
Martini 3 Model of Cellulose Microfibrils: On the Route to Capture Large Conformational Changes of Polysaccharides


Abstract
:1. Introduction
2. Materials and Methods
2.1. All-Atom MD Simulation
2.2. Coarse-Grained Model: Martini 3
3. Results
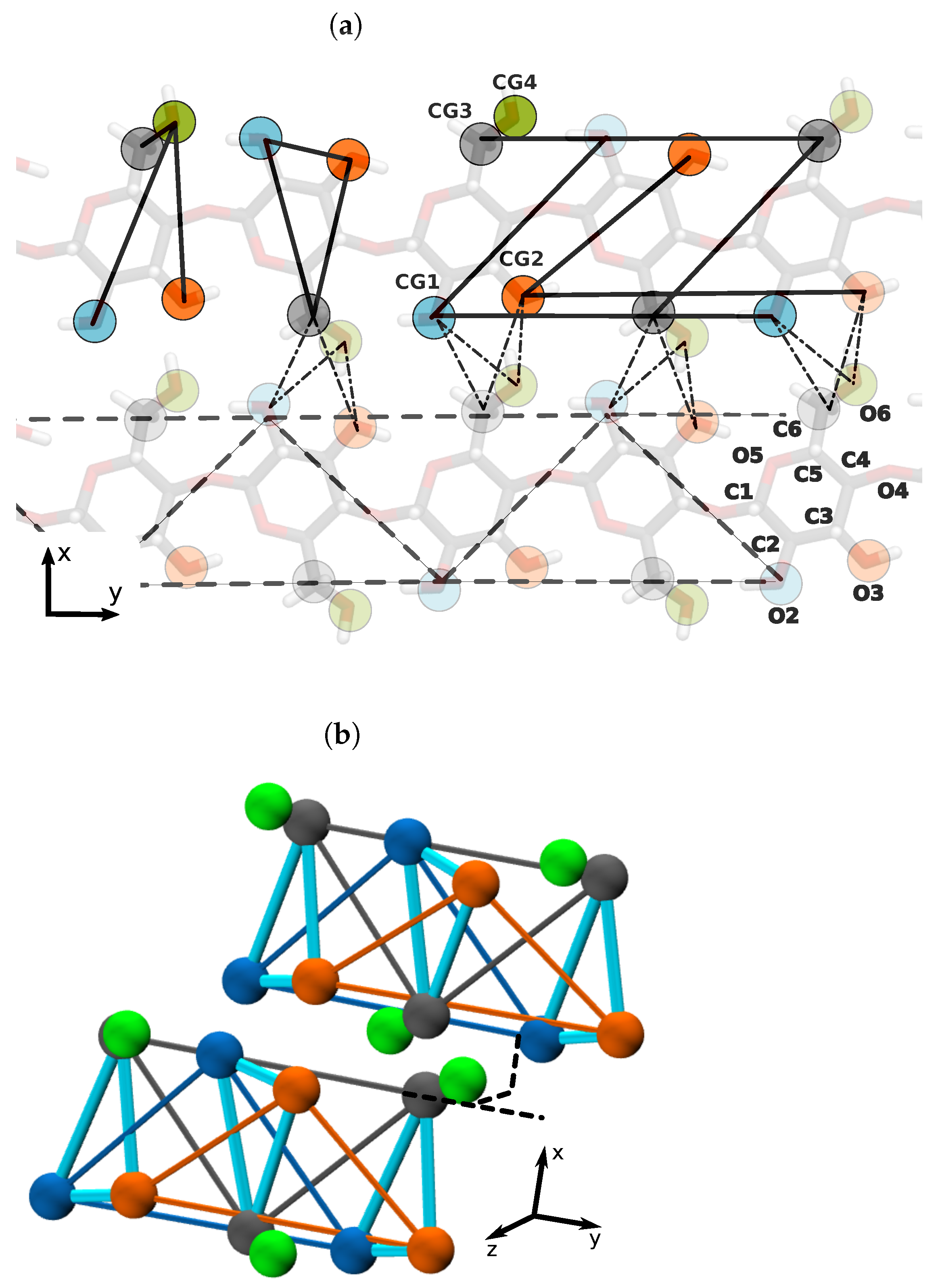
3.1. Mapping CG Structure by All-Atom MD
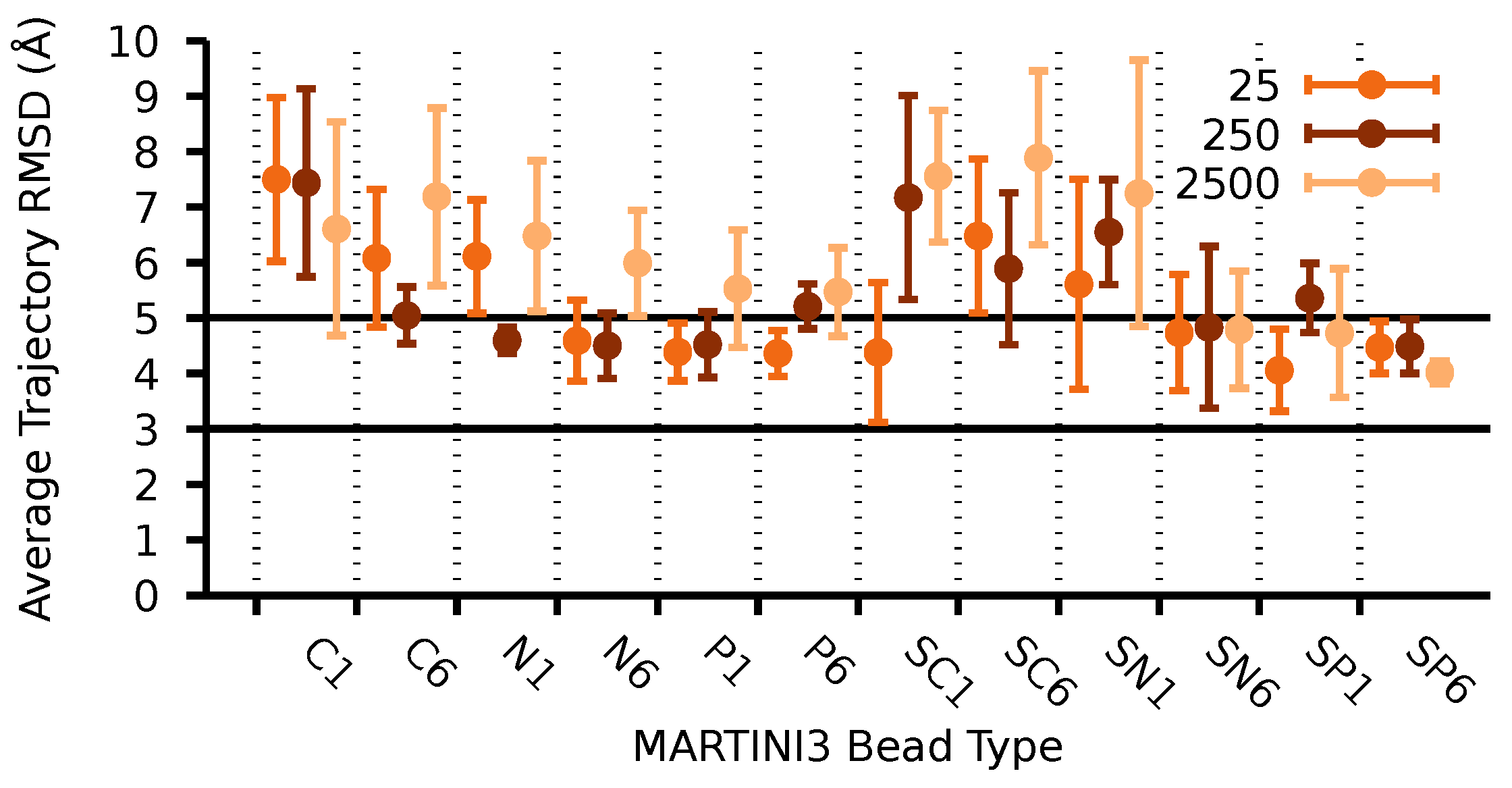
3.2. Parameterization of the CG Model for Several Cellulose Fibrils
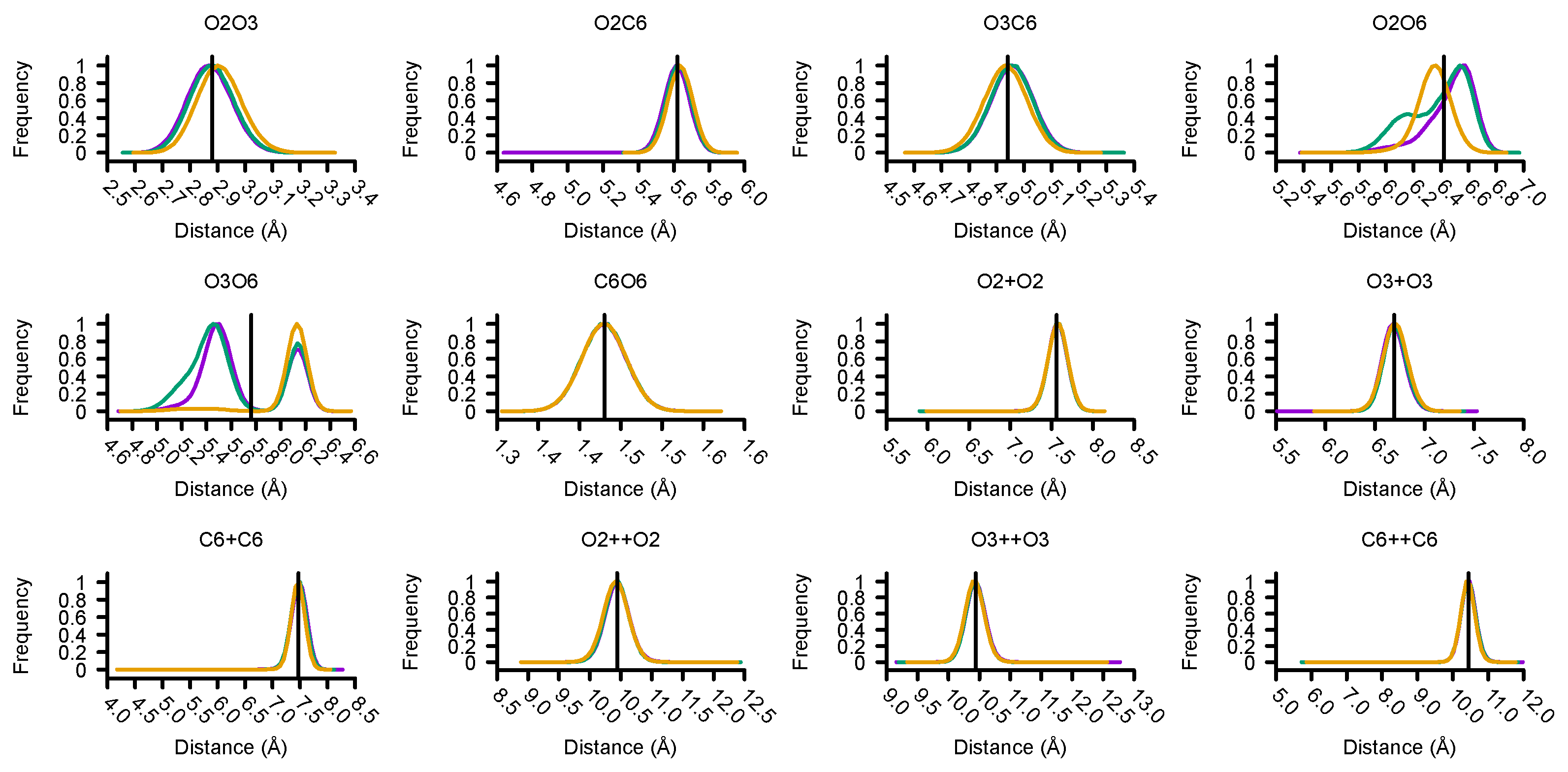
3.3. Structural Validation of the Martini 3 Model for Cellulose Fibrils
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gefen, G.; Anbar, M.; Morag, E.; Lamed, R.; Bayer, E.A. Enhanced cellulose degradation by targeted integration of a cohesin-fused β-glucosidase into the Clostridium thermocellum cellulosome. Proc. Natl. Acad. Sci. USA 2012, 109, 10298–10303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haider, T.P.; Völker, C.; Kramm, J.; Landfester, K.; Wurm, F.R. Plastics of the future? The impact of biodegradable polymers on the environment and on society. Angew. Chem. Int. Ed. 2019, 58, 50–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Wu, Y.; Chen, Q.; Yu, Z.; Wang, C.; Jin, S.; Ding, Y.; Wu, G. Dissolution of cellulose with ionic liquids and its application: A mini-review. Green Chem. 2006, 8, 325–327. [Google Scholar] [CrossRef]
- Cao, X.; Ding, B.; Yu, J.; Al-Deyab, S.S. Cellulose nanowhiskers extracted from TEMPO-oxidized jute fibers. Carbohydr. Polym. 2012, 90, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Yu, H.; Liu, Y.; Chen, W.; Wang, X.; Ouyang, M. Concentration effects on the isolation and dynamic rheological behavior of cellulose nanofibers via ultrasonic processing. Cellulose 2013, 20, 149–157. [Google Scholar] [CrossRef]
