
Population Structure and Genetic Diversity Analysis in Sugarcane (Saccharum spp. hybrids) and Six Related Saccharum Species




Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Extraction
2.3. SSR Markers and PCR Reactions
2.4. Marker Scoring
2.5. Structure and Genetic Diversity Analysis
2.6. Species-Specific and Species-Associated SSR Allele
2.7. Phylogenetic Analysis of the Sugarcane Accessions Using Species Associated SSR Alleles
3. Results
3.1. SSR Alleles
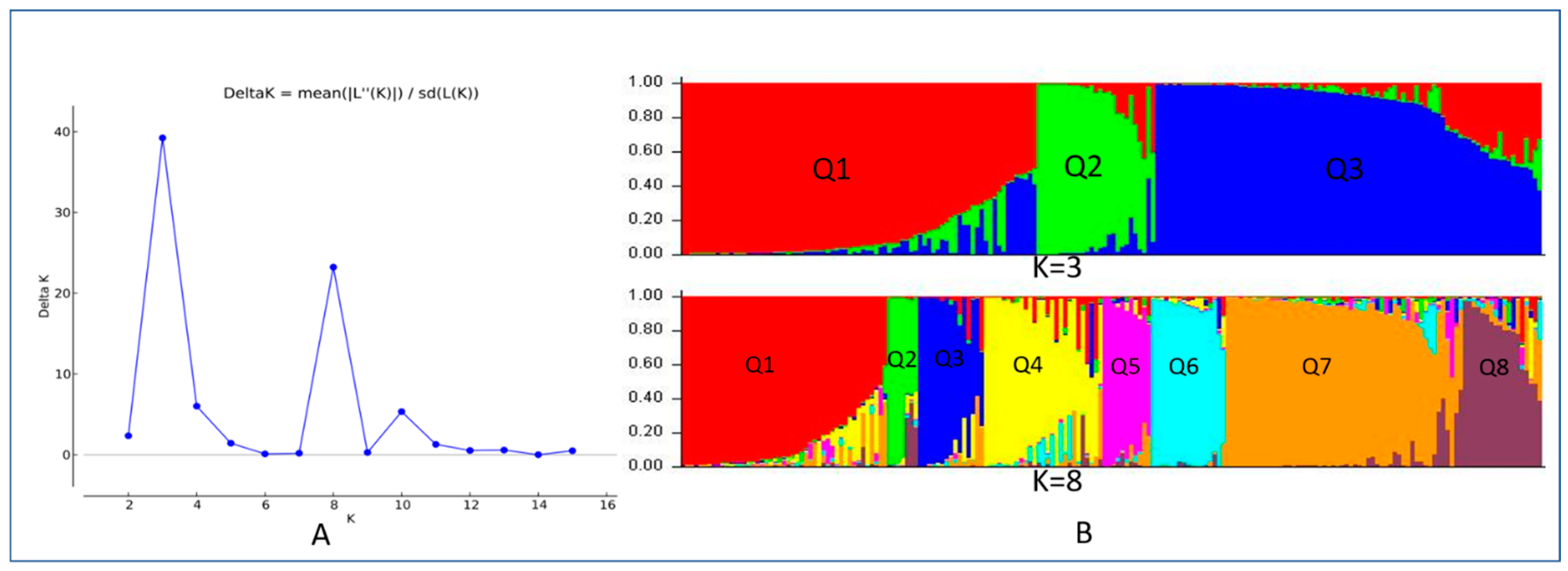
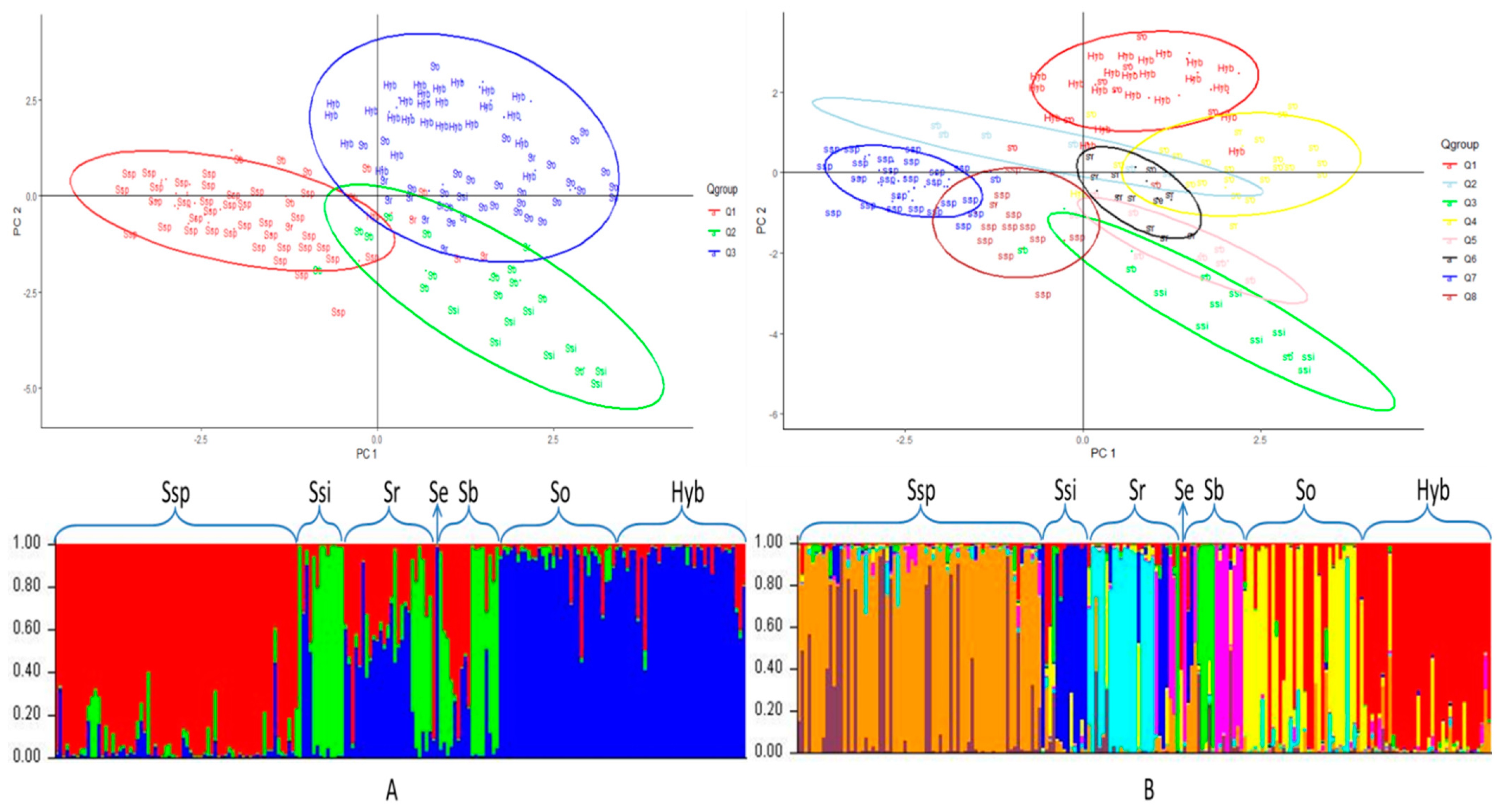
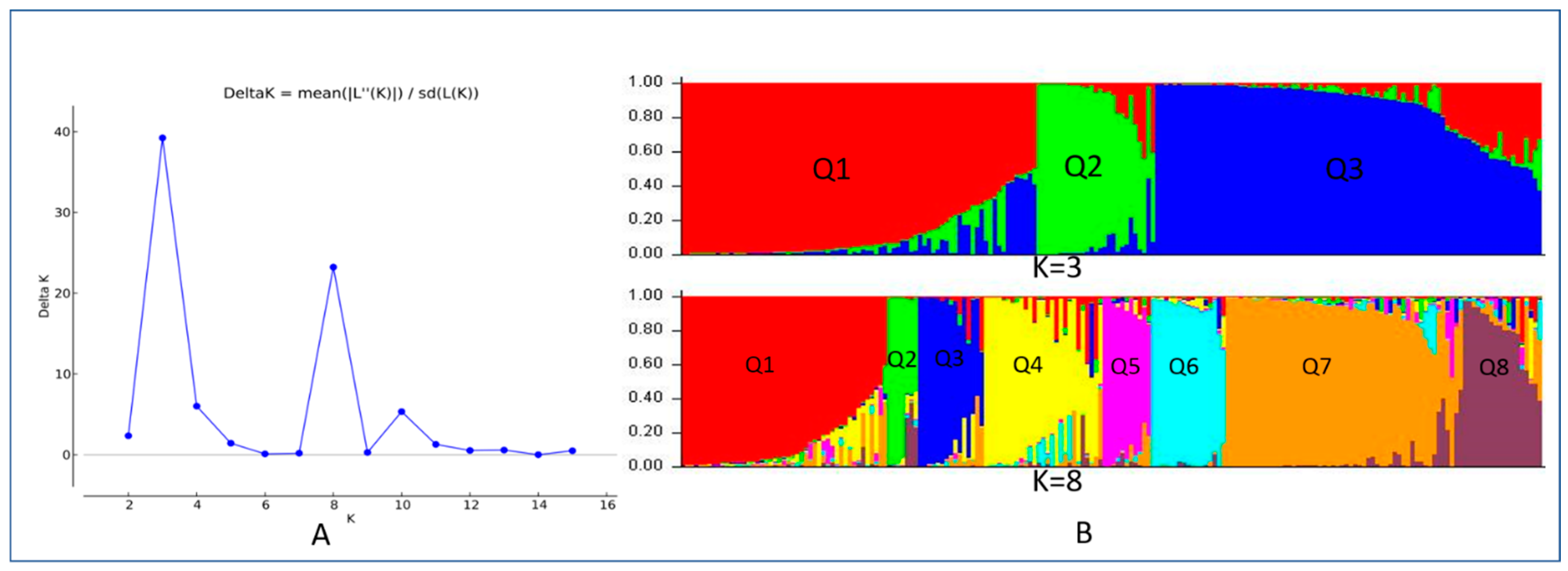
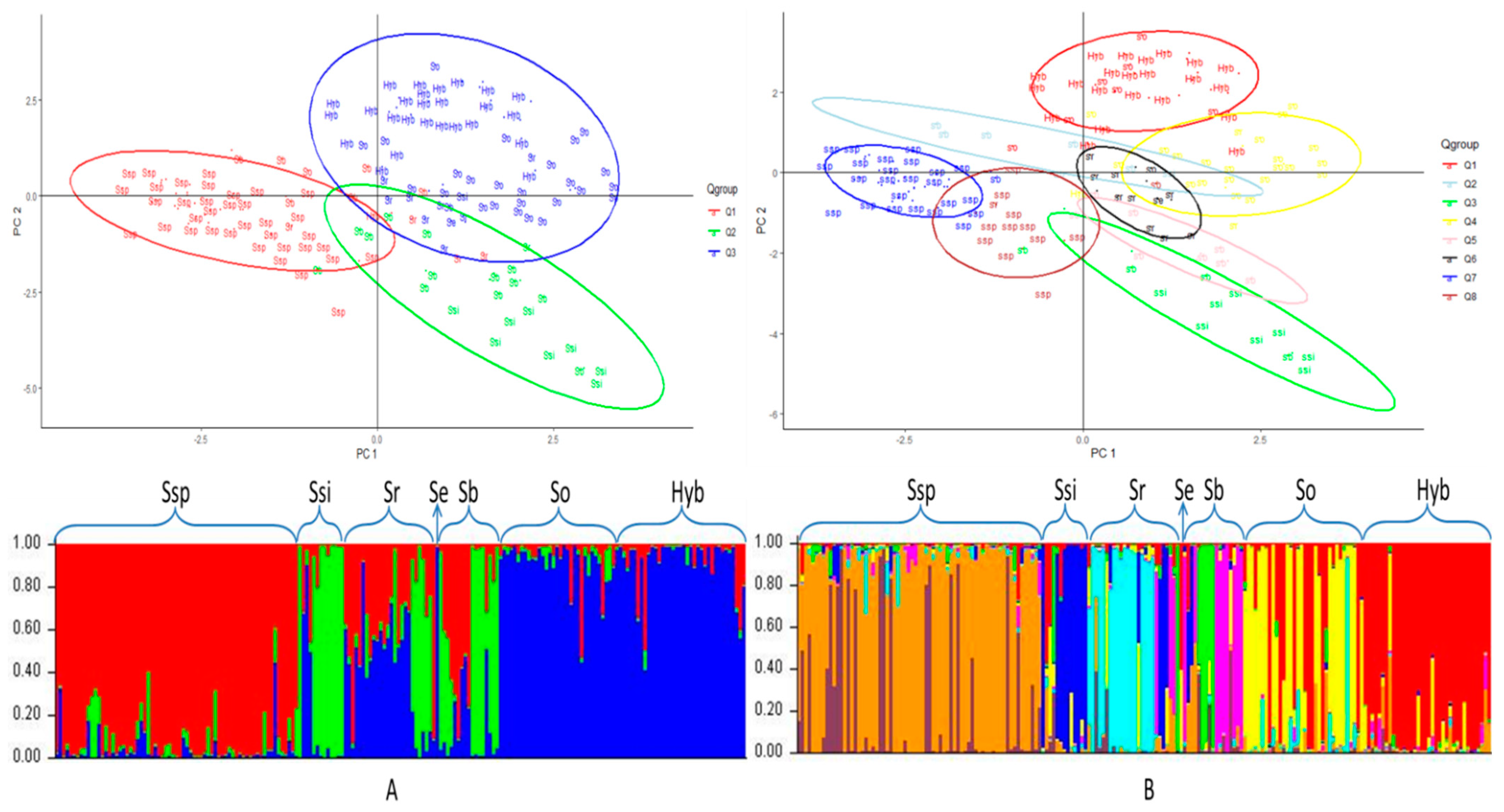
3.2. Population Structure
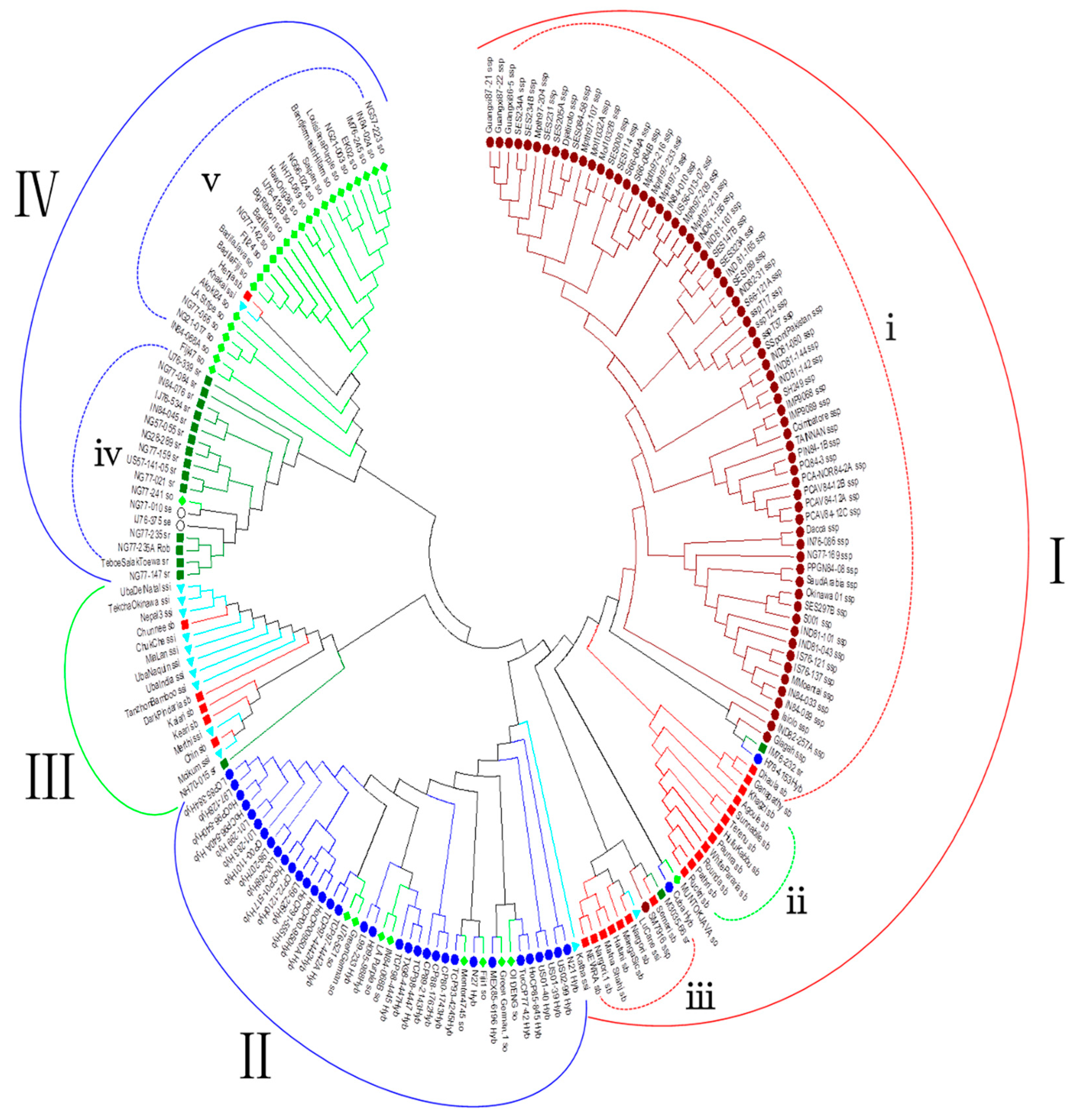
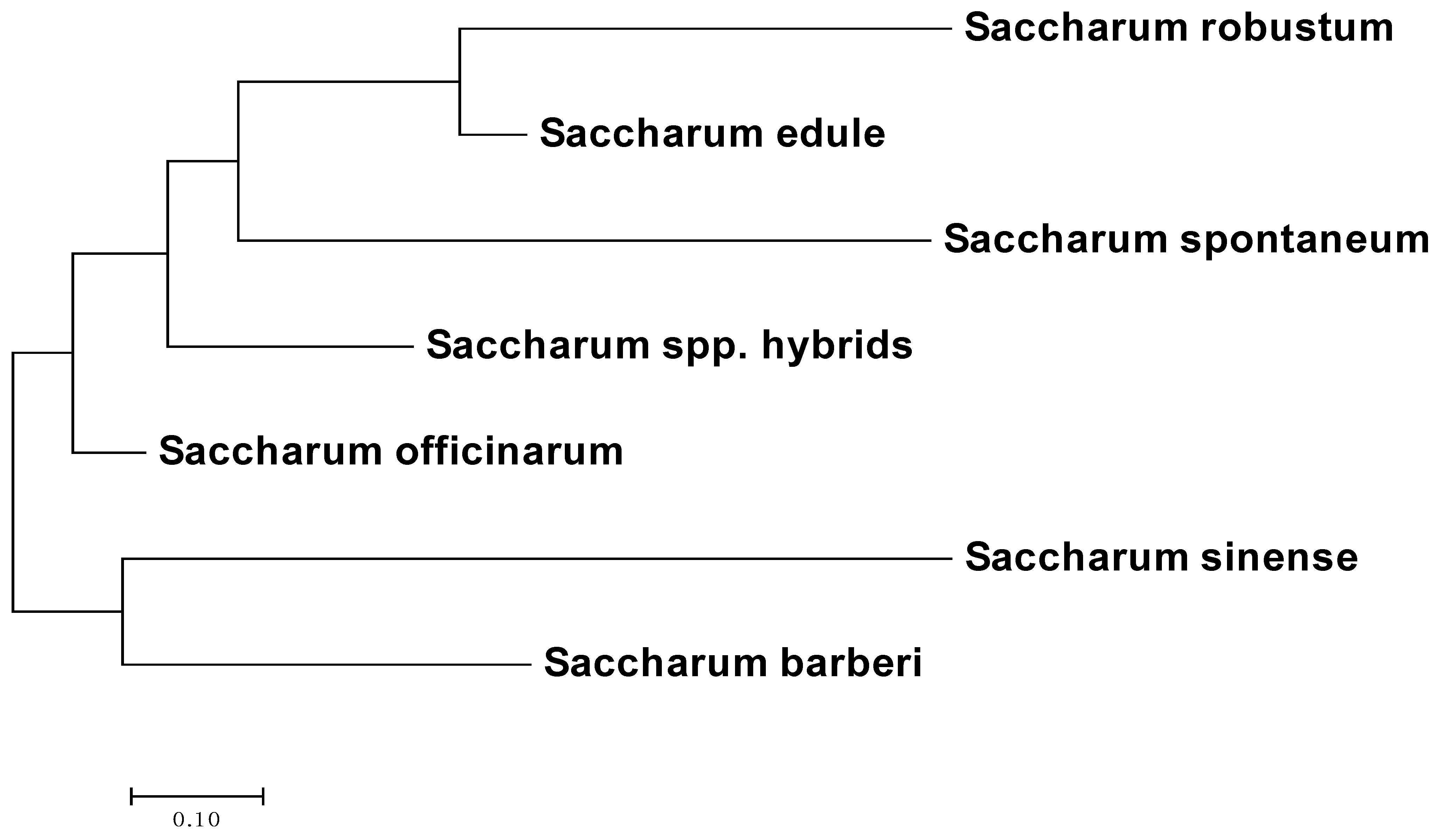
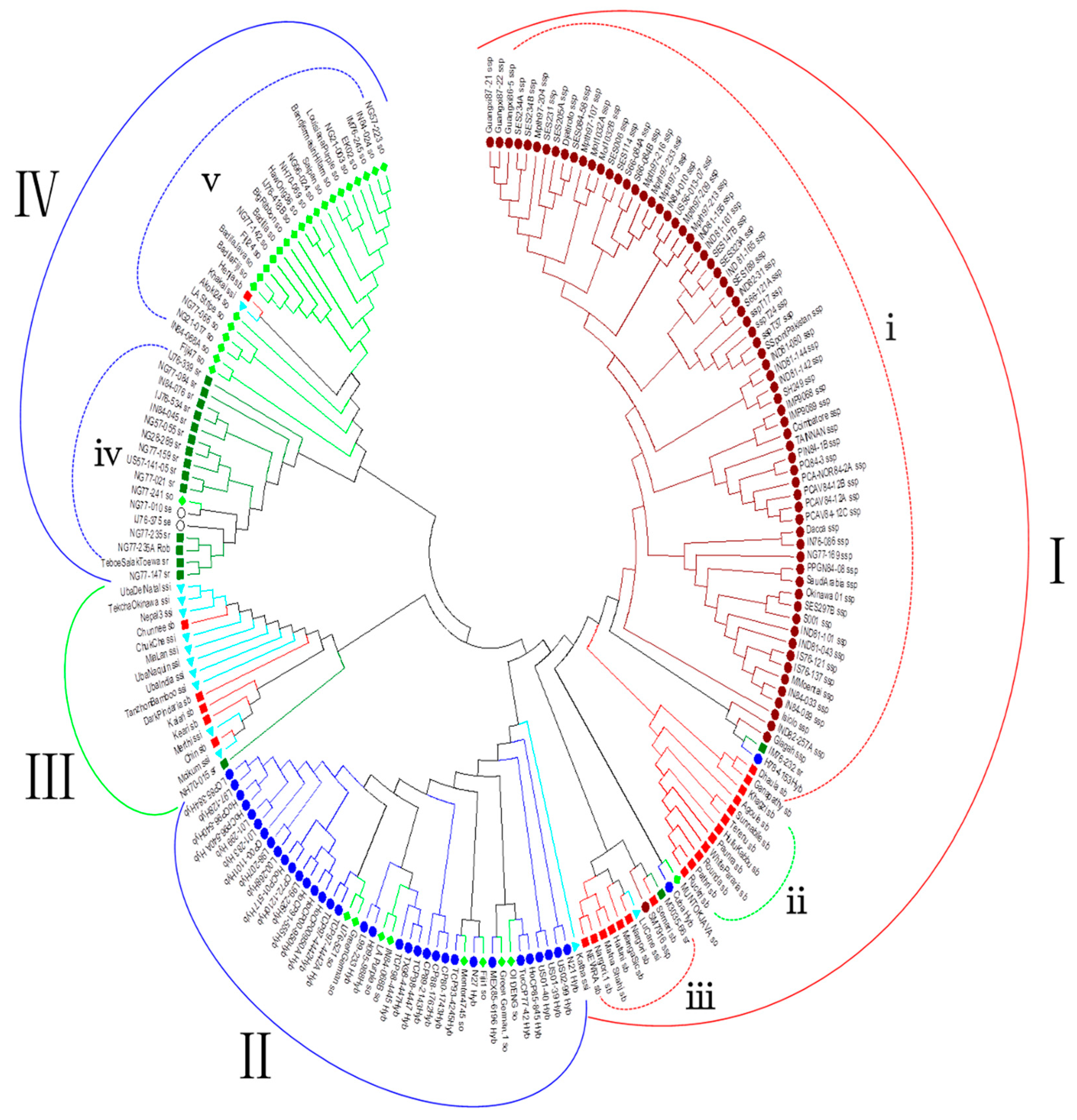
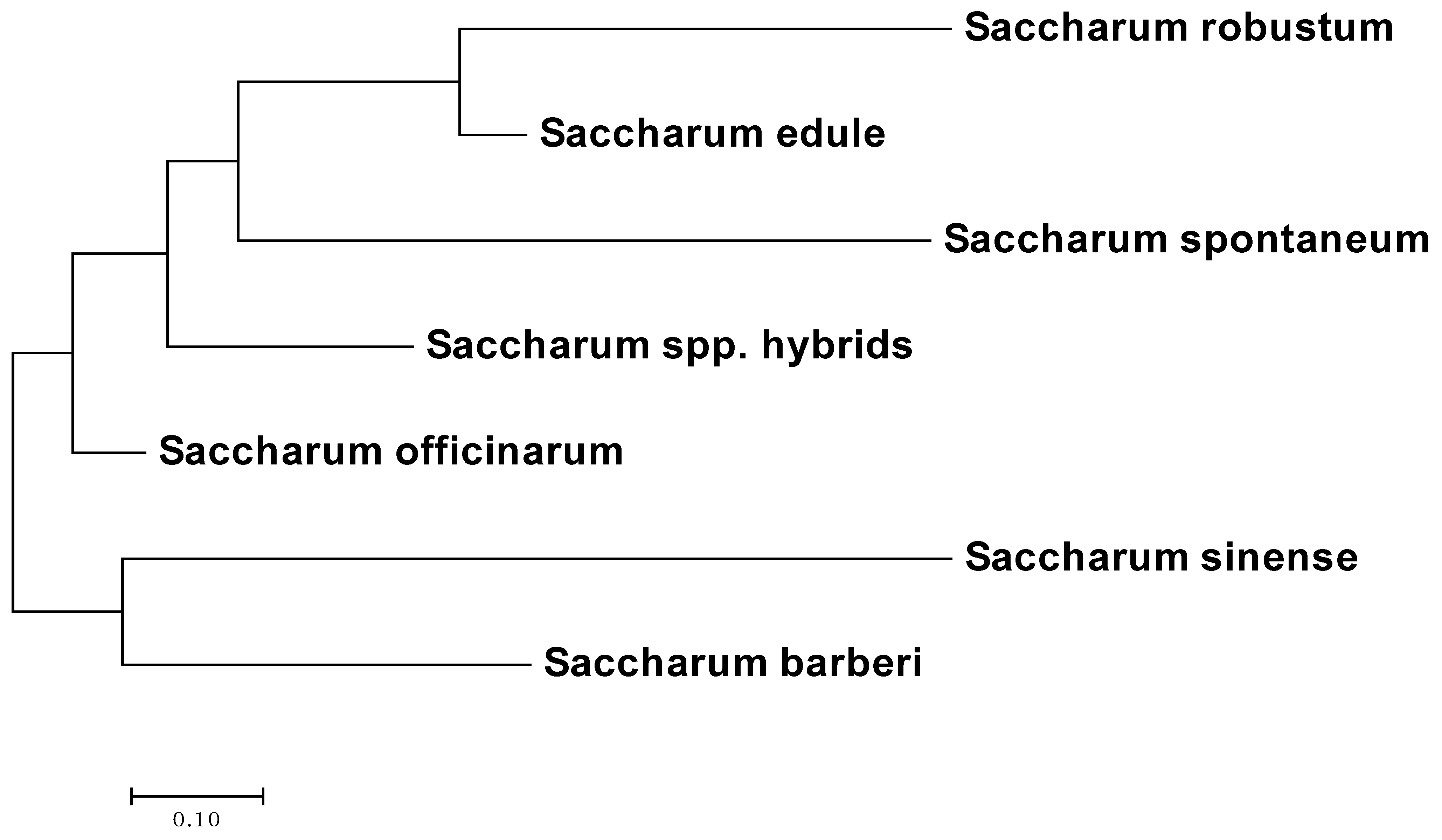
3.3. Phylogenetic Analysis
3.4. Principal Component Analysis
3.5. Species-Specific and Species-Associated SSR Alleles
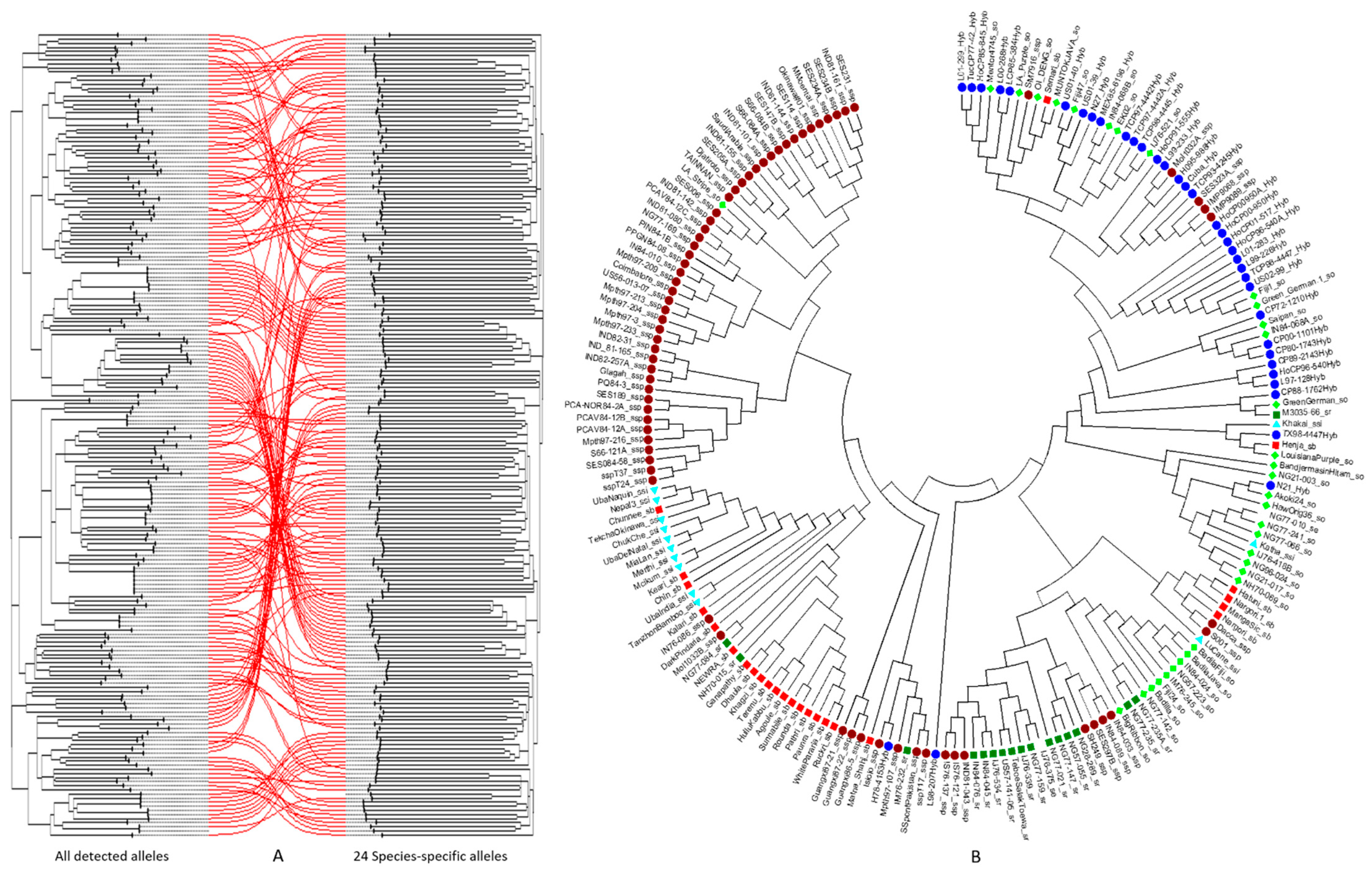
3.6. Phylogenetic Analysis of the Sugarcane Accessions Using Species-Specific or Species-Associated SSR Alleles
4. Discussion
4.1. Profile of the SSR Alleles
4.2. Population Structure and Phylogenetic Analysis
4.3. Species-Specific and Species-Associated SSR Alleles
4.4. Summary
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waclawovsky, A.J.; Sato, P.M.; Lembke, C.G.; Moore, P.H.; Souza, G. Sugarcane for bioenergy production: An assessment of yield and regulation of sucrose content. Plant Biotechnol. J. 2010, 8, 263–276. [Google Scholar] [CrossRef] [Green Version]
- Goldemberg, J.; Coelho, S.T.; Guardabassi, P. The sustainability of ethanol production from sugarcane. Energy Policy 2008, 36, 2086–2097. [Google Scholar] [CrossRef]
- Babu, K.H.; Devarumath, R.; Thorat, A.; Nalavade, V.; Saindane, M.; Appunu, C.; Suprasanna, P. Sugarcane transgenics: Developments and opportunities. In Genetically Modified Crops; Kavi Kishor, P.B., Rajam, M.V., Pullaiah, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2021; pp. 241–265. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, M.; Walsh, J.; Zhu, L.; Chen, Y.; Ming, R. Sugarcane Genetics and Genomics. In Sugarcane: Physiology, Biochemistry, and Functional Biology; Moore, P.H., Botha, F.C., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 623–643. [Google Scholar]
