
Towards Personalized Precision Oncology: A Feasibility Study of NGS-Based Variant Analysis of FFPE CRC Samples in a Chilean Public Health System Laboratory

 , , , , ,
, , , , ,
Abstract
1. Introduction
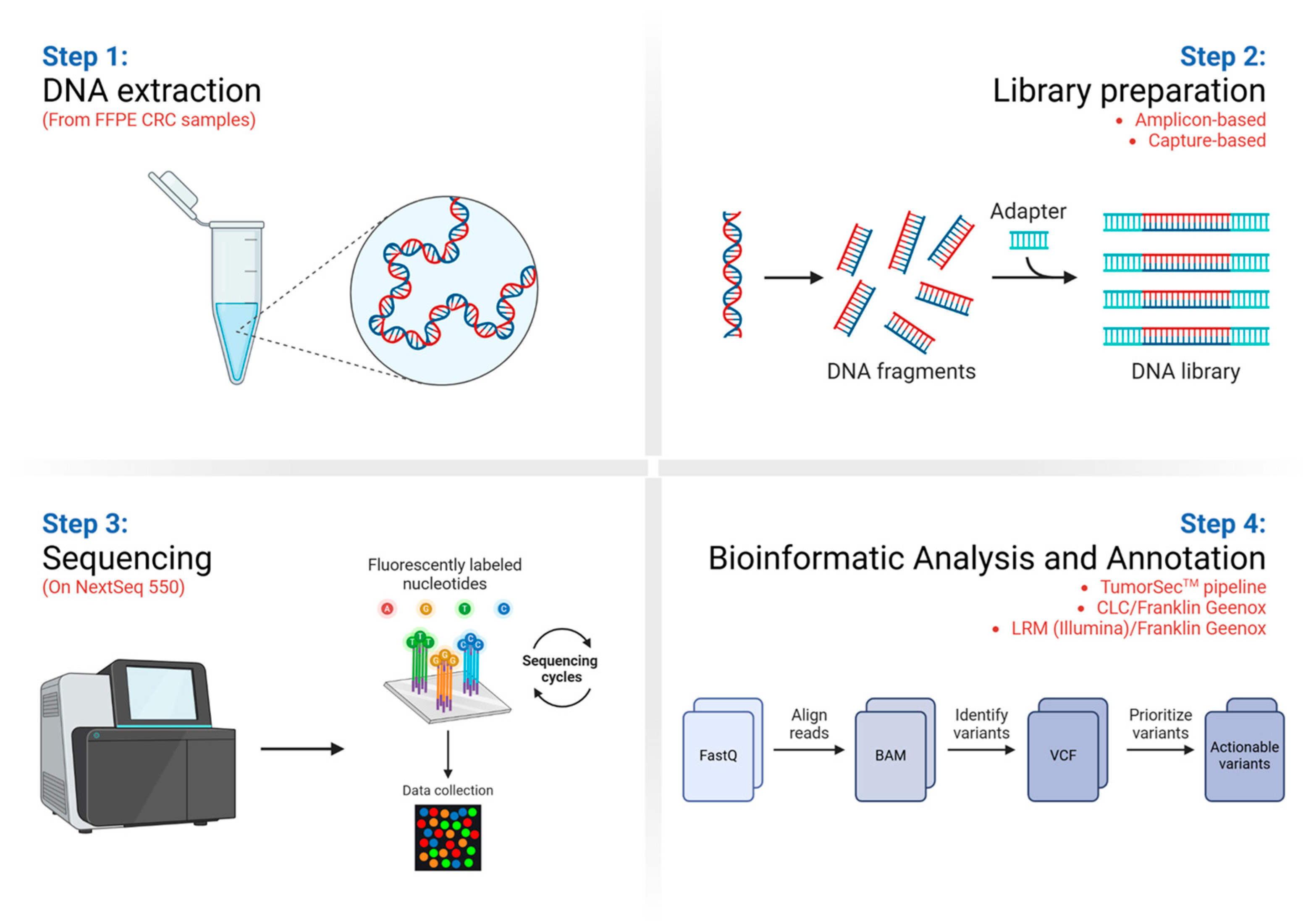
2. Materials and Methods
2.1. FFPE Tumor Specimens
