
Structural Properties and Magnetic Ground States of 100 Binary d-Metal Oxides Studied by Hybrid Density Functional Methods

 ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. General Overview of the Results
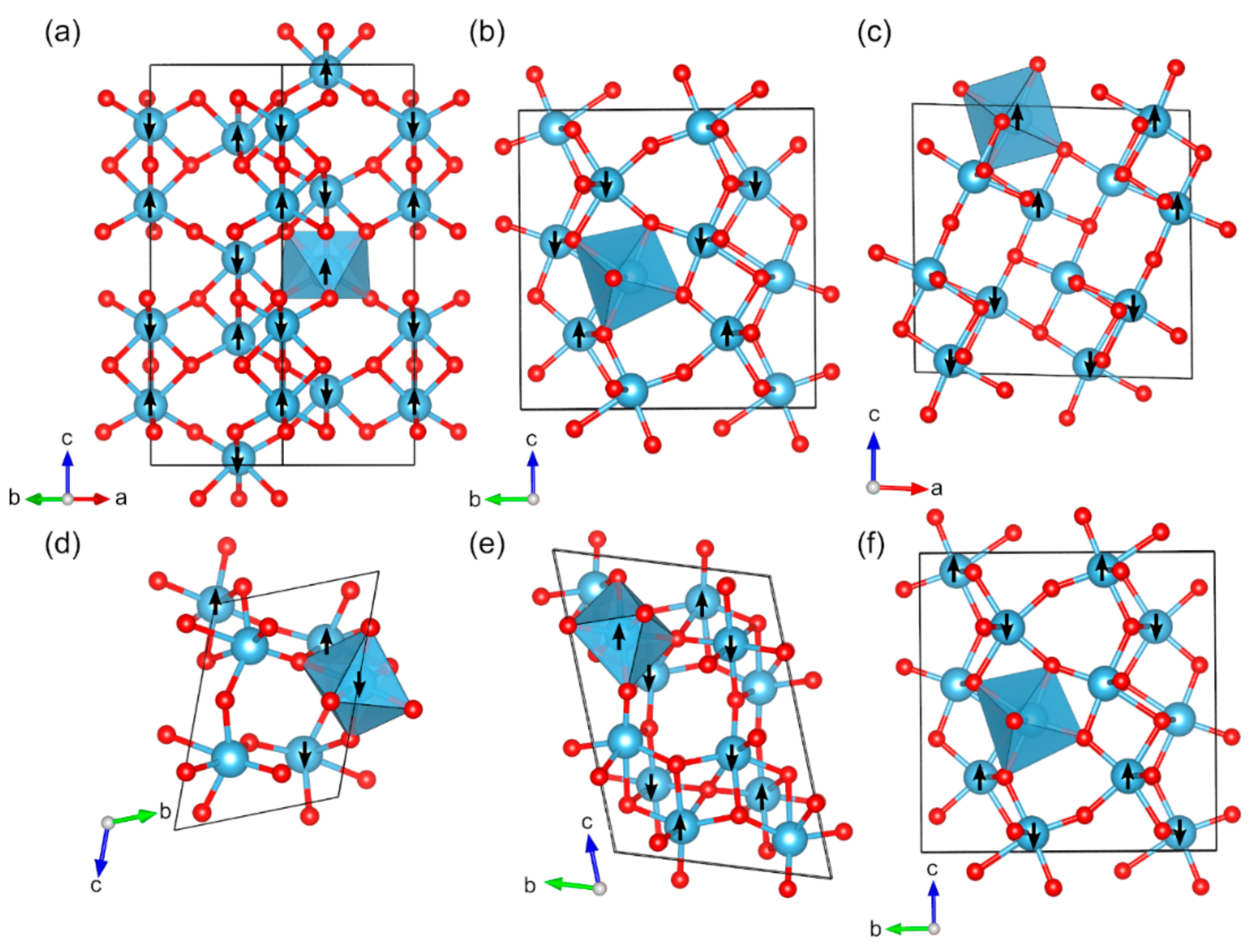
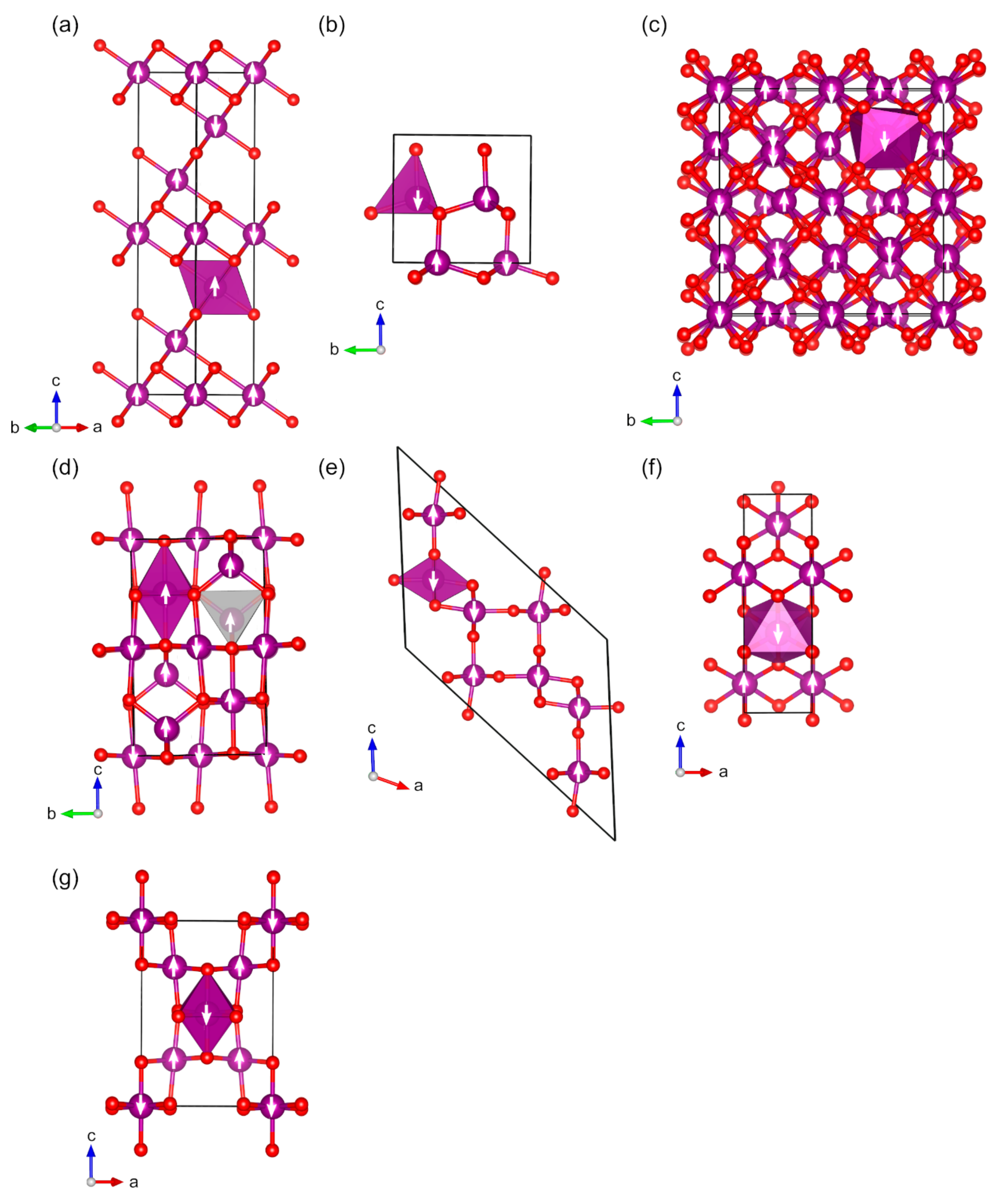
2.2. Magnetic Binary 3D-Metal Oxides
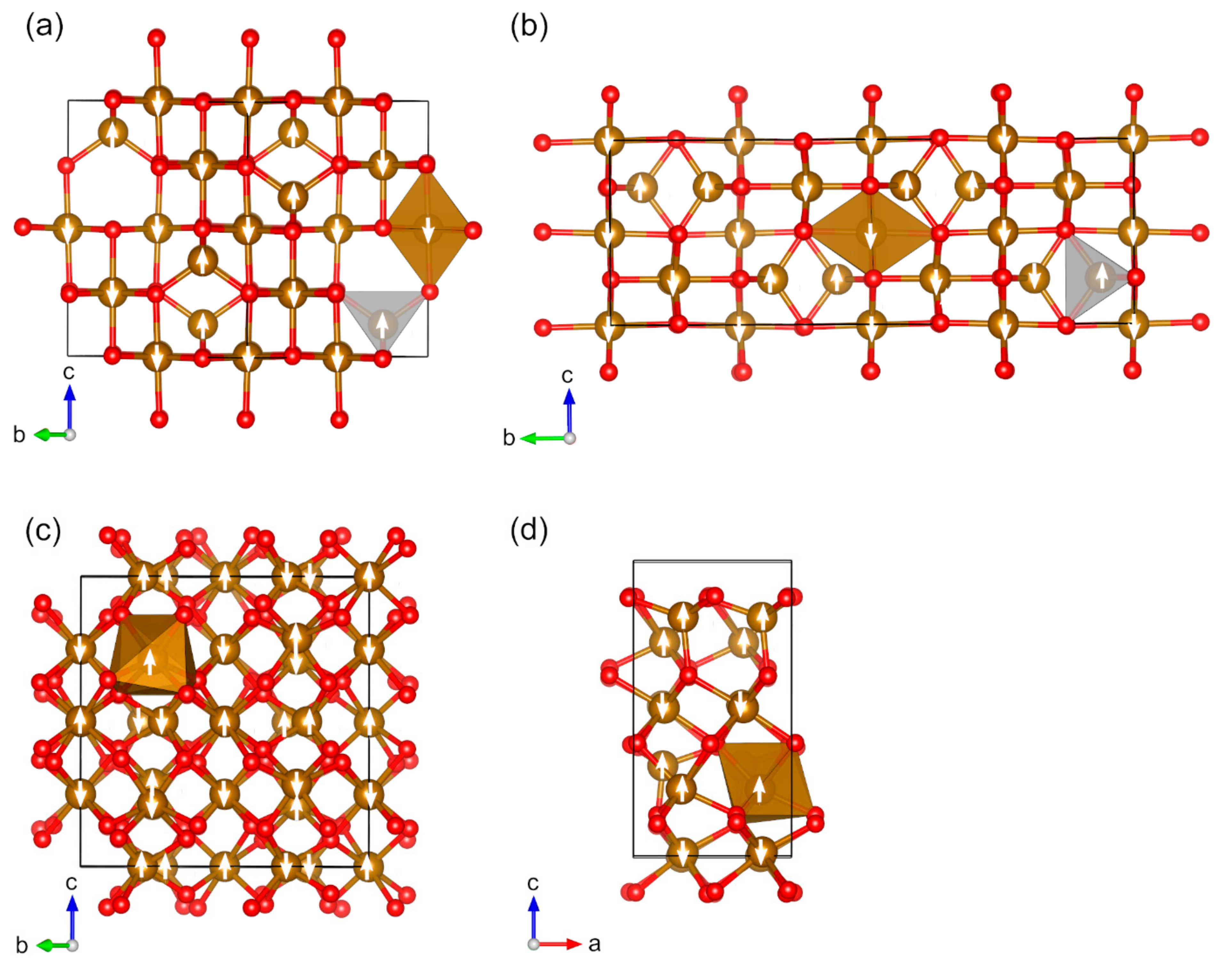
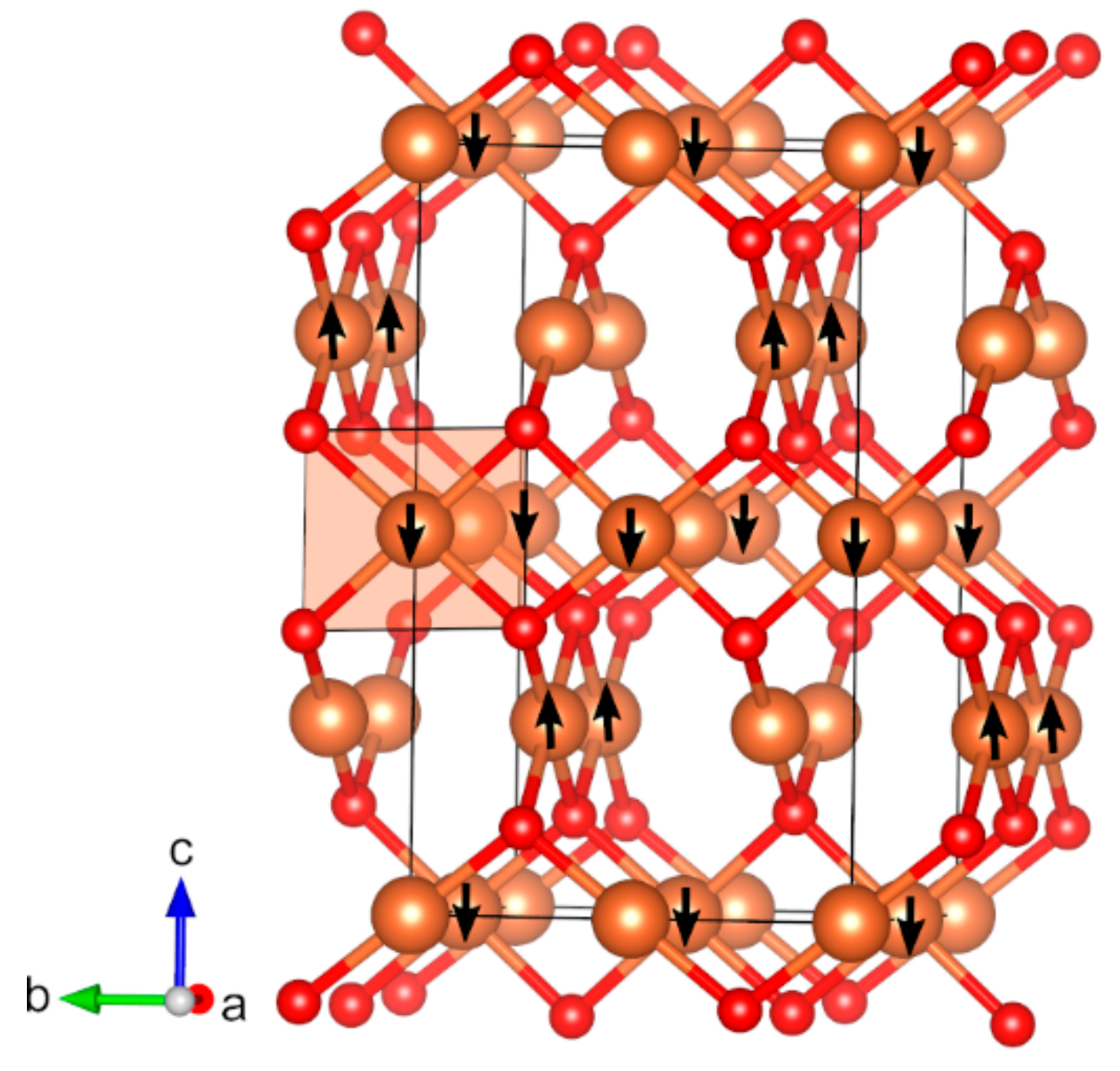
2.3. Magnetic Binary 4D-Metal Oxides
2.4. Magnetic Binary 5D-Metal Oxides
2.5. D-Metal Oxides with Molecular Structures
2.6. Mercury Oxides
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nilius, N. Properties of Oxide Thin Films and Their Adsorption Behavior Studied by Scanning Tunneling Microscopy and Conductance Spectroscopy. Surf. Sci. Rep. 2009, 64, 595–659. [Google Scholar] [CrossRef]
- Giordano, L.; Pacchioni, G. Oxide Films at the Nanoscale: New Structures, New Functions, and New Materials. Acc. Chem. Res. 2011, 44, 1244–1252. [Google Scholar] [CrossRef]
- Honkala, K. Tailoring Oxide Properties: An Impact on Adsorption Characteristics of Molecules and Metals. Surf. Sci. Rep. 2014, 69, 366–388. [Google Scholar] [CrossRef]
- Tripathi, T.S.; Karppinen, M. Atomic Layer Deposition of P-Type Semiconducting Thin Films: A Review. Adv. Mater. Interfaces 2017, 4, 1–16. [Google Scholar] [CrossRef]
- Walia, S.; Balendhran, S.; Nili, H.; Zhuiykov, S.; Rosengarten, G.; Wang, Q.H.; Bhaskaran, M.; Sriram, S.; Strano, M.S.; Kalantar-zadeh, K. Transition Metal Oxides—Thermoelectric Properties. Prog. Mater. Sci. 2013, 58, 1443–1489. [Google Scholar] [CrossRef]
- Rao, C.N.R. Transition Metal Oxides. Annu. Rev. Phys. Chem. 1989, 40, 291–326. [Google Scholar] [CrossRef]
- Dixon, S.C.; Scanlon, D.O.; Carmalt, C.J.; Parkin, I.P. N-Type Doped Transparent Conducting Binary Oxides: An Overview. J. Mater. Chem. C 2016, 4, 6946–6961. [Google Scholar] [CrossRef]
- Wang, L.; Maxisch, T.; Ceder, G. Oxidation Energies of Transition Metal Oxides within the GGA+U Framework. Phys. Rev. B 2006, 73, 195107. [Google Scholar] [CrossRef]
- Schrön, A.; Rödl, C.; Bechstedt, F. Crystalline and Magnetic Anisotropy of the 3d-Transition Metal Monoxides MnO, FeO, CoO, and NiO. Phys. Rev. B 2012, 86, 115134. [Google Scholar] [CrossRef]
- Noh, J.; Osman, O.I.; Aziz, S.G.; Winget, P.; Brédas, J.L. A Density Functional Theory Investigation of the Electronic Structure and Spin Moments of Magnetite. Sci. Technol. Adv. Mater. 2014, 15, 044202. [Google Scholar] [CrossRef]
- Lima, A.F. Density Functional Theory Study on the Magnetic Properties of Co3O4with Normal Spinel Structure. J. Phys. Chem. Solids 2016, 91, 86–89. [Google Scholar] [CrossRef]
- Singh, V.; Kosa, M.; Majhi, K.; Major, D.T. Putting DFT to the Test: A First-Principles Study of Electronic, Magnetic, and Optical Properties of Co3O4. J. Chem. Theory Comput. 2015, 11, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Li, J.; Li, S.S.; Xia, J.B.; Walsh, A.; Wei, S.H. Origin of Antiferromagnetism in CoO: A Density Functional Theory Study. Appl. Phys. Lett. 2010, 96, 162508. [Google Scholar] [CrossRef]
- Bredow, T.; Gerson, A.R. Effect of Exchange and Correlation on Bulk Properties of MgO, NiO, and CoO. Phys. Rev. B 2000, 61, 5194–5201. [Google Scholar] [CrossRef]
- Rollmann, G.; Rohrbach, A.; Entel, P.; Hafner, J. First-Principles Calculation of the Structure and Magnetic Phases of Hematite. Phys. Rev. B 2004, 69, 165107. [Google Scholar] [CrossRef]
- Linnera, J.; Karttunen, A.J. Ab Initio Study of the Lattice Thermal Conductivity of Cu2O Using the Generalized Gradient Approximation and Hybrid Density Functional Methods. Phys. Rev. B 2017, 96, 014304. [Google Scholar] [CrossRef]
- Rödl, C.; Fuchs, F.; Furthmüller, J.; Bechstedt, F. Quasiparticle Band Structures of the Antiferromagnetic Transition-Metal Oxides MnO, FeO, CoO, and NiO. Phys. Rev. B 2009, 79, 235114. [Google Scholar] [CrossRef]
- Kulik, H.J.; Marzari, N. Transition-Metal Dioxides: A Case for the Intersite Term in Hubbard-Model Functionals. J. Chem. Phys. 2011, 134, 094103. [Google Scholar] [CrossRef]
- Chen, X.; Parker, D.; Du, M.H.; Singh, D.J. Potential Thermoelectric Performance of Hole-Doped Cu2O. New J. Phys. 2013, 15, 043029. [Google Scholar] [CrossRef]
- Seo, D.H.; Urban, A.; Ceder, G. Calibrating Transition-Metal Energy Levels and Oxygen Bands in First-Principles Calculations: Accurate Prediction of Redox Potentials and Charge Transfer in Lithium Transition-Metal Oxides. Phys. Rev. B 2015, 92, 115118. [Google Scholar] [CrossRef]
- Kirchner-Hall, N.E.; Zhao, W.; Xiong, Y.; Timrov, I.; Dabo, I. Extensive Benchmarking of DFT+U Calculations for Predicting Band Gaps. Appl. Sci. 2021, 11, 2395. [Google Scholar] [CrossRef]
- Gillen, R.; Robertson, J. Accurate Screened Exchange Band Structures for the Transition Metal Monoxides MnO, FeO, CoO and NiO. J. Phys. Condens. Matter 2013, 25, 165502. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Kulik, H.J. Where Does the Density Localize in the Solid State? Divergent Behavior for Hybrids and DFT+U. J. Chem. Theory Comput. 2018, 14, 670–683. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ruzsinszky, A.; Perdew, J. Strongly Constrained and Appropriately Normed Semilocal Density Functional. Phys. Rev. Lett. 2015, 115, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.; Furness, J.W.; Buda, I.G.; Zhang, Y.; Markiewicz, R.S.; Barbiellini, B.; Sun, J.; Bansil, A. Antiferromagnetic Ground State of La2CuO4: A Parameter-Free Ab Initio Description. Phys. Rev. B 2018, 98, 125140. [Google Scholar] [CrossRef]