- Maurer, R.J.; Sax, A.F.; Ribitsch, V. Molecular simulation of surface reorganization and wetting in crystalline cellulose I and II. Cellulose 2013, 20, 25–42. [Google Scholar] [CrossRef] [Green Version]
- Poma, A.B.; Chwastyk, M.; Cieplak, M. Polysaccharide–protein complexes in a coarse-grained model. J. Phys. Chem. B 2015, 119, 12028–12041. [Google Scholar] [CrossRef]
- Poma, A.B.; Chwastyk, M.; Cieplak, M. Coarse-grained model of the native cellulose Iα and the transformation pathways to the Iβ allomorph. Cellulose 2016, 23, 1573–1591. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, Y.; Langan, P.; Chanzy, H. Crystal structure and hydrogen-bonding system in cellulose Iβ from synchrotron X-ray and neutron fiber diffraction. J. Am. Chem. Soc. 2002, 124, 9074–9082. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Sugiyama, J.; Chanzy, H.; Langan, P. Crystal structure and hydrogen bonding system in cellulose Iα from synchrotron X-ray and neutron fiber diffraction. J. Am. Chem. Soc. 2003, 125, 14300–14306. [Google Scholar] [CrossRef]
- Koehler, M.; Ray, A.; Moreira, R.A.; Juniku, B.; Poma, A.B.; Alsteens, D. Molecular insights into receptor binding energetics and neutralization of SARS-CoV-2 variants. Nat. Commun. 2021, 12, 6977. