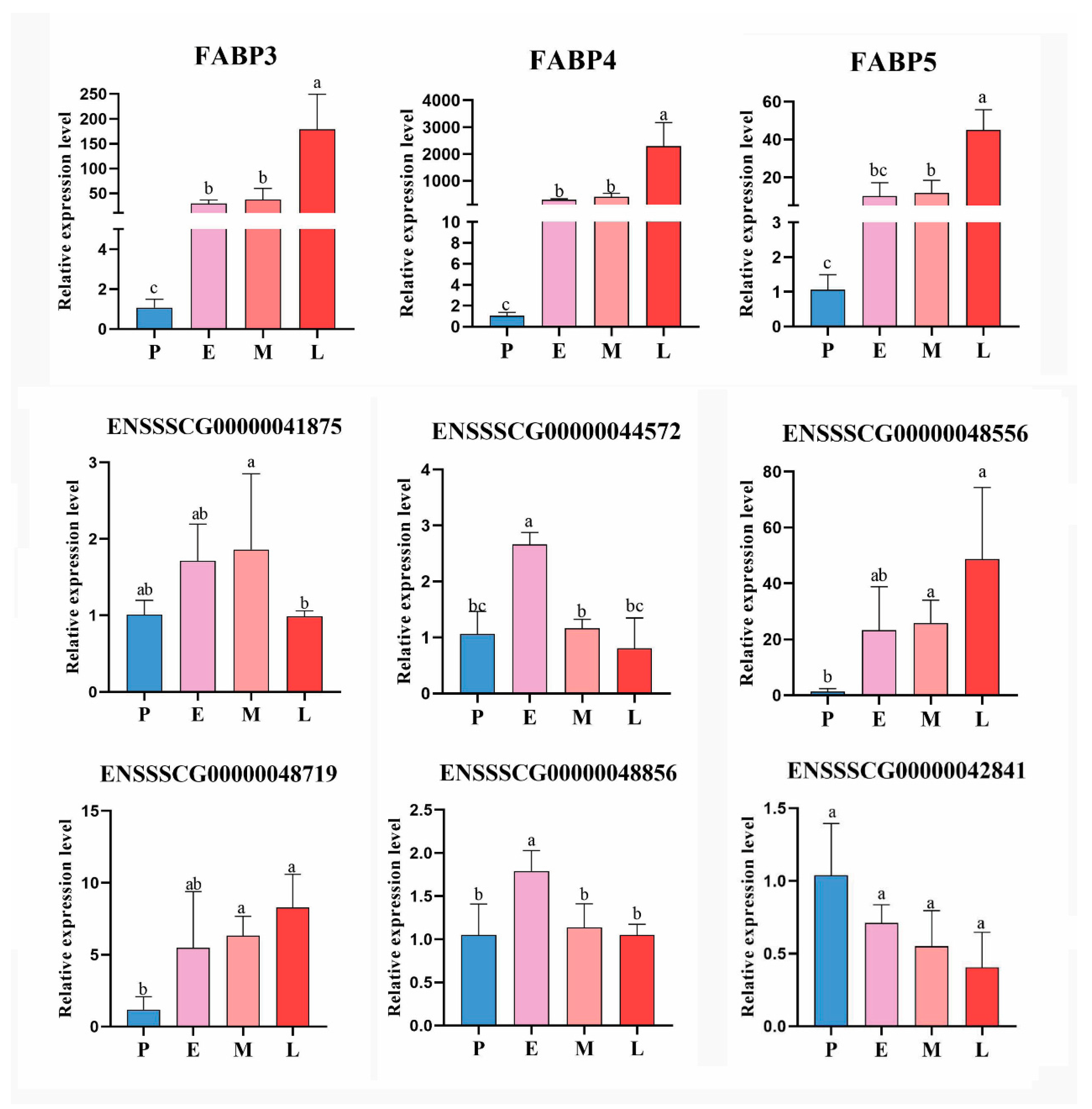
3.8. Expressions of FABP Family Genes
3.9. Verification of the Reliability of Sequencing Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Redshaw, Z.; McOrist, S.; Loughna, P. Muscle origin of porcine satellite cells affects in vitro differentiation potential. Cell Biochem. Funct. 2010, 28, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Yada, E.; Yamanouchi, K.; Nishihara, M. Adipogenic potential of satellite cells from distinct skeletal muscle origins in the rat. J. Vet. Med. Sci. 2006, 68, 479–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.J.; Lee, H.J.; Kamli, M.R.; Pokharel, S.; Bhat, A.R.; Lee, Y.H.; Choi, B.H.; Chun, T.; Kang, S.W.; Lee, Y.S.; et al. Depot-specific gene expression profiles during differentiation and transdifferentiation of bovine muscle satellite cells, and differentiation of preadipocytes. Genomics 2012, 100, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Coppi, P.; Milan, G.; Scarda, A.; Boldrin, L.; Centobene, C.; Piccoli, M.; Pozzobon, M.; Pilon, C.; Pagano, C.; Gamba, P.; et al. Rosiglitazone modifies the adipogenic potential of human muscle satellite cells. Diabetologia 2006, 49, 1962–1973. [Google Scholar] [CrossRef] [Green Version]
- Vettor, R.; Milan, G.; Franzin, C.; Sanna, M.; De Coppi, P.; Rizzuto, R.; Federspil, G. The origin of intermuscular adipose tissue and its pathophysiological implications. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E987–E998. [Google Scholar] [CrossRef]
- Asakura, A.; Komaki, M.; Rudnicki, M. Muscle satellite cells are multipotential stem cells that exhibit myogenic, osteogenic, and adipogenic differentiation. Differentiation 2001, 68, 245–253. [Google Scholar] [CrossRef]
- Teboul, L.; Gaillard, D.; Staccini, L.; Inadera, H.; Amri, E.Z.; Grimaldi, P.A. Thiazolidinediones and fatty acids convert myogenic cells into adipose-like cells. J. Biol. Chem. 1995, 270, 28183–28187. [Google Scholar] [CrossRef] [Green Version]
- Scarda, A.; Franzin, C.; Milan, G.; Sanna, M.; Dal Pra, C.; Pagano, C.; Boldrin, L.; Piccoli, M.; Trevellin, E.; Granzotto, M.; et al. Increased adipogenic conversion of muscle satellite cells in obese Zucker rats. Int. J. Obes. 2010, 34, 1319–1327. [Google Scholar] [CrossRef] [Green Version]
- Plikus, M.V.; Guerrero-Juarez, C.F.; Ito, M.; Li, Y.R.; Dedhia, P.H.; Zheng, Y.; Shao, M.; Gay, D.L.; Ramos, R.; Hsi, T.C.; et al. Regeneration of fat cells from myofibroblasts during wound healing. Science 2017, 355, 748–752. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Li, P.; Ling, H.; Xu, Z.; Yi, B.; Zhu, S. MiR-499/PRDM16 axis modulates the adipogenic differentiation of mouse skeletal muscle satellite cells. Hum. Cell 2018, 31, 282–291. [Google Scholar] [CrossRef]