- Kylänpää, I.; Balachandran, J.; Ganesh, P.; Heinonen, O.; Kent, P.R.C.; Krogel, J.T. Accuracy of Ab Initio Electron Correlation and Electron Densities in Vanadium Dioxide. Phys. Rev. Mater. 2017, 1, 065408. [Google Scholar] [CrossRef]
- Tran, F.; Stelzl, J.; Blaha, P. Rungs 1 to 4 of DFT Jacob’s Ladder: Extensive Test on the Lattice Constant, Bulk Modulus, and Cohesive Energy of Solids. J. Chem. Phys. 2016, 144. [Google Scholar] [CrossRef]
- Isaacs, E.B.; Wolverton, C. Performance of the Strongly Constrained and Appropriately Normed Density Functional for Solid-State Materials. Phys. Rev. Mater. 2018, 2, 063801. [Google Scholar] [CrossRef]
- Charles, N.; Rondinelli, J.M. Assessing Exchange-Correlation Functional Performance for Structure and Property Predictions of Oxyfluoride Compounds from First Principles. Phys. Rev. B 2016, 94. [Google Scholar] [CrossRef]
- Kuklin, M.S.; Karttunen, A.J. Crystal Structure Prediction of Magnetic Transition Metal Oxides by Using Evolutionary Algorithm and Hybrid DFT Methods. J. Phys. Chem. C 2018, 122, 24949–24957. [Google Scholar] [CrossRef]
- Brugnoli, L.; Ferrari, A.M.; Civalleri, B.; Pedone, A.; Menziani, M.C. Assessment of Density Functional Approximations for Highly Correlated Oxides: The Case of CeO2 and Ce2O3. J. Chem. Theory Comput. 2018, 14, 4914–4927. [Google Scholar] [CrossRef] [PubMed]
- Civalleri, B.; Presti, D.; Dovesi, R.; Savin, A. On Choosing the Best Density Functional Approximation. In Chemical Modelling; Springborg, M., Ed.; Royal Society of Chemistry: Cambridge, UK, 2012; Volume 9, pp. 168–185. ISBN 978-1-84973-412-7. [Google Scholar]
- Moreira, I.d.P.R.; Illas, F.; Martin, R.L. Effect of Fock Exchange on the Electronic Structure and Magnetic Coupling in NiO. Phys. Rev. B Condens. Matter Mater. Phys. 2002, 65, 1551021. [Google Scholar] [CrossRef]
- Linnera, J.; Sansone, G.; Maschio, L.; Karttunen, A.J. Thermoelectric Properties of P-Type Cu2O, CuO, and NiO from Hybrid Density Functional Theory. J. Phys. Chem. C 2018, 122, 15180–18189. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Liberto, D.G.; Tosoni, S.; Pacchioni, G. Band Gap of 3d Metal Oxides and Quasi-2D Materials from Hybrid Density Functional Theory: Are Dielectric-Dependent Functionals Superior ? J. Chem. Theory Comput. 2019, 16, 3786–3798. [Google Scholar] [CrossRef] [PubMed]
- Cipriano, L.A.; Liberto, D.G.; Tosoni, S.; Pacchioni, G. Band Gap in Magnetic Insulators from a Charge Transition Level Approach. J. Chem. Theory Comput. 2019, 15, 6294–6312. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Walther, C.F.J.; Kuc, A.; Heine, T. Density Functional Theory and Beyond for Band-Gap Screening: Performance for Transition-Metal Oxides and Dichalcogenides. J. Chem. Theory Comput. 2013, 9, 2950–2958. [Google Scholar] [CrossRef] [PubMed]
- Posysaev, S.; Miroshnichenko, O.; Alatalo, M.; Le, D.; Rahman, T.S. Oxidation States of Binary Oxides from Data Analytics of the Electronic Structure. Comput. Mater. Sci. 2019, 161, 403–414. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-Mechanical Condensed Matter Simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, 1–36. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Honig, J.M.; Reed, T.B. Electrical Properties of Ti2O3 Single Crystals. Phys. Rev. 1968, 174, 1020–1026. [Google Scholar] [CrossRef]
- Ohkoshi, S.; Tsunobuchi, Y.; Matsuda, T.; Hashimoto, K.; Namai, A.; Hakoe, F.; Tokoro, H. Synthesis of a Metal Oxide with a Room-Temperature Photoreversible Phase Transition. Nat. Chem. 2010, 2, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Nasu, T.; Miyamoto, Y.; Ozaki, N.; Tanaka, S.; Nagata, T.; Hakoe, F.; Yoshikiyo, M.; Nakagawa, K.; Umeta, Y.; et al. Structural Phase Transition between Lambda-Ti3O5 and Delta-Ti3O5 by Breaking of a One-Dimensionally Conducting Pathway. Cryst. Growth Des. 2015, 15, 653–657. [Google Scholar] [CrossRef]
- Moon, R.M. Antiferromagnetism in V2O3. Phys. Rev. Lett. 1970, 25, 527–529. [Google Scholar] [CrossRef]
- Thomas, G.A.; Rapkine, D.H.; Carter, S.A.; Millis, A.J. Observation of the Gap and Kinetic Energy in a Correlated Insulator. Phys. Rev. Lett. 1994, 73, 1529–1532. [Google Scholar] [CrossRef]
- Berglund, C.N.; Guggenheim, H.J. Electronic Properties of VO2 near the Semiconductor-Metal Transition. Phys. Rev. 1969, 185, 1022–1033. [Google Scholar] [CrossRef]
- Hill, A.H.; Harrison, A.; Dickinson, C.; Zhou, W.; Kockelmann, W. Crystallographic and Magnetic Studies of Mesoporous Eskolaite, Cr2O3. Microporous Mesoporous Mater. 2010, 130, 280–286. [Google Scholar] [CrossRef]
- Crawford, J.A.; Vest, R.W. Electrical Conductivity of Single-Crystal Cr2O3. J. Appl. Phys. 1964, 35, 2413–2418. [Google Scholar] [CrossRef]
- Cheng, C.-S.; Gomi, H.; Sakata, H. Electrical and Optical Properties of Cr2O3 Films Prepared by Chemical Vapour Deposition. Phys. Stat. Sol. 1996, 155, 417–425. [Google Scholar] [CrossRef]
- Kubota, B. Preparation of Ferromagnetic Chromium Dioxide. J. Phys. Soc. Jpn. 1960, 15, 1706. [Google Scholar] [CrossRef]
- Cheetham, A.K.; Hope, D.A.O. Magnetic Ordering and Exchange Effects in the Antiferromagnetic Solid Solutions MnxNi1-XO. Phys. Rev. B 1983, 27, 6964–6967. [Google Scholar] [CrossRef]
- Drabkin, I.A.; Emel’yanova, L.T.; Iskenderov, R.N.; Ksendzov, Y.M. Photoconductivity of Single Crystals of MnO. Sov. Phys. Solid State 1969, 10, 2428. [Google Scholar]