- Wang, T.; Fang, J.; Zhang, J. Advances in sugarcane genomics and genetics. Sugar Tech 2022, 24, 354–368. [Google Scholar] [CrossRef]
- Scortecci, K.C.; Creste, S.; Calsa, T., Jr.; Xavier, M.A.; Landell, M.G.; Figueira, A.; Benedito, V.A. Challenges, opportunities and recent advances in sugarcane breeding. Plant Breed. 2012, 1, 267–296. [Google Scholar]
- Zan, F.; Zhang, Y.; Wu, Z.; Zhao, J.; Wu, C.; Zhao, Y.; Chen, X.; Zhao, L.; Qin, W.; Yao, L.J. Genetic analysis of agronomic traits in elite sugarcane (Saccharum spp.) germplasm. PLoS ONE 2020, 15, e0233752. [Google Scholar] [CrossRef] [PubMed]
- Lao, F.; He, H.; Liu, R.; Qi, Y.J. AFLP Analysis of genetic stability of sugarcane tissue culture clones. Agric. Biotechnol. 2020, 9, 1–5. [Google Scholar]
- Aitken, K.; Jackson, P.; McIntyre, C. A combination of AFLP and SSR markers provides extensive map coverage and identification of homo (eo) logous linkage groups in a sugarcane cultivar. Theor. Appl. Genet. 2005, 110, 789–801. [Google Scholar] [CrossRef]
- Cursi, D.E.; Castillo, R.O.; Tarumoto, Y.; Umeda, M.; Tippayawat, A.; Ponragdee, W.; Racedo, J.; Perera, M.F.; Hoffmann, H.P.; Carneiro, M.S. Origin, genetic diversity, conservation, and traditional and molecular breeding approaches in sugarcane. In Cash Crops; Springer: Cham, Switzerland, 2022; pp. 83–116. [Google Scholar] [CrossRef]
- Kawar, P.; Devarumath, R.; Nerkar, Y. Use of RAPD markers for assessment of genetic diversity in sugarcane cultivars. Indian J. Biotechnol. 2009, 8, 67–71. [Google Scholar]
- Aitken, K.S. History and development of molecular markers for sugarcane breeding. Sugar Tech 2021, 24, 341–353. [Google Scholar] [CrossRef]
- You, Q.; Xu, L.; Zheng, Y.; Que, Y. Genetic diversity analysis of sugarcane parents in Chinese breeding programmes using gSSR markers. Sci. World J. 2013, 2013, 613062. [Google Scholar] [CrossRef] [Green Version]