2.2. Control Samples
2.3. DNA Extraction, Quantification, and Quality Control
2.4. Library Preparation and Target Enrichment
2.5. Next-Generation Sequencing
2.6. Bioinformatics Analyses
2.7. Variants Prioritization
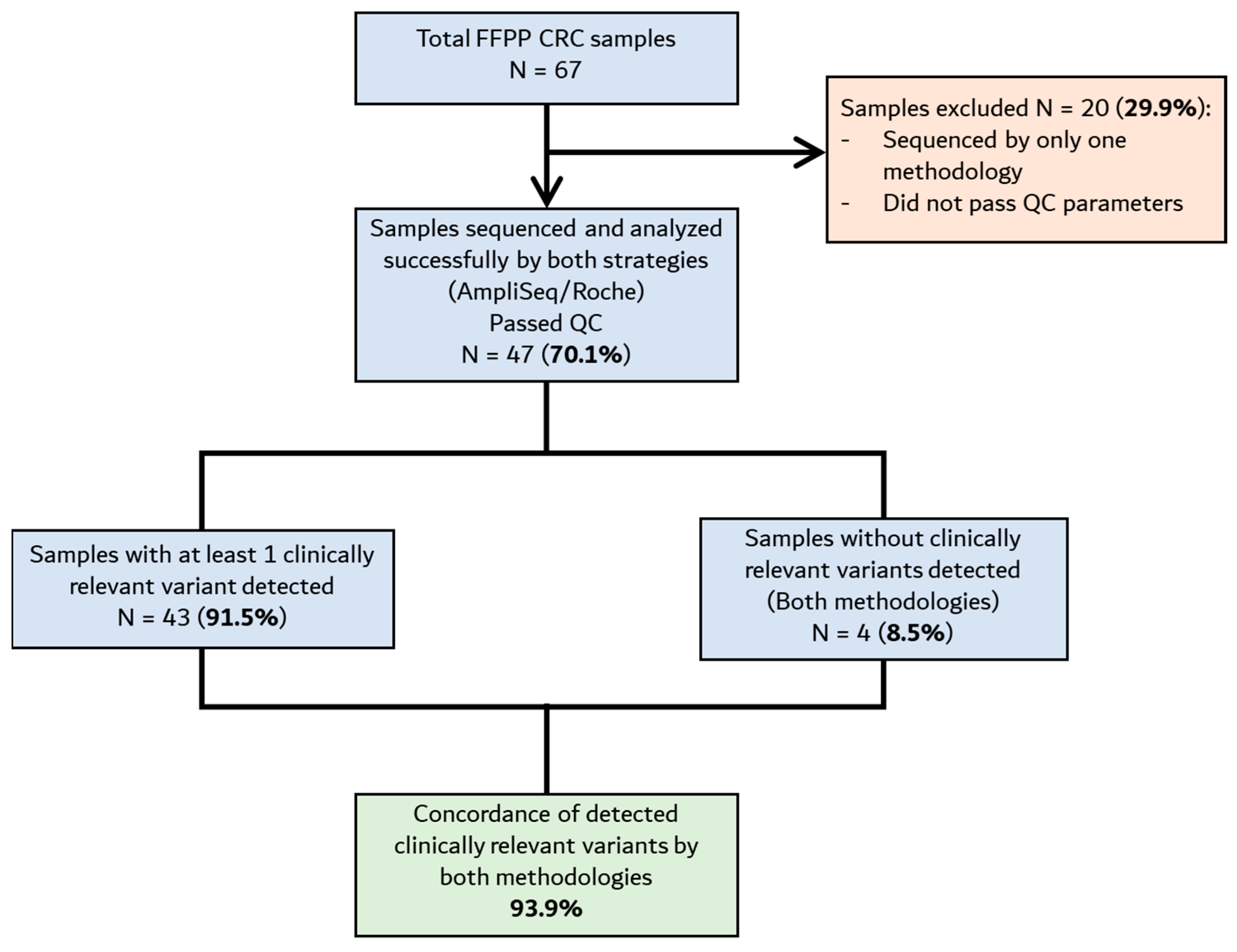
3. Results
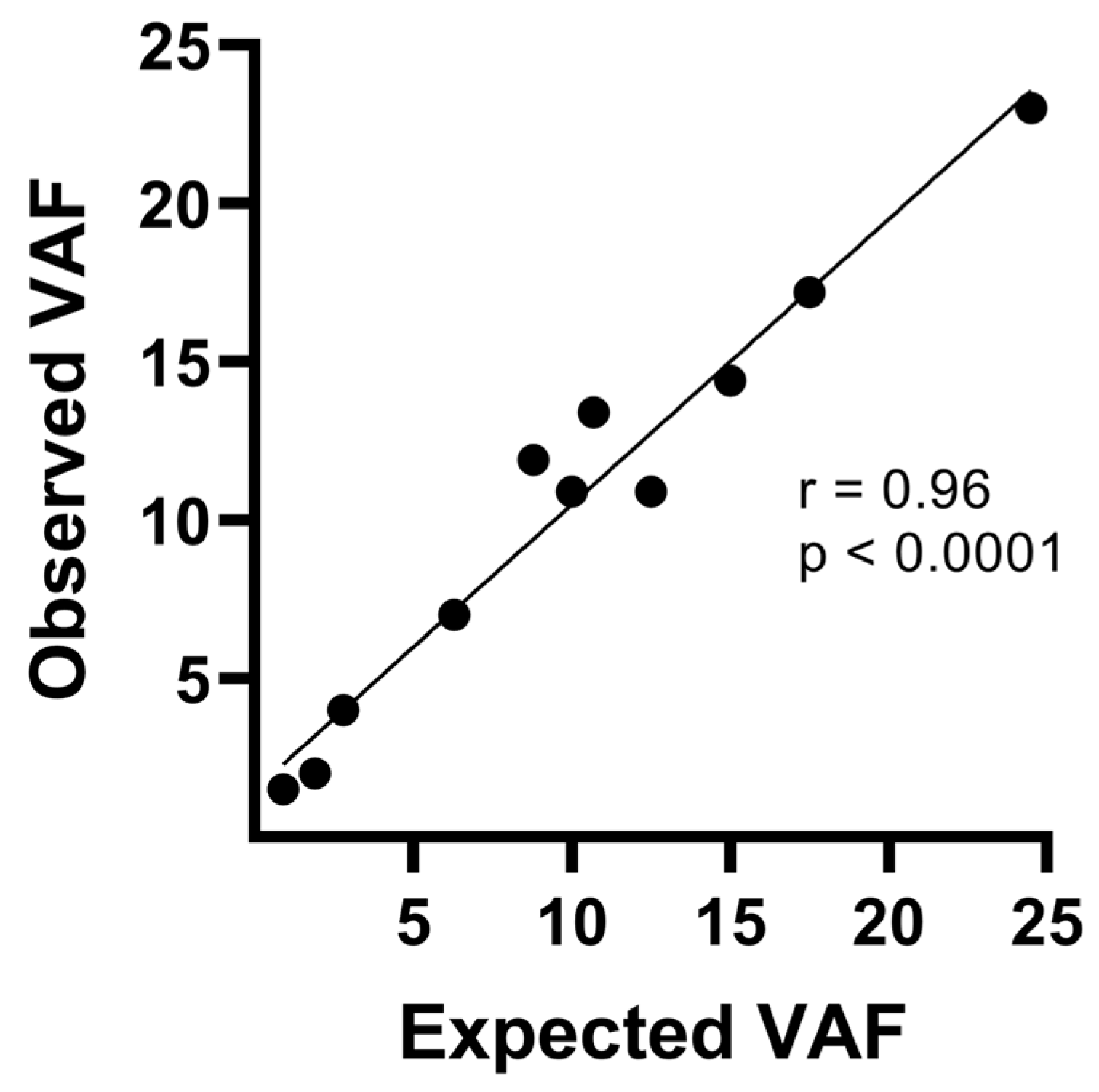