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Cooper, C.D.; Poma, A.B.; Vargas Guzman, H. A Free Energies of the Disassembly of Viral Capsids from a Multiscale Molecular Simulation Approach. J. Chem. Inf. Model 2020, 60, 974–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gkeka, P.; Stoltz, G.; Barati Farimani, A.; Belkacemi, Z.; Ceriotti, M.; Chodera, J.D.; Dinner, A.R.; Ferguson, A.L.; Maillet, J.B.; Minoux, H.; et al. Machine learning force fields and coarse-grained variables in molecular dynamics: Application to materials and biological systems. J. Chem. Theory Comput. 2020, 16, 4757–4775. [Google Scholar] [CrossRef]
- Fan, B.; Maranas, J.K. Coarse-grained simulation of cellulose Iβ with application to long fibrils. Cellulose 2015, 22, 31–44. [Google Scholar] [CrossRef]
- Srinivas, G.; Cheng, X.; Smith, J.C. Coarse-grain model for natural cellulose fibrils in explicit water. J. Phys. Chem. B 2014, 118, 3026–3034. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; De Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [Green Version]
- Liwo, A.; Czaplewski, C.; Sieradzan, A.K.; Lipska, A.G.; Samsonov, S.A.; Murarka, R.K. Theory and practice of coarse-grained molecular dynamics of biologically important systems. Biomolecules 2021, 11, 1347. [Google Scholar] [CrossRef]
- Ouldridge, T.E.; Louis, A.A.; Doye, J.P. DNA nanotweezers studied with a coarse-grained model of DNA. Phys. Rev. Lett. 2010, 104, 178101. [Google Scholar] [CrossRef]
- Gopal, S.M.; Mukherjee, S.; Cheng, Y.M.; Feig, M. PRIMO/PRIMONA: A coarse-grained model for proteins and nucleic acids that preserves near-atomistic accuracy. Proteins Struct. Funct. Bioinform. 2010, 78, 1266–1281. [Google Scholar] [CrossRef]