- You, Q.; Pan, Y.-B.; Xu, L.-P.; Gao, S.-W.; Wang, Q.-N.; Su, Y.-C.; Yang, Y.-Q.; Wu, Q.-B.; Zhou, D.-G.; Que, Y.-X. Genetic diversity analysis of sugarcane germplasm based on fluorescence-labeled simple sequence repeat markers and a capillary electrophoresis-based genotyping platform. Sugar Tech 2016, 18, 380–390. [Google Scholar] [CrossRef]
- Devarumath, R.M.; Kalwade, S.B.; Kawar, P.G.; Sushir, K.V. Assessment of genetic diversity in sugarcane germplasm using ISSR and SSR markers. Sugar Tech 2012, 14, 334–344. [Google Scholar] [CrossRef]
- Xiao, N.; Wang, H.; Yao, W.; Zhang, M.; Ming, R.; Zhang, J.J. Development and evaluation of SSR markers based on large scale full-length transcriptome sequencing in sugarcane. Trop. Plant Biol. 2020, 13, 343–352. [Google Scholar] [CrossRef]
- Singh, R.K.; Jena, S.N.; Khan, S.; Yadav, S.; Banarjee, N.; Raghuvanshi, S.; Bhardwaj, V.; Dattamajumder, S.K.; Kapur, R.; Solomon, S. Development, cross-species/genera transferability of novel EST-SSR markers and their utility in revealing population structure and genetic diversity in sugarcane. Gene 2013, 524, 309–329. [Google Scholar] [CrossRef] [PubMed]
- Ukoskit, K.; Posudsavang, G.; Pongsiripat, N.; Chatwachirawong, P.; Klomsa-Ard, P.; Poomipant, P.; Tragoonrung, S. Detection and validation of EST-SSR markers associated with sugar-related traits in sugarcane using linkage and association mapping. Genomics 2019, 111, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.A.; Benchimol, L.L.; Barbosa, A.M.; Geraldi, I.O.; Souza, C.L., Jr.; Souza, A.P.d. Comparison of RAPD, RFLP, AFLP and SSR markers for diversity studies in tropical maize inbred lines. Genet. Mol. Biol. 2004, 27, 579–588. [Google Scholar] [CrossRef]
- Liu, X.; Li, X.; Xu, C.; Lin, X.; Deng, Z. Genetic diversity of populations of Saccharum spontaneum with different ploidy levels using SSR molecular markers. Sugar Tech 2016, 18, 365–372. [Google Scholar] [CrossRef]
- Fickett, N.D.; Ebrahimi, L.; Parco, A.P.; Gutierrez, A.V.; Hale, A.L.; Pontif, M.J.; Todd, J.; Kimbeng, C.A.; Hoy, J.W.; Ayala-Silva, T. An enriched sugarcane diversity panel for utilization in genetic improvement of sugarcane. Sci. Rep. 2020, 10, 13390. [Google Scholar] [CrossRef]