3.1. Resequencing Analysis of FFPE CRC Samples Using the TumorSecTM Pipeline
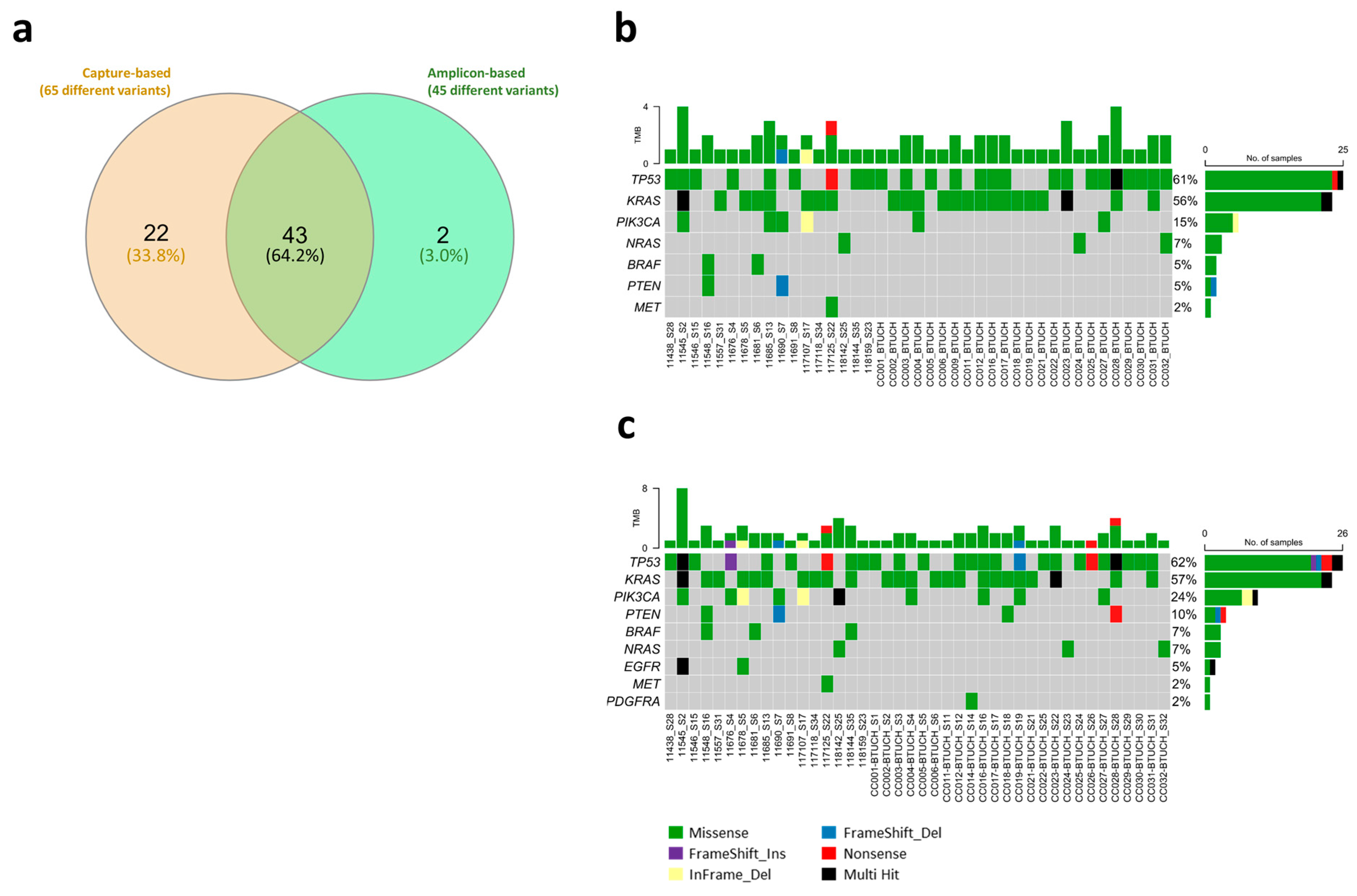
3.2. Comparison Between Library Preparation Strategies in the Identification of Actionable Variants
- Total prioritized variants detected by amplicon-based and capture-based approaches (TumorSecTM bioinformatics): 61 (93.8%).
- Amplicon-based-only detected variants (TumorSecTM bioinformatics): 4 (6.2%).
- Total prioritized variants detected by capture-based and amplicon-based approach (TumorSecTM bioinformatics): 61 (73.5%).
- Capture-based-only detected variants: 22 (26.5%).