- Iskenderov, R.N.; Drabkin, I.A.; Evel’yanova, L.T.; Ksendzov, Y.M. Absorption Spectrum of MnO Single Crystals. Sov. Phys. Solid State 1969, 10, 2031. [Google Scholar]
- Cockayne, E.; Levin, I.; Wu, H.; Llobet, A. The Magnetic Structure of Bixbyite α-Mn2O3: A Combined Density Functional Theory DFT+U and Neutron Diffraction Study. Phys. Rev. B 2012, 87, 184413. [Google Scholar] [CrossRef]
- Regulski, M.; Przeniosło, R.; Sosnowska, I.; Hohlwein, D.; Schneider, R. Neutron Diffraction Study of the Magnetic Structure of α-Mn2O3. J. Alloys Compd. 2004, 362, 236–240. [Google Scholar] [CrossRef]
- Sharma, S.; Chauhan, P.; Husain, S. Structural and Optical Properties of Mn2O3 Nanoparticles & Its Gas Sensing Applications. Adv. Mater. Proc. 2016, 2, 220–225. [Google Scholar] [CrossRef]
- Awad, H.D.; Elttayef, A.K.; Ressen, A.L.; Ali, K.A. The Effect of Annealing on the Structural and Optical Properties of Mn2O3 Thin Film Prepared by Chemical Spray Pyrolysis. Int. J. Sci. Res. 2017, 6, 291–294. [Google Scholar] [CrossRef]
- Bose, V.C.; Maniammal, K.; Madhu, G.; Veenas, C.L.; Aiswarya, R.; Biju, V. DC Electrical Conductivity of Nanocrystalline Mn3O4 Synthesized through a Novel Sol-Gel Route. Int. Conf. Mater. Sci. Technol. 2015, 73, 012084. [Google Scholar] [CrossRef]
- Gao, T.; Glerup, M.; Krumeich, F.; Nesper, R.; Fjellvåg, H.; Norby, P. Microstructures and Spectroscopic Properties of Cryptomelane-Type Manganese Dioxide Nanofibers. J. Phys. Chem. C 2008, 112, 13134–13140. [Google Scholar] [CrossRef]
- Li, T.; Wu, J.; Xiao, X.; Zhang, B.; Hu, Z.; Zhou, J. Band Gap Engineering of MnO2 through in Situ Al-Doping for Applicable Pseudocapacitors. RCS Adv. 2016, 6, 13914–13919. [Google Scholar] [CrossRef]
- Druilhe, R.; Suchet, J.P. Electron Transport in CrO2 and MnxCr1-XO2. Czech. J. Phys. B 1967, 17, 337–346. [Google Scholar] [CrossRef]
- Greedan, J.E.; Raju, N.P.; Wills, A.S.; Morin, C.; Shaw, S.M.; Reimers, J.N. Structure and Magnetism in λ-MnO2. Geometric Frustration in a Defect Spinel. Chem. Mater. 1998, 10, 3058–3067. [Google Scholar] [CrossRef]
- Bhargande, S.K.; Patil, P.S. Structural, Optical Investigation of Manganese Oxide Thin Films by Spray Pyrolysis Technique. Res. Rev. J. Pure Appl. Phys. Struct 2013, 1, 19–23. [Google Scholar]
- Rakhecha, V.C.; Murthy, N.S.S. Spin-Transfer Due to Covalency for the Tetrahedral-Site Fe3+ Ions in Fe3O4. J. Phys. C Solid State Phys. 1978, 11, 4389–4404. [Google Scholar] [CrossRef]
- Wright, J.P.; Attfield, J.P.; Radaelli, P.G. Charge Ordered Structure of Magnetite Fe3O4 below the Verwey Transition. Phys. Rev. B 2002, 66, 214422. [Google Scholar] [CrossRef]
- Krén, E.; Szabó, P.; Konczos, G. Neutron Diffraction Studies on the (1−x) Fe2O3−xRh2O3 System. Phys. Lett. 1965, 19, 103–104. [Google Scholar] [CrossRef]
- Maslen, E.N.; Streltsov, V.A.; Streltsova, N.R.; Ishizawa, N. Synchrotron X-Ray Study of the Electron Density in α-Fe2O3. Acta Crystallogr. Sect. B Struct. Sci. 1994, 50, 435–441. [Google Scholar] [CrossRef]
- Finger, L.W.; Hazen, R.M. Crystal Structure and Isothermal Compression of Fe2O3, Cr2O3, and V2O3 to 50 kbars. J. Appl. Phys. 1980, 51, 5362–5367. [Google Scholar] [CrossRef]
- Malina, O.; Tuček, J.; Jakubec, P.; Kašlík, J.; Medřík, I.; Tokoro, H.; Yoshikiyo, M.; Namai, A.; Ohkoshi, S.; Zbořil, R. Magnetic Ground State of Nanosized β-Fe2O3 and Its Remarkable Electronic Features. RSC Adv. 2015, 5, 49719–49727. [Google Scholar] [CrossRef]
- Yoshikiyo, M.; Yamada, K.; Namai, A.; Ohkoshi, S.-I. Study of the Electronic Structure and Magnetic Properties of ε-Fe2O3 by First-Principles Calculation and Molecular Orbital Calculations. J. Phys. Chem. C 2012, 116, 8688–8691. [Google Scholar] [CrossRef]
- Khan, D.C.; Erickson, R.A. Magnetic Form Factor of Co++ Ion in Cobaltous Oxide. Phys. Rev. B 1970, 1, 2243–2249. [Google Scholar] [CrossRef]
- Herrmann-Ronzaud, D.; Burlet, P.; Rossat-Mignod, J. Equivalent Type-II Magnetic Structures: CoO, a Collinear Antiferromagnet. J. Phys. C Solid State Phys. 1978, 11, 2123–2137. [Google Scholar] [CrossRef]
- Sasaki, S.; Fujino, K.; Takéuchi, Y. X-Ray Determination of Electron-Density Distributions in Oxides, MgO, MnO, CoO, and NiO, and Atomic Scattering Factors of Their Constituent Atoms. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 1979, 55, 43–48. [Google Scholar] [CrossRef]
- Ikedo, Y.; Sugiyama, J.; Nozaki, H.; Itahara, H.; Brewer, J.H.; Ansaldo, E.J.; Morris, G.D.; Andreica, D.; Amato, A. Spatial Inhomogeneity of Magnetic Moments in the Cobalt Oxide Spinel Co3O4. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 75, 1–8. [Google Scholar] [CrossRef]
- Roth, W.L. The Magnetic Structure of Co3O4. J. Phys. Chem. Solids 1964, 25, 1–10. [Google Scholar] [CrossRef]
- Qiao, L.; Xiao, H.Y.; Meyer, H.M.; Sun, J.N.; Rouleau, C.M.; Puretzky, A.A.; Geohegan, D.B.; Ivanov, I.N.; Yoon, M.; Weber, W.J.; et al. Nature of the Band Gap and Origin of the Electro-/Photo-Activity of Co3O4. J. Mater. Chem. C 2013, 1, 4628–4633. [Google Scholar] [CrossRef]
- Alperin, H.A. The Magnetic Form Factor of Nickel Oxide. J. Phys. Soc. Jpn. Suppl. B. 1962, 17, 12–15. [Google Scholar]
- Fender, B.E.F.; Jacobson, A.J.; Wedgwood, F.A. Covalency Parameters in MnO, α-MnS, and NiO. J. Chem. Phys. 1968, 48, 990–994. [Google Scholar] [CrossRef]
- Hüfner, S.; Osterwalder, J.; Riesterer, T.; Hulliger, F. Photoemission and Inverse Photoemission Spectroscopy of NiO. Solid State Commun. 1984, 52, 793–796. [Google Scholar] [CrossRef]
- Sawatzky, G.A.; Allen, J.W. Magnitude and Origin of the Band Gap in NiO. Phys. Rev. Lett. 1984, 53, 2339–2342. [Google Scholar] [CrossRef]