- Tai, P.; Miller, J. Germplasm diversity among four sugarcane species for sugar composition. Crop Sci. 2002, 42, 958–964. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Mishra, S.K.; Singh, S.P.; Mishra, N.; Sharma, M. Evaluation of microsatellite markers for genetic diversity analysis among sugarcane species and commercial hybrids. Aust. J. Crop Sci. 2010, 4, 116–125. [Google Scholar]
- Hoang, N.V.; Furtado, A.; Botha, F.C.; Simmons, B.A.; Henry, R.J. Potential for genetic improvement of sugarcane as a source of biomass for biofuels. Front. Bioeng. Biotechnol. 2015, 3, 182. [Google Scholar] [CrossRef] [Green Version]
- Porras-Hurtado, L.; Ruiz, Y.; Santos, C.; Phillips, C.; Carracedo, Á.; Lareu, M. An overview of STRUCTURE: Applications, parameter settings, and supporting software. Front. Genet. 2013, 4, 98. [Google Scholar] [CrossRef] [Green Version]
- Smiullah, F.; Afzal, A.; Abdullah, U.I.; Iftikhar, R. Genetic diversity assessment in sugarcane using principal component analysis (PCA). Int. J. Mod. Agric. 2013, 2, 34–38. [Google Scholar] [CrossRef]
- Sharma, R.; Gupta, P.; Sharma, V.; Sood, A.; Mohapatra, T.; Ahuja, P.S. Evaluation of rice and sugarcane SSR markers for phylogenetic and genetic diversity analyses in bamboo. Genome 2008, 51, 91–103. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, P.G.; Rossi, M.; de Jesus, E.M.; Saccaro, N.L., Jr.; Kajihara, D.; Massa, R.; de Felix, J.M.; Drummond, R.D.; Falco, M.C.; Chabregas, S.M.; et al. Transcriptionally active transposable elements in recent hybrid sugarcane. Plant J. 2005, 44, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Racedo, J.; Gutierrez, L.; Perera, M.F.; Ostengo, S.; Pardo, E.M.; Cuenya, M.I.; Welin, B.; Castagnaro, A.P. Genome-wide association mapping of quantitative traits in a breeding population of sugarcane. BMC Plant Biol. 2016, 16, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, S.N.; Song, J.; Villa, A.; Pathak, B.; Ayala-Silva, T.; Yang, X.; Todd, J.; Glynn, N.C.; Kuhn, D.N.; Glaz, B. Promoting utilization of Saccharum spp. genetic resources through genetic diversity analysis and core collection construction. PLoS ONE 2014, 9, e110856. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.-B. Highly polymorphic microsatellite DNA markers for sugarcane germplasm evaluation and variety identity testing. Sugar Tech 2006, 8, 246–256. [Google Scholar] [CrossRef]