- There was 96.7% concordance between CLC/Franklin vs. TumorSecTM annotated variants (59 out of 61 variants—3rd and 4th columns; amplicon-based library preparation);
- There was 100% concordance between CLC/Franklin vs. LRM/Franklin (4th and 5th columns; amplicon-based library preparation);
- There was 87.9% concordance between amplicon-based library preparation + Franklin variants annotation vs. capture-based library preparation + TumorSecTM annotation analysis (80 out of 91 variants).
- ✔ indicate the variant was detected by that particular pipeline
- ✕ indicate the variant wasn’t detected by that particular pipeline
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BC | breast cancer |
| BTUCH | Biobank from the Universidad de Chile |
| CGL | Cancer Genomics Laboratory |
| CRC | colorectal cancer |
| DNA | deoxyribonucleic acid |
| FFPE | formalin-fixed, paraffin-embedded |
| GC | gastric cancer |
| GLOBOCAN | Global Cancer Observatory |
| IARC | International Agency for Research on Cancer |
| LC | lung cancer |
| NGS | next-generation sequencing |
| PC | prostate cancer |
| PCR | polymerase chain reaction |
| PVDs | Population Variant’s Databases |
| QC | quality control |
| ROIs | regions of interest |
| SNVs | single-nucleotide variants |
| TBLC | trachea–bronchus and lung |
| TCGA | The Cancer Genome Atlas |
| UK | United Kingdom |
| USA | United States of America |
| VAF | variant allele frequency |
| VCF | variant call format |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Patterson, S.E.; Liu, R.; Statz, C.M.; Durkin, D.; Lakshminarayana, A.; Mockus, S.M. The clinical trial landscape in oncology and connectivity of somatic mutational profiles to targeted therapies. Hum. Genom. 2016, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Siu, L.L.; Conley, B.A.; Boerner, S.; LoRusso, P.M. Next-Generation Sequencing to Guide Clinical Trials. Clin. Cancer Res. 2015, 21, 4536–4544. [Google Scholar] [CrossRef]
- Kringelbach, T.; Højgaard, M.; Rohrberg, K.; Spanggaard, I.; Laursen, B.E.; Ladekarl, M.; Haslund, C.A.; Harsløf, L.; Belcaid, L.; Gehl, J.; et al. ProTarget: A Danish Nationwide Clinical Trial on Targeted Cancer Treatment based on genomic profiling—A national, phase 2, prospective, multi-drug, non-randomized, open-label basket trial. BMC Cancer 2023, 23, 182. [Google Scholar] [CrossRef]
- Singh, R.R. Target Enrichment Approaches for Next-Generation Sequencing Applications in Oncology. Diagnostics 2022, 12, 1539. [Google Scholar] [CrossRef]
- Salvo, M.; González-Feliú, E.; Toro, J.; Gallegos, I.; Maureira, I.; Miranda-González, N.; Barajas, O.; Bustamante, E.; Ahumada, M.; Colombo, A.; et al. Validation of an NGS Panel Designed for Detection of Actionable Mutations in Tumors Common in Latin America. J. Pers. Med. 2021, 11, 899. [Google Scholar] [CrossRef]
- Heberle, H.; Meirelles, G.V.; Da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.L.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef]
- Dobbin, K.K.; Cesano, A.; Alvarez, J.; Hawtin, R.; Janetzki, S.; Kirsch, I.; Masucci, G.V.; Robbins, P.B.; Selvan, S.R.; Streicher, H.Z.; et al. Validation of biomarkers to predict response to immunotherapy in cancer: Volume II—Clinical validation and regulatory considerations. J. Immunother. Cancer 2016, 4, 77. [Google Scholar] [CrossRef]
- Majewski, I.J.; Bernards, R. Taming the dragon: Genomic biomarkers to individualize the treatment of cancer. Nat. Med. 2011, 17, 304–312. [Google Scholar] [CrossRef]