- Hou, Y.; Fu, L.; Li, J.; Li, J.; Zhao, Y.; Luan, Y.; Liu, A.; Liu, H.; Li, X.; Zhao, S.; et al. Transcriptome Analysis of Potential miRNA Involved in Adipogenic Differentiation of C2C12 Myoblasts. Lipids 2018, 53, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Tang, H.; Liu, X.; Zhou, M.; Zhang, J.; Tang, K. Dickkopf1 Up-Regulation Induced by a High Concentration of Dexamethasone Promotes Rat Tendon Stem Cells to Differentiate into Adipocytes. Cell. Physiol. Biochem. 2015, 37, 1738–1749. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, H.; Liu, R.; Jin, L.; Tang, Q.; Wang, X.; Jiang, A.; Hu, Y.; Li, Z.; Zhu, L.; et al. The miRNA Transcriptome Directly Reflects the Physiological and Biochemical Differences between Red, White, and Intermediate Muscle Fiber Types. Int. J. Mol. Sci. 2015, 16, 9635–9653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, D.; Zou, T.; Gai, X.; Ma, J.; Li, M.; Huang, Z.; Chen, D. MicroRNA expression profiles differ between primary myofiber of lean and obese pig breeds. PLoS ONE 2017, 12, e0181897. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.; Qiu, X.; Zhang, Y.; Wang, J.; Wang, Q.; Wu, M.; Huang, J.; Yang, F. Comparison of LncRNA Expression Profiles during Myogenic Differentiation and Adipogenic Transdifferentiation of Myoblasts. Int. J. Mol. Sci. 2019, 20, 3725. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; He, L.; Zhao, Y.; Li, Y.; Zhang, S.; Sun, K.; So, K.; Chen, F.; Zhou, L.; Lu, L.; et al. Malat1 regulates myogenic differentiation and muscle regeneration through modulating MyoD transcriptional activity. Cell Discov. 2017, 3, 17002. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, Y.; Li, T.; Ma, Z.; Jia, H.; Chen, Q.; Zhao, Y.; Zhai, L.; Zhong, R.; Li, C.; et al. Long non-coding RNA Linc-RAM enhances myogenic differentiation by interacting with MyoD. Nat. Commun. 2017, 8, 14016–14028. [Google Scholar] [CrossRef] [Green Version]
- Musaro, A.; Carosio, S. Isolation and Culture of Satellite Cells from Mouse Skeletal Muscle. Methods Mol. Biol. 2017, 1553, 155–167. [Google Scholar] [CrossRef]
- Ernst, J.; Nau, G.J.; Bar-Joseph, Z. Clustering short time series gene expression data. Bioinformatics 2005, 21 (Suppl. S1), i159–i168. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob. H. Reactome pathway analysis: A highperformance in-memory approach. BMC Bioinform. 2017, 18, 142–158. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.L.; Liu, B.; Halls, M.L.; Wagstaff, K.M.; Patil, R.; Velkov, T.; Jans, D.A.; Bunnett, N.W.; Scanlon, M.J.; Porter, C.J. Fatty Acid-binding Proteins 1 and 2 Differentially Modulate the Activation of Peroxisome Proliferator-activated Receptor alpha in a Ligand-selective Manner. J. Biol. Chem. 2015, 290, 13895–13906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajda, A.M.; Storch, J. Enterocyte fatty acid-binding proteins (FABPs): Different functions of liver and intestinal FABPs in the intestine. Prostaglandins Leukot. Essent. Fat. Acids 2015, 93, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Song, X.; Glass, C.K.; Rosenfeld, M.G. The long arm of long noncoding RNAs: Roles as sensors regulating gene transcriptional programs. Cold Spring Harb. Perspect. Biol. 2011, 3, 3756–3772. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhao, Y.; Bao, X.; Zhu, X.; Kwok, Y.K.; Sun, K.; Chen, X.; Huang, Y.; Jauch, R.; Esteban, M.A.; et al. LncRNA Dum interacts with Dnmts to regulate Dppa2 expression during myogenic differentiation and muscle regeneration. Cell Res. 2015, 25, 335–350. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.; Li, Z.; Ramanujan, K.; Clay, I.; Zhang, Y.; Lemire-Brachat, S.; Glass, D.J. A long non-coding RNA, LncMyoD, regulates skeletal muscle differentiation by blocking IMP2-mediated mRNA translation. Dev. Cell 2015, 34, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Mueller, A.C.; Cichewicz, M.A.; Dey, B.K.; Layer, R.; Reon, B.J.; Gagan, J.R.; Dutta, A. MUNC, a long noncoding RNA that facilitates the function of MyoD in skeletal myogenesis. Mol. Cell. Biol. 2015, 35, 498–513. [Google Scholar] [CrossRef] [Green Version]
- Dong, A.; Preusch, C.B.; So, W.K.; Lin, K.; Luan, S.; Yi, R.; Wong, J.W.; Wu, Z.; Cheung, T.H. A long noncoding RNA, LncMyoD, modulates chromatin accessibility to regulate muscle stem cell myogenic lineage progression. Proc. Natl. Acad. Sci. USA 2020, 117, 32464–32475. [Google Scholar] [CrossRef]
- Zhou, Y.; Cheunsuchon, P.; Nakayama, Y.; Lawlor, M.W.; Zhong, Y.; Rice, K.A.; Zhang, L.; Zhang, X.; Gordon, F.E.; Lidov, H.G.; et al. Activation of paternally expressed genes and perinatal death caused by deletion of the Gtl2 gene. Development 2010, 137, 2643–2652. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Li, M.; Sun, Y.; Cai, H.; Lan, X.; Huang, Y.; Bai, Y.; Qi, X.; Chen, H. The developmental transcriptome sequencing of bovine skeletal muscle reveals a long noncoding RNA, lncMD, promotes muscle differentiation by sponging miR-125b. Biochim. Biophys. Acta 2016, 1863, 2835–2845. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.F.; Li, Y.; Ding, X.B.; Li, X.; Zhang, L.L.; Liu, X.F.; Guo, H. lnc133b, a novel, long non-coding RNA, regulates bovine skeletal muscle satellite cell proliferation and differentiation by mediating miR-133b. Gene 2017, 630, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Cai, B.; Jiang, L.; Abdalla, B.A.; Li, Z.; Nie, Q.; Zhang, X. lncRNA-Six1 Is a Target of miR-1611 that Functions as a ceRNA to Regulate Six1 Protein Expression and Fiber Type Switching in Chicken Myogenesis. Cells 2018, 7, 243. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Wu, M.; Wang, C.; Liu, R.; Zhao, H.; Yang, L.; Liu, J.; Wang, Y.; Zhang, S.; Yuan, Z.; et al. Long Noncoding RNA Lnc-SEMT Modulates IGF2 Expression by Sponging miR-125b to Promote Sheep Muscle Development and Growth. Cell. Physiol. Biochem. 2018, 49, 447–462. [Google Scholar] [CrossRef]
- Li, Z.; Cai, B.; Abdalla, B.A.; Zhu, X.; Zheng, M.; Han, P.; Nie, Q.; Zhang, X. LncIRS1 controls muscle atrophy via sponging miR-15 family to activate IGF1-PI3K/AKT pathway. J. Cachexia Sarcopenia Muscle 2019, 10, 391–410. [Google Scholar] [CrossRef] [Green Version]