- Gomes, T.C.; Skaf, M.S. Cellulose-Builder: A toolkit for building crystalline structures of cellulose. J. Comput. Chem. 2012, 33, 1338–1346. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, E.; Guvench, O.; MacKerell, A.D., Jr. CHARMM additive all-atom force field for aldopentofuranoses, methyl-aldopentofuranosides, and fructofuranose. J. Phys. Chem. B 2009, 113, 12466–12476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Harvey, M.; De Fabritiis, G. An implementation of the smooth particle mesh Ewald method on GPU hardware. J. Chem. Theory Comput. 2009, 5, 2371–2377. [Google Scholar] [CrossRef]
- Langan, P.; Nishiyama, Y.; Chanzy, H. X-ray structure of mercerized cellulose II at 1 Å resolution. Biomacromolecules 2001, 2, 410–416. [Google Scholar] [CrossRef]
- Souza, P.C.; Alessandri, R.; Barnoud, J.; Thallmair, S.; Faustino, I.; Grünewald, F.; Patmanidis, I.; Abdizadeh, H.; Bruininks, B.M.; Wassenaar, T.A.; et al. Martini 3: A general purpose force field for coarse-grained molecular dynamics. Nat. Methods 2021, 18, 382–388. [Google Scholar] [CrossRef]
- Souza, P.C.; Thallmair, S.; Marrink, S.J.; Mera-Adasme, R. An Allosteric Pathway in Copper, Zinc Superoxide Dismutase Unravels the Molecular Mechanism of the G93A Amyotrophic Lateral Sclerosis-Linked Mutation. J. Phys. Chem. Lett. 2019, 10, 7740–7744. [Google Scholar] [CrossRef]
- Liu, Z.; Moreira, R.A.; Dujmović, A.; Liu, H.; Yang, B.; Poma, A.B.; Nash, M.A. Mapping mechanostable pulling geometries of a therapeutic anticalin/CTLA-4 protein complex. Nano Lett. 2022, 22, 179–187. [Google Scholar] [CrossRef]