- Forsyth, J.B.; Brown, P.J.; Wanklyn, B.M. Magnetism in Cupric Oxide. J. Phys. C Solid State Phys. 1988, 21, 2917–2929. [Google Scholar] [CrossRef]
- Yang, B.X.; Thurston, T.R.; Tranquada, J.M.; Shirane, G. Magnetic Neutron Scattering Study of Single-Crystal Cupric Oxide. Phys. Rev. B 1989, 39, 4343–4349. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Tranquada, J.; Shirane, G. Neutron Scattering Studies of the Magnetic Structure of Cupric Oxide. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 38, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Marabelli, F.; Parravicini, G.B.; Salghetti-Drioli, F. Optical Gap of CuO. Phys. Rev. B 1995, 52, 1433–1436. [Google Scholar] [CrossRef] [PubMed]
- Pinsard-Gaudart, L.; Rodriguez-Carvajal, J.; Gukasov, A.; Monod, P. Magnetic Properties of Paramelaconite (Cu4O3): A Pyrochlore Lattice with S=1/2. Phys. Rev. B 2004, 69, 104408. [Google Scholar] [CrossRef]
- Meyer, B.K.; Polity, A.; Reppin, D.; Becker, M.; Hering, P.; Klar, P.J.; Sander, T.; Reindl, C.; Benz, J.; Eickhoff, M.; et al. Binary Copper Oxide Semiconductors: From Materials towards Devices. Phys. Status Solidi B 2012, 249, 1487–1509. [Google Scholar] [CrossRef]
- Berlijn, T.; Snijders, P.C.; Delaire, O.; Zhou, H.; Maier, T.A.; Cao, H.; Chi, S.; Matsuda, M.; Wang, Y.; Koehler, M.R.; et al. Itinerant Antiferromagnetism in RuO2. Phys. Rev. Lett. 2017, 118, 077201. [Google Scholar] [CrossRef]
- Rice, C.E.; Robinson, W.R. Structural Changes Associated with the Semiconductor-to-Metal Transition in Ti2O3. Mater. Res. Bull. 1976, 11, 1355–1359. [Google Scholar] [CrossRef]
- Guo, Y.; Clark, S.J.; Robertson, J. Electronic and Magnetic Properties of Ti2O3, Cr2O3, and Fe2O3 Calculated by the Screened Exchange Hybrid Density Functional. J. Phys. Condens. Matter 2012, 24, 325504. [Google Scholar] [CrossRef]
- Onoda, M. Phase Transitions of Ti3O5. J. Solid State Chem. 1998, 136, 67–73. [Google Scholar] [CrossRef]
- Steiner, H.; Turrillasb, X.; Steele, B.C.H. Phase Relationships and Electrical Properties of Ti3O5, CrTi2O5 and the Pseudobrookite-Type Systems MgxTi3-XO5 and LixTi3-XO5. J. Mater. Chem. 1992, 2, 1249–1256. [Google Scholar] [CrossRef]
- Åsbrink, S.; Magnéli, A. Crystal Structure Studies on Trititanium Pentoxide, Ti3O5. Acta Crystallogr. 1959, 12, 575–581. [Google Scholar] [CrossRef]
- Hong, B.Y.S.; Sbrink, S. The Structure of Gamma-Ti3O5 at 297 K. Acta Cryst. 1982, 2570–2576. [Google Scholar] [CrossRef]
- Dernier, P.D.; Marezio, M. Crystal Structure of the Low-Temperature Antiferromagnetic Phase of V2O3. Phys. Rev. B 1970, 2, 3771–3776. [Google Scholar] [CrossRef]
- Dernier, P.D. The Crystal Structure of V2O3 and (V0.962Cr0.0382)2O3 near the Metal-Insulator Transition. J. Phys. Chem. Solids 1970, 31, 2569–2575. [Google Scholar] [CrossRef]
- Catti, M.; Sandrone, G.; Dovesi, R. Periodic Unrestricted Hartree-Fock Study of Corundum like Ti2O3 and V2O3. Phys. Rev. B Condens. Matter Mater. Phys. 1997, 55, 16122–16131. [Google Scholar] [CrossRef]
- Qazilbash, M.M.; Schafgans, A.A.; Burch, K.S.; Yun, S.J.; Chae, B.G.; Kim, B.J.; Kim, H.T.; Basov, D.N. Electrodynamics of the Vanadium Oxides VO2 and V2O3. Phys. Rev. B Condens. Matter Mater. Phys. 2008, 77, 1–10. [Google Scholar] [CrossRef]
- Rogers, K.D. An X-ray Diffraction Study of Semiconductor and Metallic Vanadium Dioxide. Poweder Diffr. 1993, 8, 240–244. [Google Scholar] [CrossRef]
- Théobald, F.; Cabala, R.; Bernard, J. Essai Sur La Structure de VO2(B). J. Solid State Chem. 1976, 17, 431–438. [Google Scholar] [CrossRef]
- Kung, H.H. Transition Metal Oxides: Surface Chemistry and Catalysis; Elsevier: Amsterdam, The Netherlands, 1989. [Google Scholar]
- Pintchovski, F.; Glaunsinger, W.S.; Navrotsky, A. Experimental Study of the Electronic and Lattice Contributions to the VO2 Transition. J. Phys. Chem. Solids 1978, 39, 941–949. [Google Scholar] [CrossRef]
- Park, J.H.; Coy, J.M.; Serkan Kasirga, T.; Huang, C.; Fei, Z.; Hunter, S.; Cobden, D.H. Measurement of a Solid-State Triple Point at the Metal-Insulator Transition in VO2. Nature 2013, 500, 431–434. [Google Scholar] [CrossRef]
- Grau-Crespo, R.; Wang, H.; Schwingenschlögl, U. Why the Heyd-Scuseria-Ernzerhof Hybrid Functional Description of VO2 Phases Is Not Correct. Phys. Rev. B Condens. Matter Mater. Phys. 2012, 86, 2–5. [Google Scholar] [CrossRef]
- Burdett, J.K.; Miller, G.J.; Richardson, J.W.; Smith, V.J. Low-Temperature Neutron Powder Diffraction Study of Chromium Dioxide and the Validity of the Jahn-Teller Viewpoint. J. Am. Chem. Soc. 1988, 110, 8064–8071. [Google Scholar] [CrossRef]
- Tan, M.; Tao, X. Ab Initio Electronic Structure of CrO2. Chin. Phys. Lett. 1999, 16, 199–201. [Google Scholar] [CrossRef]
- Li, X.W.; Gupta, A.; Xiao, G. Influence of Strain on the Magnetic Properties of Epitaxial (100) Chromium Dioxide (CrO2) Films. Appl. Phys. Lett. 1999, 75, 713–715. [Google Scholar] [CrossRef]
- Yang, F.Y.; Chien, C.L.; Li, X.W.; Xiao, G.; Gupta, A. Critical Behavior of Epitaxial Half-Metallic Ferromagnetic CrO2 films. Phys. Rev. B Condens. Matter Mater. Phys. 2001, 63, 2–5. [Google Scholar] [CrossRef]
- Blech, I.A.; Averbach, B.L. Spin Correlations in MnO. Physics 1964, 1, 31–44. [Google Scholar] [CrossRef]
- Nam, K.M.; Kim, I.Y.; Jo, Y.; Lee, S.M.; Kim, B.G.; Choi, R.; Choi, I.S.; Song, H.; Park, J.T. New Crystal Structure: Synthesis and Characterization of Hexagonal Wurtzite MnO. J. Am. Chem. Soc. 2012, 134, 8392–8395. [Google Scholar] [CrossRef]