- Ali, A.; Jin-Da, W.; Yong-Bao, P.; Zu-Hu, D.; Zhi-Wei, C.; Ru-Kai, C.; San-Ji, G. Molecular identification and genetic diversity analysis of Chinese sugarcane (Saccharum spp. hybrids) varieties using SSR markers. Trop. Plant Biol. 2017, 10, 194–203. [Google Scholar] [CrossRef]
- Chen, P.; Pan, Y.-B.; Chen, R.-K.; Xu, L.-P.; Chen, Y.-Q. SSR marker-based analysis of genetic relatedness among sugarcane cultivars (Saccharum spp. hybrids) from breeding programs in China and other countries. Sugar Tech 2009, 11, 347–354. [Google Scholar] [CrossRef]
- Pan, Y.; Scheffler, B.; Richard, E., Jr. High-throughput molecular genotyping of commercial sugarcane clones with microsatellite (SSR) markers. Sugar Tech 2007, 9, 176–181. [Google Scholar]
- Pan, Y.-B. Databasing molecular identities of sugarcane (Saccharum spp.) clones constructed with microsatellite (SSR) DNA markers. Am. J. Plant Sci. 2010, 1, 87. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Que, Y.; Pan, Y.-B. Highly polymorphic microsatellite DNA markers for sugarcane germplasm evaluation and variety identity testing. Sugar Tech 2011, 13, 129–136. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A.; VonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Shi, A.; Qin, J.; Mou, B.; Correll, J.; Weng, Y.; Brenner, D.; Feng, C.; Motes, D.; Yang, W.; Dong, L. Genetic diversity and population structure analysis of spinach by single-nucleotide polymorphisms identified through genotyping-by-sequencing. PLoS ONE 2017, 12, e0188745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Srichant, N.; Chankaew, S.; Monkham, T.; Thammabenjapone, P.; Sanitchon, J. Development of Sakon Nakhon rice variety for blast resistance through marker assisted backcross breeding. Agronomy 2019, 9, 67. [Google Scholar] [CrossRef] [Green Version]
- Berding, N.; Roach, B. Germplasm collection, maintenance and use. In ‘Sugarcane improvement through breeding. Dev. Crop Sci. 1987, 4, 143–210. [Google Scholar] [CrossRef]