- Liaci, A.M.; Steigenberger, B.; de Souza, P.C.T.; Tamara, S.; Gröllers-Mulderij, M.; Ogrissek, P.; Marrink, S.J.; Scheltema, R.A.; Förster, F. Structure of the human signal peptidase complex reveals the determinants for signal peptide cleavage. Mol. Cell 2021, 81, 3934–3948. [Google Scholar] [CrossRef]
- Croom, F.H. Basic Concepts of Algebraic Topology; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Tironi, I.G.; Sperb, R.; Smith, P.E.; van Gunsteren, W.F. A generalized reaction field method for molecular dynamics simulations. J. Chem. Phys. 1995, 102, 5451–5459. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Crystal structure and pair potentials: A molecular-dynamics study. Phys. Rev. Lett. 1980, 45, 1196. [Google Scholar] [CrossRef]
- Poma, A.B.; Cieplak, M.; Theodorakis, P.E. Combining the MARTINI and structure-based coarse-grained approaches for the molecular dynamics studies of conformational transitions in proteins. J. Chem. Theory Comput. 2017, 13, 1366–1374. [Google Scholar] [CrossRef]
- Chwastyk, M.; Bernaola, A.P.; Cieplak, M. Statistical radii associated with amino acids to determine the contact map: Fixing the structure of a type I cohesin domain in the Clostridium thermocellum cellulosome. Phys. Biol. 2015, 12, 046002. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, K.i.; Koga, N.; Takada, S.; Onuchic, J.N.; Wolynes, P.G. Multiple-basin energy landscapes for large-amplitude conformational motions of proteins: Structure-based molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2006, 103, 11844–11849. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, M.I.; Poma, A.B.; Okazaki, K.i. Optimizing Gō-MARTINI coarse-grained model for F-BAR protein on lipid membrane. Front. Mol. Biosci. 2021, 8, 619381. [Google Scholar] [CrossRef]
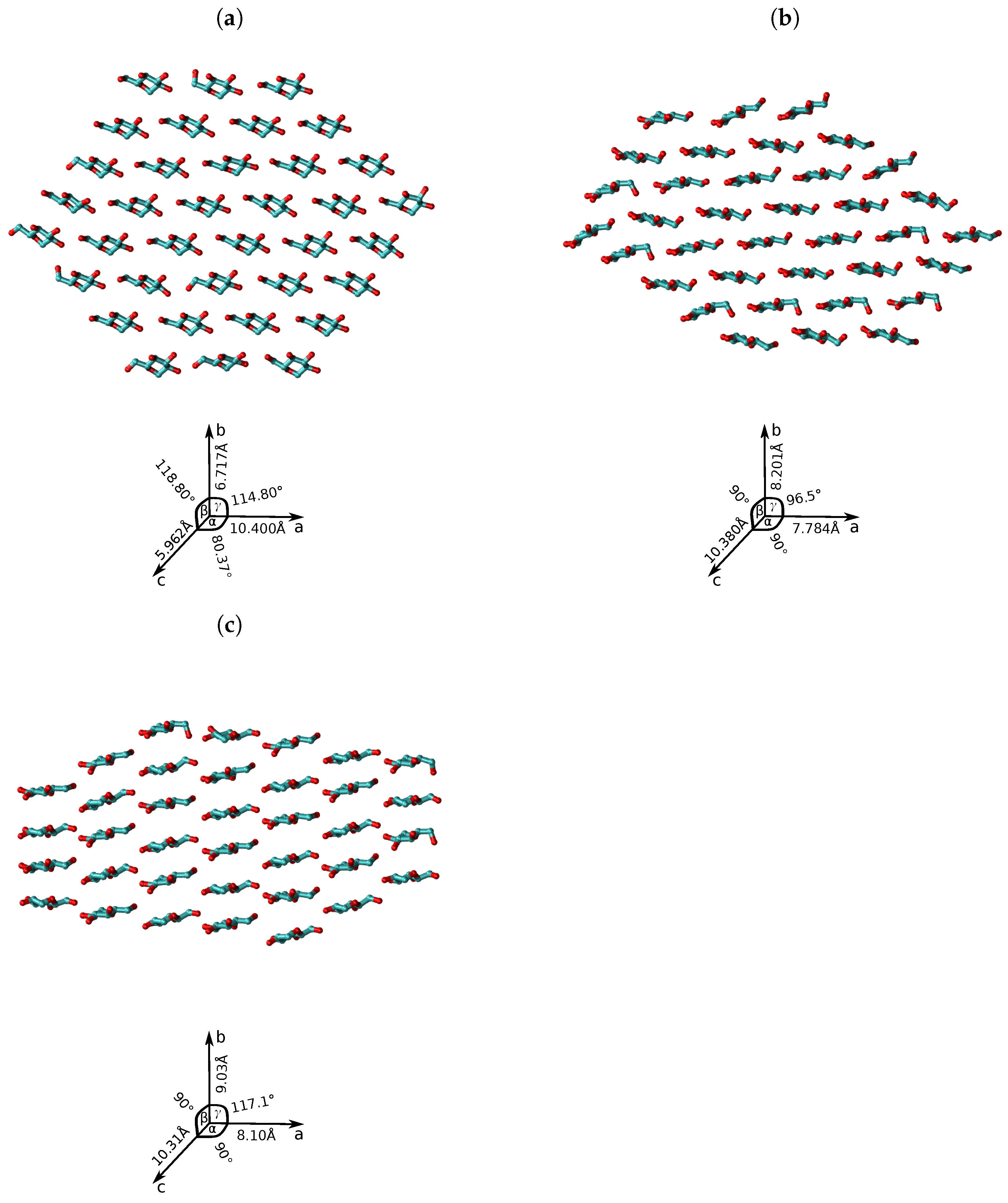
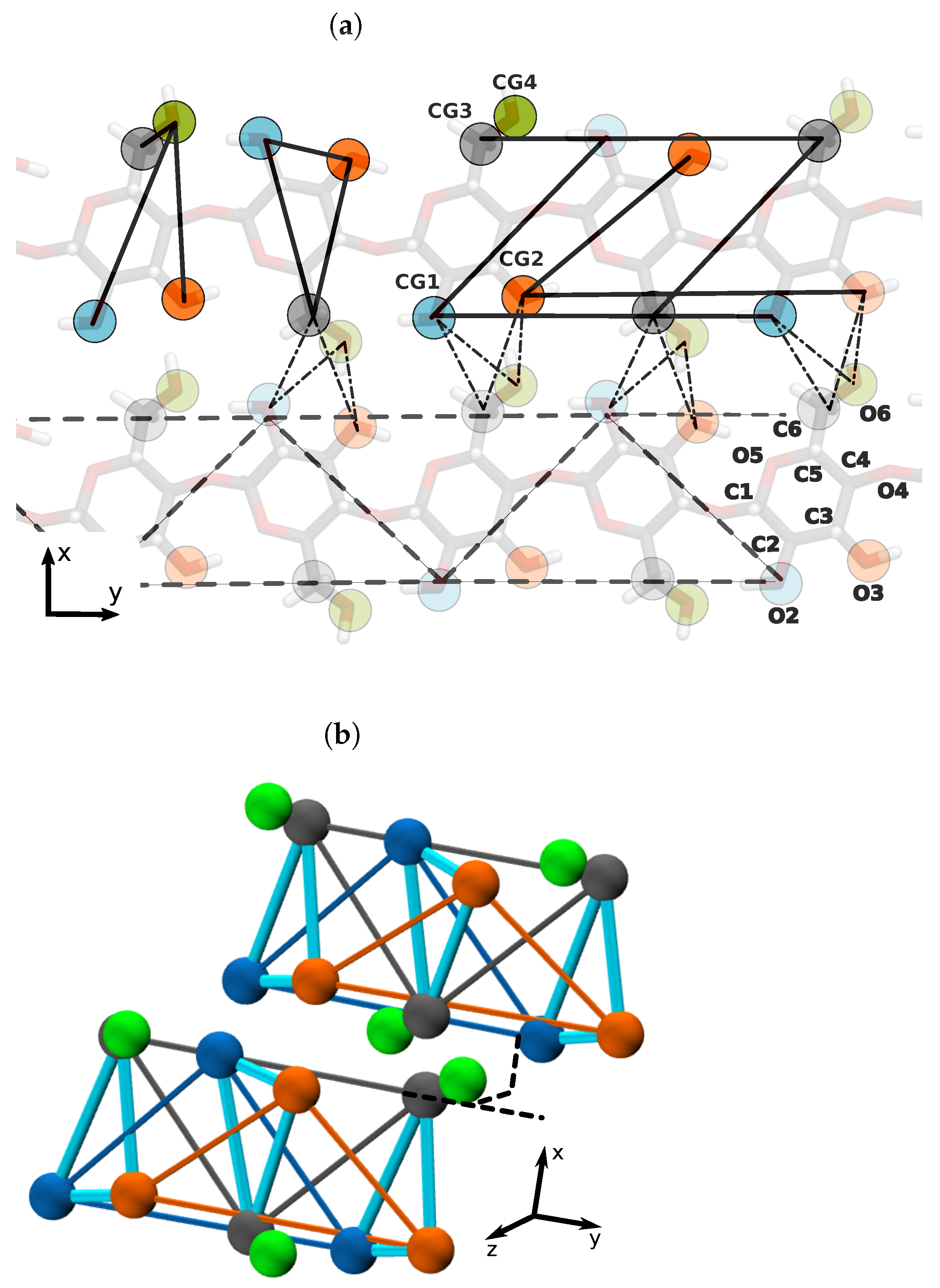

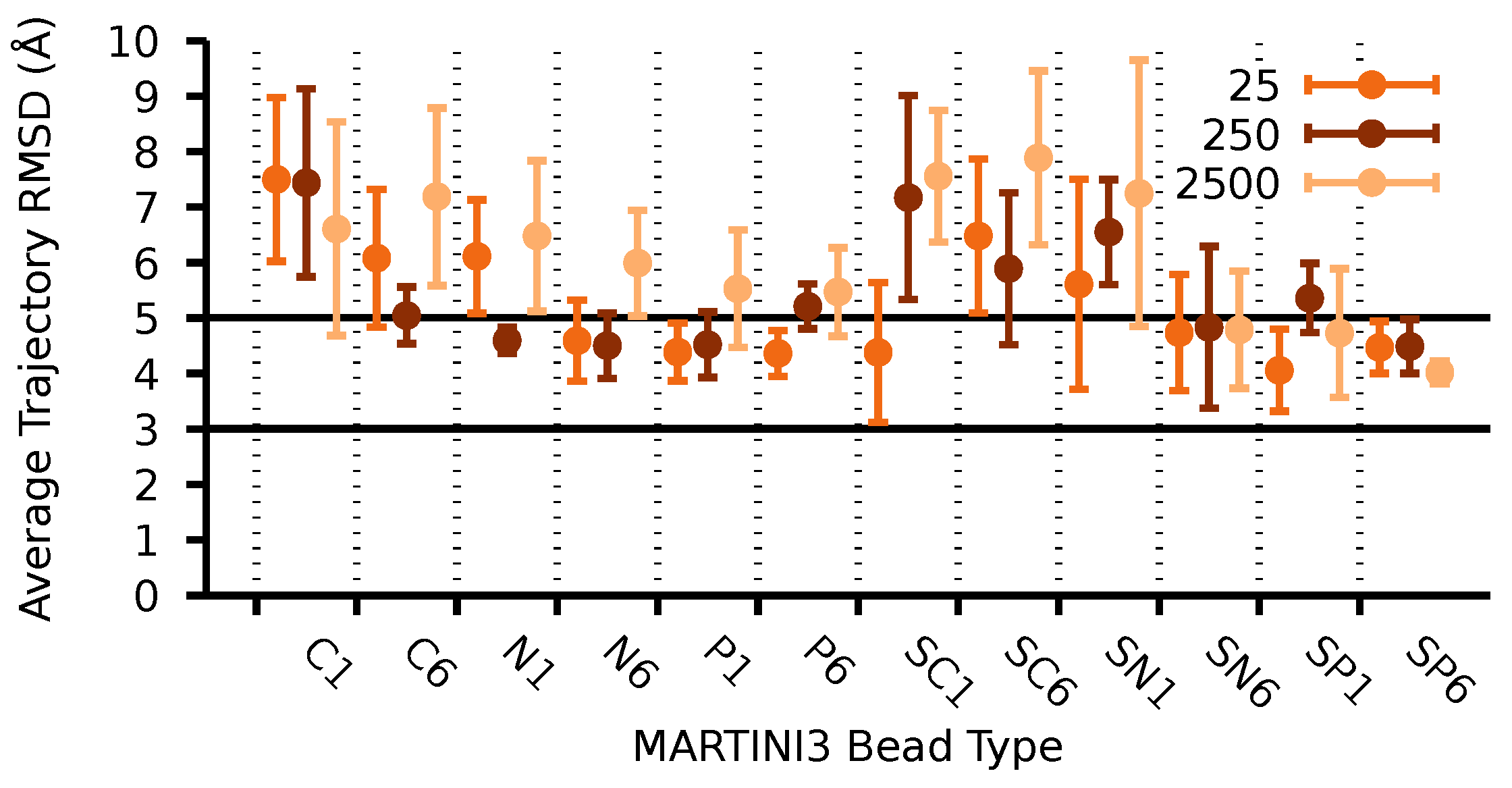


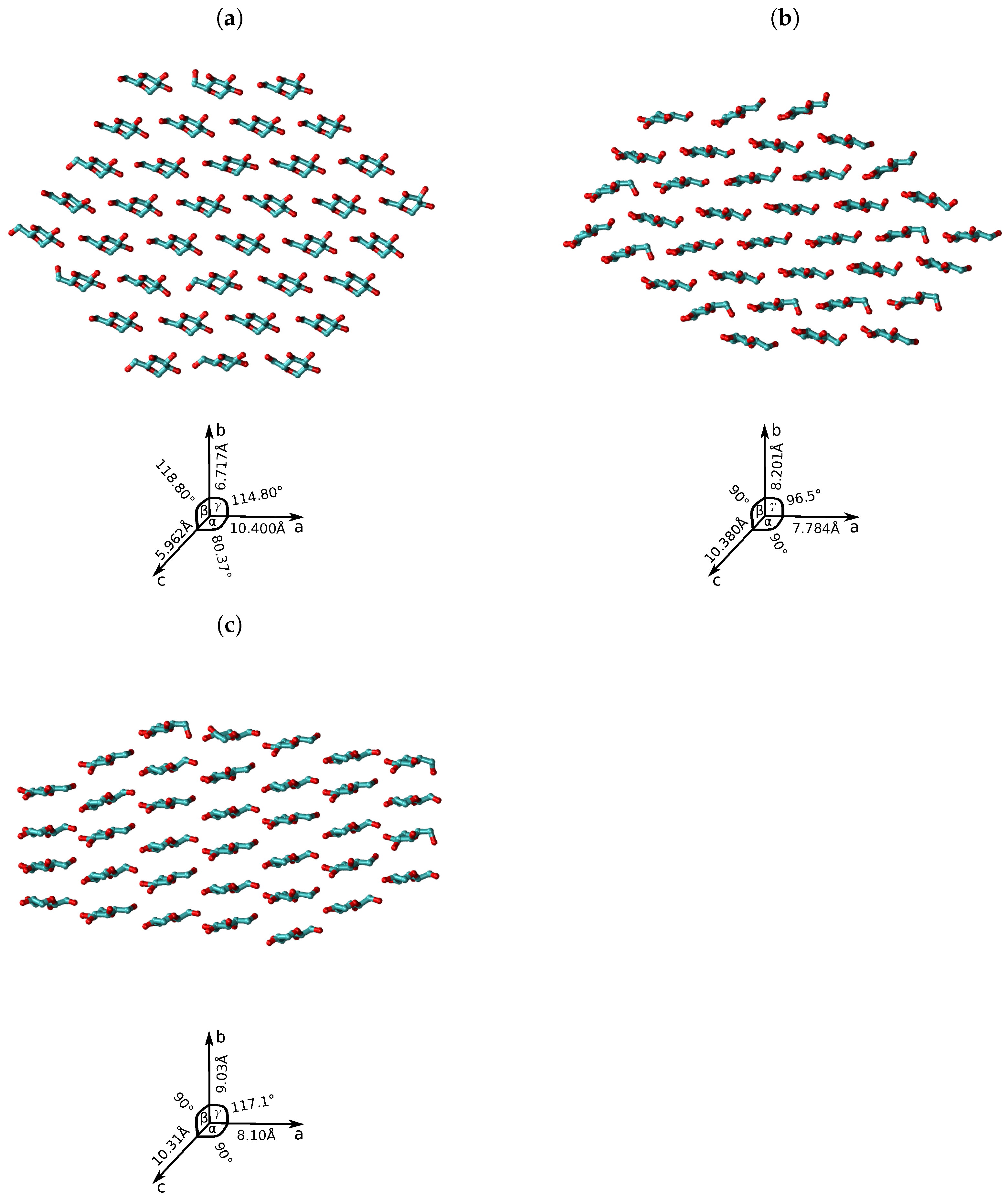{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D-Glucose Neighbours | Center Pairs | (Å) | (kJ/mol) |
|---|---|---|---|
| none | O2-O3 | 2.88 | 30,000 |
| none | O2-C6 | 5.62 | 30,000 |
| none | O3-C6 | 4.94 | 30,000 |
| none | C6-O6 | 1.43 | 30,000 |
| none | O2-O6 | 6.42 | 2500 |
| none | O3-O6 | 5.76 | 2500 |
| 1st | O2-O2 | 7.56 | 30,000 |
| 1st | O3-O3 | 6.69 | 30,000 |
| 1st | C6-C6 | 7.47 | 30,000 |
| 2nd | O2-O2 | 10.44 | 30,000 |
| 2nd | O3-O3 | 10.44 | 30,000 |
| 2nd | C6-C6 | 10.44 | 30,000 |
| Interaction between CG Bead Types | (Å) | (kJ/mol) | |
|---|---|---|---|
| W | SP6 | 4.250 | 4.530 |
| SP6 | SP6 | 4.100 | 4.290 |
| W | TC1 | 4.150 | 0.550 |
| TC1 | TC1 | 3.400 | 1.510 |
| SP6 | TC1 | 4.840 | 0.890 |
| D-glucose Neighbours | Pairs | Fibril | Distances (Å) | |
|---|---|---|---|---|
| AA-MD | CG-MD SP6/2500 | |||
| None | O2-O3 | I | 2.87 ± 0.28 | 2.88 ± 0.23 |
| None | O2-O3 | I | 2.88 ± 0.28 | 2.88 ± 0.23 |
| None | O2-O3 | II | 2.91 ± 0.28 | 2.88 ± 0.23 |
| None | O2-C6 | I | 5.62 ± 0.27 | 5.63 ± 0.25 |
| None | O2-C6 | I | 5.63 ± 0.27 | 5.63 ± 0.25 |
| None | O2-C6 | II | 5.64 ± 0.27 | 5.62 ± 0.25 |