- Schrön, A.; Rödl, C.; Bechstedt, F. Energetic Stability and Magnetic Properties of MnO in the Rocksalt, Wurtzite, and Zinc-Blende Structures: Influence of Exchange and Correlation. Phys. Rev. B Condens. Matter Mater. Phys. 2010, 82, 165109. [Google Scholar] [CrossRef]
- Geller, S. Structure of Alpha-Mn2O3, (Mn0.983Fe0.017)2O3 and (Mn0.37Fe0.63)2O3 and Relation to Magnetic Ordering. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1971, 27, 821–828. [Google Scholar] [CrossRef]
- Norrestam, R. Alpha-Manganese (III) Oxide-a C-Type Sesquioxide of Orthorhombic Symmetry. Acta Chem. Scand. 1967, 21, 2871–2884. [Google Scholar] [CrossRef]
- Chevalier, R.R.; Roult, G.; Bertaut, E.F. Etude Par Effet Mossbauer Du Systeme Mn2-XFexO3-et Transitions Magnetiques Dans Mn2O3 Par Diffraction Neutronique. Solid State Commun. 1967, 5, 7–11. [Google Scholar] [CrossRef]
- Sharrouf, M.; Awad, R.; Roumié, M.; Marhaba, S. Structural, Optical and Room Temperature Magnetic Study of Mn2O3 Nanoparticles. Mater. Sci. Appl. 2015, 850–859. [Google Scholar] [CrossRef]
- Satomi, K. Oxygen Positional Parameters of Tetragonal Mn3O4. J. Phys. Soc. Jpn. 1961, 16, 258–266. [Google Scholar] [CrossRef]
- Paris, E.; Ross, I.; Charles, R.; Olijnyk, H. Mn3O4 at High Pressure: A Diamond-Anvil Cell Study and a Structural Modelling. Eur. J. Mineral. 1992, 4, 87–94. [Google Scholar] [CrossRef]
- Franchini, C.; Podloucky, R.; Paier, J.; Marsman, M.; Kresse, G. Ground-State Properties of Multivalent Manganese Oxides: Density Functional and Hybrid Density Functional Calculations. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 75, 195128. [Google Scholar] [CrossRef]
- Chartier, A.; Arco, P.D.; Dovesi, R.; Giuria, P.; Torino, I.-; Saunders, V.R. Ab Initio Hartree-Fock Investigation of the Structural, Electronic, and Magnetic Properties of Mn3O4. Phys. Rev. B 1999, 60, 42–48. [Google Scholar] [CrossRef]
- Rossouw, M.H.; Liles, D.C.; Thackeray, M.M. Alpha Manganese Dioxide for Lithium Batteries: A Structural and Electrochemical Study. Mat. Res. Bull. 1992, 27, 221–230. [Google Scholar] [CrossRef]
- Fong, C.; Kennedy, B.J.; Elcombe, M.M. A Powder Neutron Diffraction Study of Lambda and Gamma Manganese Dioxide and of LiMn2O4. Z. Krist. 1994, 209, 941–945. [Google Scholar] [CrossRef]
- Bolzan, A.A.; Fong, C.; Kennedy, B.J.; Howard, C.J. Powder Neutron Diffraction Study of Pyrolusite, Beta-MnO2. Aust. J. Chem. 1993, 46, 939–944. [Google Scholar] [CrossRef]
- Vadlamani, B.; An, K.; Jagannathan, M.; Chandran, K.S.R. An In-Situ Electrochemical Cell for Neutron Diffraction Studies of Phase Transitions in Small Volume Electrodes of Li-Ion Batteries. J. Electrochem. Soc. 2014, 161, A1731–A1741. [Google Scholar] [CrossRef]
- Yoshimori, A. A New Type of Antiferromagnetic Structure in the Rutile Type Crystal. J. Phys. Soc. Jpn. 1959, 14, 807–821. [Google Scholar] [CrossRef]
- Sato, H.; Wakiya, K.; Enoki, T.; Kiyama, T.; Wakabayashi, Y.; Nakao, H.; Murakami, Y. Magnetic Structure of β-MnO2: X-Ray Magnetic Scattering Study. J. Phys. Soc. Jpn. 2001, 70, 37–40. [Google Scholar] [CrossRef]
- Yamamoto, N.; Endo, T.; Shimada, M.; Takada, T. Single Crystal Growth of Alpha-MnO2. Jpn. J. Appl. Phys. 1974, 13, 723–724. [Google Scholar] [CrossRef]
- Guo, L.W.; Peng, D.L.; Makino, H.; Hanada, T.; Hong, S.K.; Sumiyama, K.; Yao, T.; Inaba, K. Structural Characteristics and Magnetic Properties of λ-MnO2 Films Grown by Plasma-Assisted Molecular Beam Epitaxy. J. Appl. Phys. 2001, 90, 351–354. [Google Scholar] [CrossRef]
- Mazzocchi, V.L.; Parente, C.B.R. Refinement of the Ferri- and Paramagnetic Phases of Magnetite from Neutron Multiple Diffraction Data. J. Appl. Crystallogr. 1998, 31, 718–725. [Google Scholar] [CrossRef]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurences, and Uses; Wiley: Weinheim, Germany, 2003; ISBN 978-3-527-60644-3. [Google Scholar]
- Masrour, R.; Hlil, E.K.; Hamedoun, M.; Benyoussef, A.; Mounkachi, O.; El Moussaoui, H. Electronic and Magnetic Structures of Fe3O4ferrimagnetic Investigated by First Principle, Mean Field and Series Expansions Calculations. J. Magn. Magn. Mater. 2015, 378, 37–40. [Google Scholar] [CrossRef]
- Danno, T.; Nakatsuka, D.; Kusano, Y.; Asaoka, H.; Nakanishi, M.; Fujii, T.; Ikeda, Y.; Takada, J. Crystal Structure of β-Fe2O3 and Topotactic Phase Transformation to α-Fe2O3. Cryst. Growth Des. 2013, 13, 770–774. [Google Scholar] [CrossRef]
- Kelm, K.; Mader, W. Synthesis and Structural Analysis of ε-Fe2O3. Z. Anorg. Allg. Chem. 2005, 631, 2383–2389. [Google Scholar] [CrossRef]
- Svendsen, M.B. Beta-Fe2O3-Eine Neue Eisen(III)Oxyd-Struktur. Die Nat. 1958, 45, 542. [Google Scholar] [CrossRef]
- Dachs, V.H. Die Kristallstrucktur Des Bixbyits (Fe,Mn)2O3. Z. Krist. 1956, 107, 370–395. [Google Scholar] [CrossRef]
- Sakurai, S.; Namai, A.; Hashimoto, K.; Ohkoshi, S.-I. First Observation of Phase Transformation of All Four Fe2O3 Phases (Gamma -> Epsilon -> Beta -> Alpha-Phase). J. Am. Chem. Soc. 2009, 131, 18299–18303. [Google Scholar] [CrossRef] [PubMed]
- Linnera, J.; Karttunen, A.J. Lattice Dynamical Properties of Antiferromagnetic MnO, CoO, and NiO, and the Lattice Thermal Conductivity of NiO. Phys. Rev. B 2019, 100, 144307. [Google Scholar] [CrossRef]
- Risbud, A.S.; Snedeker, L.P.; Elcombe, M.M.; Cheetham, A.K.; Seshadri, R. Wurtzite CoO. Chem. Mater. 2005, 17, 834–838. [Google Scholar] [CrossRef]