- Ali, A.; Pan, Y.-B.; Wang, Q.-N.; Wang, J.-D.; Chen, J.-L.; Gao, S.-J. Genetic diversity and population structure analysis of Saccharum and Erianthus genera using microsatellite (SSR) markers. Sci. Rep. 2019, 9, 395. [Google Scholar] [CrossRef] [Green Version]
- Parida, S.K.; Kalia, S.K.; Kaul, S.; Dalal, V.; Hemaprabha, G.; Selvi, A.; Pandit, A.; Singh, A.; Gaikwad, K.; Sharma, T.R. Informative genomic microsatellite markers for efficient genotyping applications in sugarcane. Theor. Appl. Genet. 2009, 118, 327–338. [Google Scholar] [CrossRef]
- Marconi, T.G.; Costa, E.A.; Miranda, H.R.; Mancini, M.C.; Cardoso-Silva, C.B.; Oliveira, K.M.; Pinto, L.R.; Mollinari, M.; Garcia, A.A.; Souza, A.P. Functional markers for gene mapping and genetic diversity studies in sugarcane. BMC Res. Notes 2011, 4, 264. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Zhou, H.; Pan, Y.; Chen, C.; Zhu, J.; Chen, P.; Li, Y.; Cai, Q.; Chen, R. Segregation analysis of microsatellite (SSR) markers in sugarcane polyploids. Genet. Mol. Res 2015, 14, 18384–18395. [Google Scholar] [CrossRef]
- Singh, R.B.; Mahenderakar, M.D.; Jugran, A.K.; Singh, R.K.; Srivastava, R.K. Assessing genetic diversity and population structure of sugarcane cultivars, progenitor species and genera using microsatellite (SSR) markers. Gene 2020, 753, 144800. [Google Scholar] [CrossRef]
- Wu, J.; Wang, Q.; Xie, J.; Pan, Y.-B.; Zhou, F.; Guo, Y.; Chang, H.; Xu, H.; Zhang, W.; Zhang, C. SSR marker-assisted management of parental germplasm in sugarcane (Saccharum spp. hybrids) breeding programs. Agronomy 2019, 9, 449. [Google Scholar] [CrossRef] [Green Version]
- Creste, S.; Sansoli, D.; Tardiani, A.; Silva, D.; Gonçalves, F.; Fávero, T.; Medeiros, C.; Festucci, C.; Carlini-Garcia, L.; Landell, M. Comparison of AFLP, TRAP and SSRs in the estimation of genetic relationships in sugarcane. Sugar Tech 2010, 12, 150–154. [Google Scholar] [CrossRef]
- Banumathi, G.; Krishnasamy, V.; Maheswaran, M.; Samiyappan, R.; Govindaraj, P.; Kumaravadivel, N. Genetic diversity analysis of sugarcane (Saccharum spp.) clones using simple sequence repeat markers of sugarcane and rice. Electron. J. Plant Breed. 2010, 1, 517–526. [Google Scholar]