| None | O3-C6 | I | 4.96 ± 0.28 | 4.94 ± 0.25 |
| None | O3-C6 | I | 4.95 ± 0.28 | 4.93 ± 0.25 |
| None | O3-C6 | II | 4.93 ± 0.28 | 4.94 ± 0.25 |
| None | C6-O6 | I | 1.43 ± 0.16 | 1.43 ± 0.27 |
| None | C6-O6 | I | 1.43 ± 0.16 | 1.42 ± 0.27 |
| None | C6-O6 | II | 1.43 ± 0.16 | 1.42 ± 0.27 |
| None | O2-O6 | I | 6.47 ± 0.42 | 6.29 ± 0.57 |
| None | O2-O6 | I | 6.38 ± 0.46 | 6.31 ± 0.57 |
| None | O2-O6 | II | 6.35 ± 0.35 | 6.57 ± 0.49 |
| None | O3-O6 | I | 5.69 ± 0.58 | 5.65 ± 0.56 |
| None | O3-O6 | I | 5.62 ± 0.60 | 5.64 ± 0.56 |
| None | O3-O6 | II | 6.08 ± 0.47 | 5.49 ± 0.50 |
| 1st | O2-O2 | I | 7.57 ± 0.35 | 7.56 ± 0.25 |
| 1st | O2-O2 | I | 7.57 ± 0.34 | 7.56 ± 0.25 |
| 1st | O2-O2 | II | 7.58 ± 0.35 | 7.56 ± 0.25 |
| 1st | O3-O3 | I | 6.69 ± 0.34 | 6.70 ± 0.24 |
| 1st | O3-O3 | I | 6.70 ± 0.34 | 6.71 ± 0.24 |
| 1st | O3-O3 | II | 6.70 ± 0.35 | 6.70 ± 0.24 |
| 1st | C6-C6 | I | 7.48 ± 0.38 | 7.46 ± 0.26 |
| 1st | C6-C6 | I | 7.47 ± 0.37 | 7.47 ± 0.26 |
| 1st | C6-C6 | II | 7.46 ± 0.36 | 7.47 ± 0.26 |
| 2nd | O2-O2 | I | 10.44 ± 0.45 | 10.44 ± 0.24 |
| 2nd | O2-O2 | I | 10.44 ± 0.44 | 10.43 ± 0.24 |
| 2nd | O2-O2 | II | 10.42 ± 0.45 | 10.44 ± 0.24 |
| 2nd | O3-O3 | I | 10.44 ± 0.40 | 10.43 ± 0.24 |
| 2nd | O3-O3 | I | 10.44 ± 0.39 | 10.43 ± 0.24 |
| 2nd | O3-O3 | II | 10.42 ± 0.40 | 10.44 ± 0.24 |
| 2nd | C6-C6 | I | 10.44 ± 0.46 | 10.44 ± 0.26 |
| 2nd | C6-C6 | I | 10.44 ± 0.46 | 10.44 ± 0.26 |
| 2nd | C6-C6 | II | 10.42 ± 0.45 | 10.44 ± 0.26 |
| AA-MD | CG-MD | Trajectory RMSD(Å) |
|---|---|---|
| I | I | 4.264 ± 0.751 |
| I | I | 16.860 ± 0.127 |