- Will, G.; Masciocchi, N.; Parrish, W.; Hart, M. Refinement of Simple Crystal Structures from Synchrotron Radiation Powder Diffraction Data. J. Appl. Crystallogr. 1987, 20, 394–401. [Google Scholar] [CrossRef]
- Jiang, Z.; Rappe, A.M. Mechanistic Study of the Li–Air Battery with a Co3O4 Cathode and Dimethyl Sulfoxide Electrolyte. J. Phys. Chem. C 2021, 125, 21873–21881. [Google Scholar] [CrossRef]
- Zasada, F.; Piskorz, W.; Sojka, Z. Cobalt Spinel at Various Redox Conditions: DFT+U Investigations into the Structure and Surface Thermodynamics of the (100) Facet. J. Phys. Chem. C 2015, 119, 19180–19191. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 6th ed.; John Wiley & Sons: New York, NY, USA, 1986; ISBN 978-0-471-87474-4. [Google Scholar]
- Lide, D. CRC Handbook of Chemistry and Physics, 79th ed.; CRC: Boca Raton, FL, USA, 2005; ISBN 0-8493-0485-7. [Google Scholar]
- Åsbrink, S.; Norrby, L.J. A Refinement of the Crystal Structure of Copper(II) Oxide with a Discussion of Some Exceptional e.s.d.’s. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1970, 26, 8–15. [Google Scholar] [CrossRef]
- Rödl, C.; Sottile, F.; Reining, L. Quasiparticle Excitations in the Photoemission Spectrum of CuO from First Principles: A GW Study. Phys. Rev. B Condens. Matter Mater. Phys. 2015, 91, 045102. [Google Scholar] [CrossRef]
- O’Keeffe, M.; Bovin, J.-O. The Crystal Structure of Paramelaconite, Cu4O3. Am. Mineral. 1978, 63, 180–185. [Google Scholar]
- Heinemann, M.; Eifert, B.; Heiliger, C. Band Structure and Phase Stability of the Copper Oxides Cu2O, CuO, and Cu4O3. Phys. Rev. B Condens. Matter Mater. Phys. 2013, 87, 3–7. [Google Scholar] [CrossRef]
- Bolzan, A.; Kennedy, B.; Howard, C. Neutron Powder Diffraction Study of Molybdenum and Tungsten Dioxides. Aust. J. Chem. 1995, 48, 1473. [Google Scholar] [CrossRef]
- Rogers, D.B.; Shannon, R.D.; Sleight, A.W.; Gillson, J.L. Crystal Chemistry of Metal Dioxides with Rutile-Related Structures. Inorg. Chem. 1969, 8, 841–849. [Google Scholar] [CrossRef]
- Ben-Dor, L.; Shimony, Y. Crystal Structure, Magnetic Susceptibility and Electrical Conductivity of Pure and NiO-Doped MoO2 and WO2. Mat. Res. Bull. 1974, 9, 837–844. [Google Scholar] [CrossRef]
- Rodriguez, E.E.; Poineau, F.; Llobet, A.; Sattelberger, A.P.; Bhattacharjee, J.; Waghmare, V.U.; Hartmann, T.; Cheetham, A.K. Structural Studies of TcO2 by Neutron Powder Diffraction and First-Principles Calculations. J. Am. Chem. Soc. 2007, 129, 10244–10248. [Google Scholar] [CrossRef] [PubMed]
- Boman, C.-E.; Danielsen, J.; Haaland, A.; Jerslev, B.; Schäffer, C.E.; Sunde, E.; Sørensen, N.A. Refinement of the Crystal Structure of Ruthenium Dioxide. Acta Chem. Scand. 1970, 24, 116–122. [Google Scholar] [CrossRef]
- Ryden, W.D.; Lawson, A.W. Magnetic Susceptibility of IrO2 and RuO2. J. Chem. Phys. 1970, 52, 6058–6061. [Google Scholar] [CrossRef]
- Shannon, R.D. Synthesis and Properties of Two New Members of the Rutile Family RhO2 and PtO2. Solid State Commun. 1968, 6, 139–143. [Google Scholar] [CrossRef]
- Shirako, Y.; Wang, X.; Tsujimoto, Y.; Tanaka, K.; Guo, Y.; Matsushita, Y.; Nemoto, Y.; Katsuya, Y.; Shi, Y.; Mori, D.; et al. Synthesis, Crystal Structure, and Electronic Properties of High-Pressure PdF2-Type Oxides MO2(M = Ru, Rh, Os, Ir, Pt). Inorg. Chem. 2014, 53, 11616–11625. [Google Scholar] [CrossRef]
- Richards, R. Surface and Nanomolecular Catalysis, 1st ed.; CRC Press: Boca Raton, FL, USA, 2006; ISBN 978-0-367-39081-5. [Google Scholar]
- Standke, B.; Jansen, M. Darstellung Und Kristallstruktur von Ag3O4. J. Solid State Chem. 1987, 67, 278–284. [Google Scholar] [CrossRef]
- Tudela, D. Silver(II) Oxide or Silver(I,III) Oxide? J. Chem. Educ. 2008, 85, 863. [Google Scholar] [CrossRef]
- Standke, B.; Jansen, M. Ag3O4, the First Silver(II,III) Oxide. Angew. Chem. Int. Ed. Engl. 1986, 25, 77–78. [Google Scholar] [CrossRef]
- Palmer, D.; Dickens, P. Tungsten Dioxide: Structure Refinement by Powder Neutron Diffraction. Acta. Cryst. 1979, B35, 2199–2201. [Google Scholar] [CrossRef]
- Goodenough, J.B.; Pierre, G.; Jean, B. Magnetic and Electric Properties of ReO2: Theoretical Investigation. Compt. Rend. 1965, 261, 2331–2335. [Google Scholar]
- Gibart, P. Physico-Chemical Properties of Rhenium Dioxides. Bull. Soc. Chim. Fr. 1967, 2, 444. [Google Scholar]
- Wang, S.; Liu, Y.; Yu, Z.; Sheng, X.; Yang, S.A. Hourglass Dirac Chain Metal in Rhenium Dioxide. Nat. Commun. 2017, 8, 1844. [Google Scholar] [CrossRef]
- Corrêa, H.P.S.; Cavalcante, I.P.; Martinez, L.G.; Orlando, C.G.P.; Orlando, M.T.D. Refinement of Monoclinic ReO2 Structure from XRD by Rietveld Method Rietveld. Braz. J. Phys. 2004, 34, 1208–1210. [Google Scholar] [CrossRef][Green Version]
- Magnéli, A.; Siitonen, S.; Skrifvars, B.; Schliack, J.; Reio, L. Studies on Rhenium Oxides. Acta Chem. Scand. 1957, 11, 28–33. [Google Scholar] [CrossRef][Green Version]
- Tribalat, S.; Jungfleisch, J.; Delafosse, D. Sur Les Structures Cristallines Du Dioxyde de Rhenium. C. R. Acad. Sc. Paris 1964, 259, 2109–2112. [Google Scholar]
- Ivanovskii, A.L.; Chupakhina, T.I.; Zubkov, V.G.; Tyutyunnik, A.P.; Krasilnikov, V.N.; Bazuev, V.G.; Okatov, V.S.; Lichtenstein, A.I. Structure and Electronic Properties of New Rutile-like Rhenium (IV) Dioxide ReO2. Phys. Lett. Sect. A Gen. At. Solid State Phys. 2005, 348, 66–70. [Google Scholar] [CrossRef]
- Bolzan, A.A.; Fong, C.; Kennedy, B.J.; Howard, C.J. Structural Studies of Rutile-Type Metal Dioxides. Acta Crystallogr. Sect. B Struct. Sci. 1997, 53, 373–380. [Google Scholar] [CrossRef]