- Cordeiro, G.M.; Pan, Y.-B.; Henry, R.J. Sugarcane microsatellites for the assessment of genetic diversity in sugarcane germplasm. Plant Sci. 2003, 165, 181–189. [Google Scholar] [CrossRef]
- Cruz, G.B.D.; Gerardo, B.D.; Tanguilig, B.T., III. Optimization. Agricultural crops classification models based on PCA-GA implementation in data mining. Int. J. Model. Optim. 2014, 4, 375–382. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, S.A.; Prasanna, B. Analysis of genetic diversity in crop plants—Salient statistical tools and considerations. Crop Sci. 2003, 43, 1235–1248. [Google Scholar] [CrossRef] [Green Version]
- Bossart, J.; Prowell, D.P. Genetic estimates of population structure and gene flow: Limitations, lessons and new directions. Trends Ecol. Evol. 1998, 13, 202–206. [Google Scholar] [CrossRef]
- Ibrahim, A.K.; Zhang, L.; Niyitanga, S.; Afzal, M.Z.; Xu, Y.; Zhang, L.; Zhang, L.; Qi, J. Principles and approaches of association mapping in plant breeding. Trop. Plant Biol. 2020, 13, 212–224. [Google Scholar] [CrossRef]
- Korte, A.; Farlow, A.J.P.m. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Aranzana, M.J.; Kim, S.; Lister, C.; Shindo, C.; Tang, C.; Toomajian, C.; Zheng, H.; Dean, C.; Marjoram, P. An Arabidopsis example of association mapping in structured samples. PLoS Genet. 2007, 3, e4. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | SSR Markers | Size Range (bp) | Number of Detected Peaks (Alleles) | Number of Original Alleles | Number of New Alleles | New SSR Alleles (bp) Detected in This Study |
|---|---|---|---|---|---|---|
| 1 | SMC119CG | 80~239 | 32 | 6 | 26 | 80, 92, 95, 99, 109, 115, 121, 125, 136, 138, 141, 143, 145, 149, 152, 155, 168, 172, 187, 190, 195, 204, 210, 229, 231, 239 |
| 2 | SMC1604SA | 91~140 | 26 | 19 | 7 | 92, 97, 104, 107, 126, 129, 140 |
| 3 | SMC1751CL | 122~169 | 26 | 23 | 3 | 156, 168, 169 |
| 4 | SMC18SA | 127~165 | 14 | 13 | 1 | 128 |
| 5 | SMC22DUQ | 123~177 | 21 | 20 | 1 | 141 |
| 6 | SMC24DUQ | 119~144 | 17 | 15 | 2 | 119, 126 |