- Yeow, K.; Phillips, B.; Dani, C.; Cabane, C.; Amri, E.Z.; Derijard, B. Inhibition of myogenesis enables adipogenic trans-differentiation in the C2C12 myogenic cell line. FEBS Lett. 2001, 506, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Tachibana, H.; Morinaga, Y.; Fujimura, Y.; Yamada, K. Modulation of proliferation and differentiation of C2C12 skeletal muscle cells by fatty acids. Life Sci. 2009, 84, 415–420. [Google Scholar] [CrossRef]
- O’Neill, B.T.; Lauritzen, H.P.; Hirshman, M.F.; Smyth, G.; Goodyear, L.J.; Kahn, C.R. Differential Role of Insulin/IGF-1 Receptor Signaling in Muscle Growth and Glucose Homeostasis. Cell Rep. 2015, 11, 1220–1235. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Hao, J.; Zeng, J.; Sauter, E.R. SnapShot: FABP Functions. Cell 2020, 182, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.H.; Kim, M.J.; Jeon, G.J.; Chung, H.Y. Association of genetic variants for FABP3 gene with back fat thickness and intramuscular fat content in pig. Mol. Biol. Rep. 2011, 38, 2161–2166. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Borrmann, C.M.; Borchers, T.; Spener, F. Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors alpha—And gamma-mediated gene expression via liver fatty acid binding protein: A signaling path to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 2323–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, R.; Miquel, S.; Ulmer, J.; Langella, P.; Bermudez-Humaran, L.G. Gut ecosystem: How microbes help us. Benef. Microbes 2014, 5, 219–233. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, X.; Gao, G.; Du, L.; Wang, J.; Wang, Q.; Yang, F.; Zhou, X.; Long, D.; Huang, J.; Liu, Z.; et al. Time-Series Clustering of lncRNA-mRNA Expression during the Adipogenic Transdifferentiation of Porcine Skeletal Muscle Satellite Cells. Curr. Issues Mol. Biol. 2022, 44, 2038-2053. https://doi.org/10.3390/cimb44050138
Qiu X, Gao G, Du L, Wang J, Wang Q, Yang F, Zhou X, Long D, Huang J, Liu Z, et al. Time-Series Clustering of lncRNA-mRNA Expression during the Adipogenic Transdifferentiation of Porcine Skeletal Muscle Satellite Cells. Current Issues in Molecular Biology. 2022; 44(5):2038-2053. https://doi.org/10.3390/cimb44050138
Chicago/Turabian StyleQiu, Xiaoyu, Guangliang Gao, Lei Du, Jing Wang, Qi Wang, Feiyun Yang, Xiaorong Zhou, Dingbiao Long, Jinxiu Huang, Zuohua Liu, and et al. 2022. "Time-Series Clustering of lncRNA-mRNA Expression during the Adipogenic Transdifferentiation of Porcine Skeletal Muscle Satellite Cells" Current Issues in Molecular Biology 44, no. 5: 2038-2053. https://doi.org/10.3390/cimb44050138
APA StyleQiu, X., Gao, G., Du, L., Wang, J., Wang, Q., Yang, F., Zhou, X., Long, D., Huang, J., Liu, Z., & Qi, R. (2022). Time-Series Clustering of lncRNA-mRNA Expression during the Adipogenic Transdifferentiation of Porcine Skeletal Muscle Satellite Cells. Current Issues in Molecular Biology, 44(5), 2038-2053. https://doi.org/10.3390/cimb44050138