- Ping, Y.; Galli, G.; Goddard, W.A. Electronic Structure of IrO2: The Role of the Metal d Orbitals. J. Phys. Chem. C 2015, 119, 11570–11577. [Google Scholar] [CrossRef]
- Steinmann, S.N.; Corminboeuf, C. Comprehensive Benchmarking of a Density-Dependent Dispersion Correction. J. Chem. Theory Comput. 2011, 7, 3567–3577. [Google Scholar] [CrossRef] [PubMed]
- Tkatchenko, A. Current Understanding of Van Der Waals Effects in Realistic Materials. Adv. Funct. Mater. 2015, 25, 2054–2061. [Google Scholar] [CrossRef]
- Stephens, J.S.; Cruickshank, D.W.J. The Crystal Structure of (CrO3)∞. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1970, 26, 222–226. [Google Scholar] [CrossRef]
- Sitepu, H. Texture and Structural Refinement Using Neutron Diffraction Data from Molybdite (MoO3) and Calcite (CaCO3) Powders and a Ni-Rich Ni50.7Ti49.30 Alloy. Powder Diffr. 2009, 24, 315–326. [Google Scholar] [CrossRef]
- Locherer, K.R.; Swainson, I.P.; Salje, E.K.H. Transition to a New Tetragonal Phase of WO3: Crystal Structure and Distortion Parameters. J. Phys. Condens. Matter 1999, 11, 4143–4156. [Google Scholar] [CrossRef]
- Simon, V.A.; Dronskowski, R.; Krebs, B.; Hettic, B. Die Kristallstruktur von Mn2O7. Angew. Chem. 1987, 99, 160–161. [Google Scholar] [CrossRef]
- Krebs, V.B. Technetium(VIII)-Oxid: Ein Ubergangsmetalloxid Mit Molekulstruktur Im Festen Zustand. Z. Anorg. Allg. Chem. 1971, 380, 146–159. [Google Scholar] [CrossRef]
- Pley, M.; Wickleder, M.S. Two Crystalline Modifications of RuO4. J. Solid State Chem. 2005, 178, 3206–3209. [Google Scholar] [CrossRef]
- Krebs, B.; Hasse, K.D. Refinements of the Crystal Structures of KTcO4, KReO4 and OsO4. The Bond Lengths in Tetrahedral Oxoanions and Oxides of d0 Transition Metals. Acta Crystallogr. 1976, B32, 1334–1337. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Pušelj, M.; Ban, Z.; Lukačević, E.; Morvaj, J. On the Preparation of Mercuric Peroxides and Refinement of the α—HgO2 Structure. Z. Anorg. Allg. Chem. 1985, 528, 191–194. [Google Scholar] [CrossRef]
- Vannerberg, N.G. Formation and Structure of Mercuric Peroxides. Arkiv. Kemi. 1959, 13, 515–521. [Google Scholar]
- Hesse, W.; Jansen, M.; Schnick, W. Recent Results in Solid State Chemistry of Ionic Ozonides, Hyperoxides, and Peroxides. Prog. Solid State Chem. 1989, 19, 47–110. [Google Scholar] [CrossRef]
- Puselj, M.; Ban, Z.; Lukačević, E. The Crystal Structure of α-Mercury(II) Peroxide. J. Appl. Crystallogr. 1983, 16, 357. [Google Scholar] [CrossRef]
- Dovesi, R.; Orlando, R.; Erba, A.; Zicovich-Wilson, C.M.; Civalleri, B.; Casassa, S.; Maschio, L.; Ferrabone, M.; De La Pierre, M.; D’Arco, P.; et al. CRYSTAL14: A Program for the Ab Initio Investigation of Crystalline Solids. Int. J. Quantum Chem. 2014, 114, 1287–1317. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-Corrected Mean-Field Electronic Structure Methods. Chem. Rev. 2016, 116, 5105–5154. [Google Scholar] [CrossRef]
- Pascale, F.; Zicovich-Wilson, C.M.; López Gejo, F.; Civalleri, B.; Orlando, R.; Dovesi, R. The Calculation of the Vibrational Frequencies of Crystalline Compounds and Its Implementation in the CRYSTAL Code: Crystalline Compounds and the CRYSTAL Code. J. Comput. Chem. 2004, 25, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Zicovich-Wilson, C.M.; Pascale, F.; Roetti, C.; Saunders, V.R.; Orlando, R.; Dovesi, R. Calculation of the Vibration Frequencies of A-Quartz: The Effect of Hamiltonian and Basis Set. J. Comput. Chem. 2004, 25, 1873–1881. [Google Scholar] [CrossRef] [PubMed]
- Zagorac, D.; Müller, H.; Ruehl, S.; Zagorac, J.; Rehme, S. Recent Developments in the Inorganic Crystal Structure Database: Theoretical Crystal Structure Data and Related Features. J Appl Cryst. 2019, 52, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Grazulis, S.; Chateigner, D.; Downs, R.T.; Yokochi, A.F.T.; Quiros, M.; Lutterotti, L.; Manakova, E.; Butkus, J.; Moeck, P.; Le Bail, A. Crystallography Open Database—an Open-Access Collection of Crystal Structures. J. Appl. Crystallogr. 2009, 42, 726–729. [Google Scholar] [CrossRef]
- Grazulic, S.; Daskevic, A.; Merkys, A.; Chateigner, D.; Lutterotti, L.; Serebryanaya, N.R.; Moeck, P.; Downs, R.T.; Le Bail, A. Crystallography Open Database (COD): An Open-Access Collection of Crystal Structures and Platform for World-Wide Collaboration. Nucleic Acids Res. 2012, 40, 420–427. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Cryst. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Freund, H.J.; Pacchioni, G. Oxide Ultra-Thin Films on Metals: New Materials for the Design of Supported Metal Catalysts. Chem. Soc. Rev. 2008, 37, 2224–2242. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
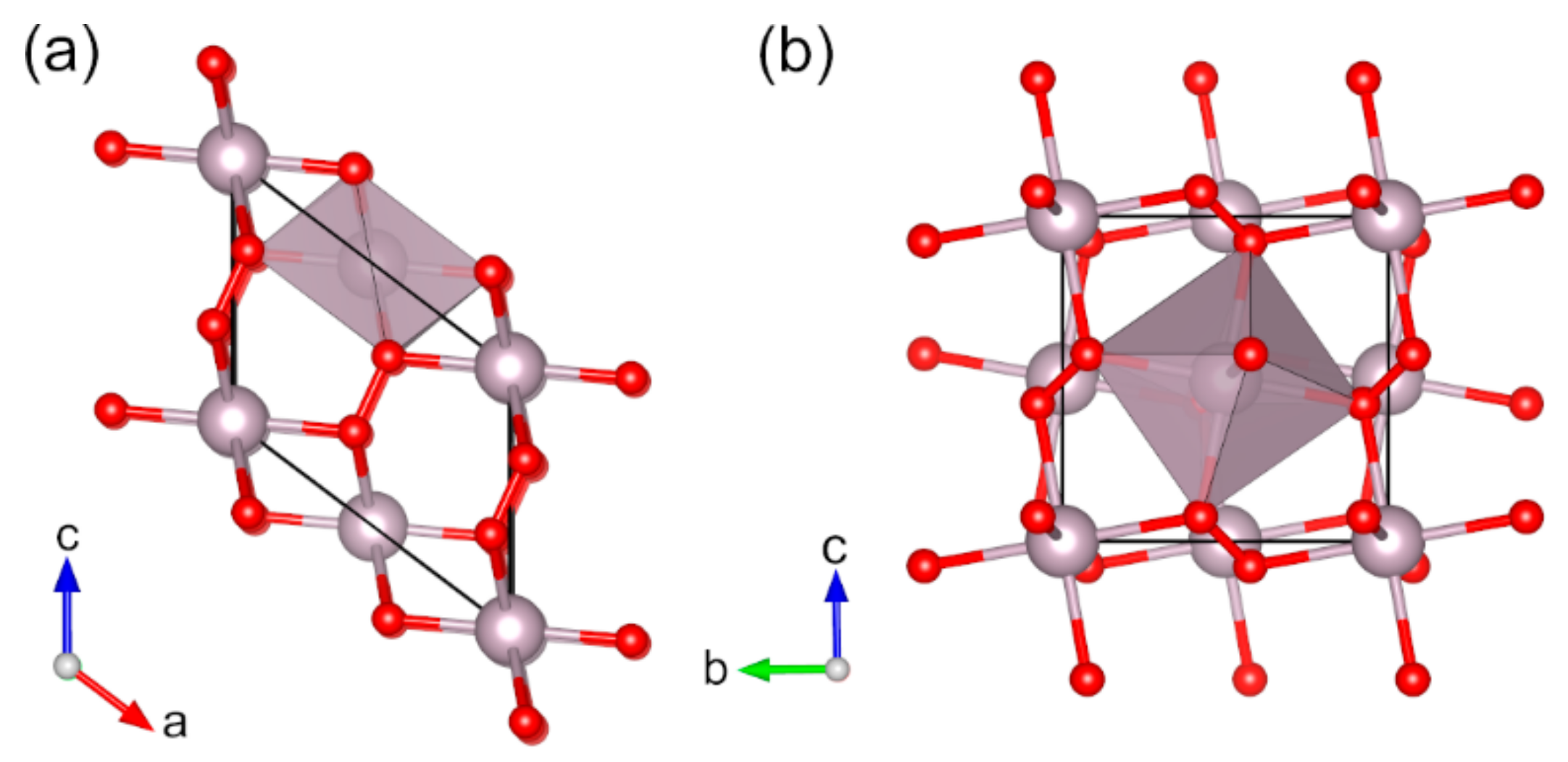{kind=link}
| Oxide | Pearson Symbol a | Space Group of Nonmagnetic Unit Cell | Space Group of Magnetic Unit Cell | Magnetic Ground State b | Magnetic Moment (µB) c | Band Gap (eV) | ||
|---|---|---|---|---|---|---|---|---|
| Calc. | Exp. | Calc. | Exp. | |||||
| 3d metals | ||||||||
| Ti2O3 | hR10 | R-3c (167) | R3c (161) | AFM | 0.9 | 2.7 | 0.1 [42] | |
| α-Ti3O5 | oS32 | Cmcm (63) | Cm (8) | FiM | 1.0 | 2.0 | ||
| β-Ti3O5 | mS32 | C2/m (12) | Cm (8) | AFM | 0.9, 1.0 | 1.3 | 0.14 [43] | |
| γ-Ti3O5 | mS32 | I2/c (15) | P1 (1) | AFM | 1.0 | 2.3 | ||
| δ-Ti3O5 | mS32 | P2/a (13) | P-1 (2) | AFM | 1.0 | 2.4 | 0.07 [44] | |
| λ-Ti3O5 | mS32 | C2/m (12) | Cm (8) | AFM | 1.0 | 1.7 | ||
| V2O3 | hR10 | R-3c (167) | R3c (161) | AFM/AFM | 2.0 | 3.0 | ||
| V2O3 | mS20 | I2/a (15) | P2/c (13) | AFM/AFM | 2.0 | 1.2 [45] | 2.8 | 0.6 [46] |