| 7 | SMC278CS | 110~238 | 32 | 30 | 2 | 112, 144 |
| 8 | SMC319CG | 79~189 | 26 | 0 | 26 | 79, 123, 143, 144, 145, 147, 149, 151, 153, 155, 158, 160, 162, 163, 164, 166, 168, 170, 172, 175, 179, 181, 183, 185, 187, 189 |
| 9 | SMC31CUQ | 108~234 | 30 | 29 | 1 | 151 |
| 10 | SMC334BS | 130~170 | 30 | 27 | 3 | 130, 141, 169 |
| 11 | SMC336BS | 112~239 | 31 | 30 | 1 | 152 |
| 12 | SMC36BUQ | 82~338 | 35 | 27 | 8 | 102, 124, 137, 147, 192, 258, 277, 338 |
| 13 | SMC486CG | 222~247 | 15 | 14 | 1 | 229 |
| 14 | SMC569CS | 165~238 | 38 | 36 | 2 | 177, 211 |
| 15 | SMC597CS | 128~188 | 46 | 45 | 1 | 177 |
| 16 | SMC703BS | 181~237 | 33 | 31 | 2 | 181, 184 |
| 17 | SMC7CUQ | 144~171 | 16 | 13 | 3 | 144, 147, 150 |
| 18 | SMC851MS | 127~158 | 31 | 31 | 0 | N/A |
| 19 | CIR3 | 148~467 | 34 | 26 | 8 | 166, 401, 424, 440, 442, 462, 464, 467 |
| 20 | CIR43 | 162~256 | 37 | 31 | 6 | 162, 164, 203, 222, 249, 251 |
| 21 | CIR66 | 102~154 | 27 | 27 | 0 | N/A |
| 22 | CIR74 | 118~245 | 27 | 22 | 5 | 118, 175, 200, 202, 245 |
| Total | N/A | N/A | 624 | 515 | 109 | 109 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, H.; Chen, Y.; Gao, S.-J.; Pan, Y.-B.; Shi, A. Population Structure and Genetic Diversity Analysis in Sugarcane (Saccharum spp. hybrids) and Six Related Saccharum Species. Agronomy 2022, 12, 412. https://doi.org/10.3390/agronomy12020412
Xiong H, Chen Y, Gao S-J, Pan Y-B, Shi A. Population Structure and Genetic Diversity Analysis in Sugarcane (Saccharum spp. hybrids) and Six Related Saccharum Species. Agronomy. 2022; 12(2):412. https://doi.org/10.3390/agronomy12020412
Chicago/Turabian StyleXiong, Haizheng, Yilin Chen, San-Ji Gao, Yong-Bao Pan, and Ainong Shi. 2022. "Population Structure and Genetic Diversity Analysis in Sugarcane (Saccharum spp. hybrids) and Six Related Saccharum Species" Agronomy 12, no. 2: 412. https://doi.org/10.3390/agronomy12020412
APA StyleXiong, H., Chen, Y., Gao, S.-J., Pan, Y.-B., & Shi, A. (2022). Population Structure and Genetic Diversity Analysis in Sugarcane (Saccharum spp. hybrids) and Six Related Saccharum Species. Agronomy, 12(2), 412. https://doi.org/10.3390/agronomy12020412