| I | II | 21.111 ± 0.235 |
| I | I | 16.867 ± 0.120 |
| I | I | 5.440 ± 0.681 |
| I | II | 18.786 ± 0.390 |
| II | I | 20.290 ± 0.091 |
| II | I | 17.840 ± 0.130 |
| II | II | 3.740 ± 0.400 |
| Average | SD | Max | Min | |
|---|---|---|---|---|
| AA-MD | ||||
| I | 3.557587 | 11.986272 | −136.20 | 123.50 |
| I | 2.550789 | 7.222558 | −167.00 | 99.80 |
| II | −0.420902 | 6.886370 | −150.00 | 173.40 |
| CG-MD | ||||
| I | 1.406122 | 6.499510 | −54.10 | 61.10 |
| I | 1.251399 | 6.292622 | −33.80 | 89.30 |
| II | −0.427939 | 9.148864 | −107.20 | 120.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreira, R.A.; Weber, S.A.L.; Poma, A.B. Martini 3 Model of Cellulose Microfibrils: On the Route to Capture Large Conformational Changes of Polysaccharides. Molecules 2022, 27, 976. https://doi.org/10.3390/molecules27030976
Moreira RA, Weber SAL, Poma AB. Martini 3 Model of Cellulose Microfibrils: On the Route to Capture Large Conformational Changes of Polysaccharides. Molecules. 2022; 27(3):976. https://doi.org/10.3390/molecules27030976
Chicago/Turabian StyleMoreira, Rodrigo A., Stefan A. L. Weber, and Adolfo B. Poma. 2022. "Martini 3 Model of Cellulose Microfibrils: On the Route to Capture Large Conformational Changes of Polysaccharides" Molecules 27, no. 3: 976. https://doi.org/10.3390/molecules27030976
APA StyleMoreira, R. A., Weber, S. A. L., & Poma, A. B. (2022). Martini 3 Model of Cellulose Microfibrils: On the Route to Capture Large Conformational Changes of Polysaccharides. Molecules, 27(3), 976. https://doi.org/10.3390/molecules27030976