| VO2 | mP12 | P21/c (14) | P21 (4) | PM/AFM | 1.1 | 3.0 | 0.6–0.7 [47] | |
| VO2 | mS12 | C2/m (12) | Cm (8) | PM/AFM | 1.1 | 3.3 | ||
| VO2 | tP6 | P42/mnm (136) | Cmmm (65) | PM/AFM | 1.1 | 2.8 | - | |
| Cr2O3 | hR10 | R-3c (167) | R3c (161) | AFM/AFM | 3.0 | ca. 2.7 [48] | 5.1 | 3.2–3.4 [49,50] |
| CrO2 | tP6 | P4/mnm (136) | P4/mnm (136) | FM/FM | 2.4 | 2.01 [51] | ||
| MnO | cF8 | Fm-3m (225) | R-3m (166) | AFM/AFM | 4.8 | 4.58 [52] | 3.9 | 3.6–4.2 [53,54] |
| MnO | hP4 | P63mc (186) | Pmc21 (26) | AFM | 4.8 | 3.0 | ||
| Mn2O3 | oP80 | Pbca (61) | Pbca (61) | AFM/AFM | 3.9, 4.0 | 2.3–3.9 [55,56] | 3.0 | 2.17 [57], 2.4 [58] |
| Mn2O3 | cI80 | Ia-3 (206) | Ia-3 (206) | PM/FM | 4.1 | |||
| Mn3O4 | tI28 | I41/amd (141) | I41/amd (141) | FiM/FiM | 3.9, 4.0, 4.9 | 3.2 | 1.77–2.72 [59] | |
| MnO2 | tI24 | I4/m (87) | C2/m (12) | AFM/AFM | 3.1 | 3.4 | 1.32 [60] | |
| MnO2 | oP12 | Pnam (62) | Pmc21 (26) | AFM/AFM | 3.0 | 3.5 | 2.57 [61] | |
| MnO2 | tP6 | P4/mnm (136) | Cmmm (65) | AFM/AFM | 3.1 | 2.1 | 0.3 [62] | |
| MnO2 | cF48 | Fd-3m (227) | Imma (74) | AFM/AFM | 3.1 | 2.78 [63] | 3.7 | 1.7–3.5 [64] |
| Fe3O4 | cF56 | Fd-3m (227) | Fd-3m (227) | FiM/FiM | 4.0, 4.2 | 3.82 [65] | ||
| Fe3O4 | mP56 | P2/c (13) | P2/c (13) | FiM/FiM | 3.7–4.3 | 4.17, 4.44 [66] | 1.6 | 0.14 [11] |
| Fe2O3 | hR10 | R-3c (167) | R-3 (148) | AFM/AFM | 4.2 | 4.6-5.2 [67] | 4.0 | 5.0 [68,69] |
| Fe2O3 | cI80 | Ia-3 (206) | I212121 (24) | AFM/AFM | 4.3 | 3.3 | 2.2 [70] | |
| Fe2O3 | oP40 | Pna21 (33) | Pna21 (33) | FiM/AFM | 4.3 | 4.2 | 1.6 [71] | |
| CoO | cF8 | Fm3m (225) | R-3m (166) | AFM/AFM | 2.7 | 3.35, 3.8 [72,73] | 4.5 | 4.3 [74] |
| CoO | hP4 | P63mc (186) | Pmc21 (26) | AFM | 2.8 | 3.2 | ||
| Co3O4 | cF56 | Fd-3m (227) | F-43m (216) | AFM/AFM | 2.8 | 3.88 [75], 3.0 [76] | 4.0 | 0.7 [77] |
| NiO | cF8 | Fm3m (225) | R-3m (166) | AFM/AFM | 1.7 | 1.64 [78], 1.77 [79], 1.90 [52] | 5.2 | 4.0 [80], 4.3 [81] |
| CuO | mS8 | C2/c (15) | P21/c (14) | AFM/AFM | 0.6 | 0.65 [82], 0.68 [83,84] | 3.8 | 1.7 [85] |
| Cu4O3 | tI28 | I41/amd | Imma (74) | AFM/AFM | 0.7 | 0.66 [86] | 2.9 | ca. 1.5 [87] |
| 4d metals | ||||||||
| MoO2 | mP12 | P21/c (14) | P21 (4) | PM/AFM | 1.1 | |||
| TcO2 | mP12 | P21/c (14) | P21 (4) | PM/AFM | 2.7 | 2.4 | ||
| RuO2 | tP6 | P4/mnm (136) | Cmmm (65) | PM/AFM | 1.5 | 0.05 [88] | ||
| RhO2 | tP6 | P42/mnm (136) | P4/mnm (136) | PM/FM | 0.6 | |||
| Ag3O4 | mP14 | P21/c (14) | P21/c (14) | PM/FM | 0.2 | |||
| 5d metals | ||||||||
| WO2 | mP12 | P21/c (14) | P21 (4) | PM/AFM | 0.4 | |||
| ReO2 | mP12 | P21/c (14) | P21 (4) | PM/AFM | 2.1 | 1.5 | ||
| ReO2 | oP12 | Pbcn (60) | P21212 (18) | PM/AFM | 1.1 | 1.6 | ||
| ReO2 | tP6 | P42/mnm (136) | Cmmm (65) | AFM | 2.7 | 1.6 | ||
| IrO2 | tP6 | P42/mnm (136) | Cmmm (65) | PM/AFM | 0.5 | |||
| Oxide | Pearson Symbol | Space Group | a (Å) | b (Å) | c (Å) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No D3 | D3 ZD a | D3 BJ b | No D3 | D3 ZD | D3 BJ | No D3 | D3 ZD | D3 BJ | |||
| CrO3 [171] | oS16 | C2cm (40) | 5.748 (0.1%) | 5.688 (−1.0%) | 5.710 (−0.6%) | 8.979 (4.9%) | 8.050 (−5.9%) | 8.218 (−4.0%) | 4.925 (2.8%) | 4.715 (−1.6%) | 4.711 (−1.6%) |
| MoO3 [172] | oP16 | Pbnm (62) | 14.477 (4.5%) | 13.380 (−3.5%) | 13.515 (−2.5%) | 3.695 (0%) | 3.697 (0%) | 3.692 (−0.1%) | 3.972 (0.2%) | 3.955 (−0.2%) | 3.941 (−0.5%) |
| WO3 [173] | tP8 | P4/nmm (129) | 5.314 (0.2%) | 5.294 (−0.2%) | 5.297 (−0.1%) | 4.020 (2.2%) | 4.018 (2.1%) | 4.014 (2.0%) | |||
| Mn2O7 [174] | mP72 | P21/c (14) | 6.986 (2.8%) | 6.693 (−1.5%) | 6.697 (−1.4%) | 17.504 (4.9%) | 16.494 (−1.2%) | 16.493 (−1.2%) | 9.598 (1.5%) | 9.023 (−4.6%) | 9.063 (−4.1%) |
| Tc2O7 [175] | oP36 | Pbca (61) | 13.852 (0.7%) | 13.543 (−1.5%) | 13.535 (−1.6%) | 7.600 (2.2%) | 6.908 (−7.1%) | 7.033 (−5.5%) | 5.762 (2.6%) | 5.337 (−5.0%) | 5.353 (−4.7%) |
| RuO4 [176] | cP40 | P-43n (218) | 8.761 (3.0%) | 8.254 (−3.0%) | 8.359 (−1.8%) | ||||||
| RuO4 [176] | mS20 | C2/c (15) | 9.562 (2.8%) | 9.092 (−2.3%) | 9.146 (−1.7%) | 4.534 (3.1%) | 4.231 (−3.8%) | 4.318 −1.8%) | 8.673 (2.6%) | 8.177 (−3.3%) | 8.285 (−2.0%) |
| OsO4 [177] | mS20 | C2/c (15) | 9.514 (1.4%) | 9.066 (−3.3%) | 9.058 (−3.4%) | 4.572 (1.3%) | 4.321 (−4.3%) | 4.327 (−4.2%) | 8.632 (0%) | 8.212 (−4.8%) | 8.250 (−4.4%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuklin, M.S.; Eklund, K.; Linnera, J.; Ropponen, A.; Tolvanen, N.; Karttunen, A.J. Structural Properties and Magnetic Ground States of 100 Binary d-Metal Oxides Studied by Hybrid Density Functional Methods. Molecules 2022, 27, 874. https://doi.org/10.3390/molecules27030874
Kuklin MS, Eklund K, Linnera J, Ropponen A, Tolvanen N, Karttunen AJ. Structural Properties and Magnetic Ground States of 100 Binary d-Metal Oxides Studied by Hybrid Density Functional Methods. Molecules. 2022; 27(3):874. https://doi.org/10.3390/molecules27030874
Chicago/Turabian StyleKuklin, Mikhail S., Kim Eklund, Jarno Linnera, Artturi Ropponen, Nikolas Tolvanen, and Antti J. Karttunen. 2022. "Structural Properties and Magnetic Ground States of 100 Binary d-Metal Oxides Studied by Hybrid Density Functional Methods" Molecules 27, no. 3: 874. https://doi.org/10.3390/molecules27030874
APA StyleKuklin, M. S., Eklund, K., Linnera, J., Ropponen, A., Tolvanen, N., & Karttunen, A. J. (2022). Structural Properties and Magnetic Ground States of 100 Binary d-Metal Oxides Studied by Hybrid Density Functional Methods. Molecules, 27(3), 874. https://doi.org/10.3390/molecules27030874