- Hung, S.S.; Meissner, B.; Chavez, E.A.; Ben-Neriah, S.; Ennishi, D.; Jones, M.R.; Shulha, H.P.; Chan, F.C.; Boyle, M.; Kridel, R.; et al. Assessment of Capture and Amplicon-Based Approaches for the Development of a Targeted Next-Generation Sequencing Pipeline to Personalize Lymphoma Management. J. Mol. Diagn. 2018, 20, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Cainap, C.; Balacescu, O.; Cainap, S.S.; Pop, L.-A. Next Generation Sequencing Technology in Lung Cancer Diagnosis. Biology 2021, 10, 864. [Google Scholar] [CrossRef] [PubMed]
- Suh, K.J.; Kim, S.H.; Kim, Y.J.; Shin, H.; Kang, E.; Kim, E.-K.; Lee, S.; Woo, J.W.; Na, H.Y.; Ahn, S.; et al. Clinical Application of Next-Generation Sequencing in Patients with Breast Cancer: Real-World Data. J. Breast Cancer 2022, 25, 366. [Google Scholar] [CrossRef] [PubMed]
- Mosele, M.; Westphalen, C.; Stenzinger, A.; Barlesi, F.; Bayle, A.; Bièche, I.; Bonastre, J.; Castro, E.; Dienstmann, R.; Krämer, A.; et al. Recommendations for the use of next-generation sequencing (NGS) for patients with advanced cancer in 2024: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2024, 35, 588–606. [Google Scholar] [CrossRef]
- Marotti, J.D.; De Abreu, F.B.; Wells, W.A.; Tsongalis, G.J. Triple-Negative Breast Cancer. Am. J. Pathol. 2017, 187, 2133–2138. [Google Scholar] [CrossRef]
- Gibbs, S.N.; Peneva, D.; Carter, G.C.; Palomares, M.R.; Thakkar, S.; Hall, D.W.; Dalglish, H.; Campos, C.; Yermilov, I. Comprehensive Review on the Clinical Impact of Next-Generation Sequencing Tests for the Management of Advanced Cancer. JCO Precis. Oncol. 2023, 7, e2200715. [Google Scholar] [CrossRef]
- McKenzie, A.J.; Dilks, H.H.; Jones, S.F.; Burris, H. Should next-generation sequencing tests be performed on all cancer patients? Expert Rev. Mol. Diagn. 2019, 19, 89–93. [Google Scholar] [CrossRef]
- Colomer, R.; Mondejar, R.; Romero-Laorden, N.; Alfranca, A.; Sanchez-Madrid, F.; Quintela-Fandino, M. When should we order a next generation sequencing test in a patient with cancer? eClinicalMedicine 2020, 25, 100487. [Google Scholar] [CrossRef]
- Union for International Cancer Control (UICC). ICCI-LA Study Collaborators. Addressing the rising burden of cancer in Chile: Challenges & opportunities. An Analysis of Chile’s Health System and Cancer Control Policies. Available online: https://www.uicc.org/resources/addressing-rising-burden-cancer-chile-challenges-opportunities (accessed on 1 March 2025).
- Marcelain, K.; Selman-Bravo, C.; García-Bloj, B.; Bustamante, E.; Fernández, J.; Gaete, F.; Moyano, L.; Bustos, J.C.; Plaza-Parroquia, F.; Godoy, J.A.; et al. Avances y desafíos locales en el diagnóstico molecular de tumores sólidos: Una mirada sanitaria hacia la oncología de precisión en Chile. Rev. Méd. Chile 2023, 151, 1344–1360. [Google Scholar] [CrossRef]
- Vacarezza, C.; Araneda, J.; Gonzalez, P.; Arteaga, O.; Marcelain, K.; Castellon, E.A.; Periera, A.; Khoury, M.; Müller, B.; Lecaros, J.A.; et al. A snapshot of cancer in Chile II: An update on research, strategies and analytical frameworks for equity, innovation and national development. Biol. Res. 2024, 57, 95. [Google Scholar] [CrossRef]
- Tan, O.; Shrestha, R.; Cunich, M.; Schofield, D.J. Application of next-generation sequencing to improve cancer management: A review of the clinical effectiveness and cost-effectiveness. Clin. Genet. 2018, 93, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, B.S.; Eaton, K.; Emond, B.; Lafeuille, M.-H.; Hilts, A.; Lefebvre, P.; Morrison, L.; Stevens, A.L.; Ewara, E.M.; Cheema, P. Cost Savings of Expedited Care with Upfront Next-Generation Sequencing Testing versus Single-Gene Testing among Patients with Metastatic Non-Small Cell Lung Cancer Based on Current Canadian Practices. Curr. Oncol. 2023, 30, 2348–2365. [Google Scholar] [CrossRef] [PubMed]
- Samorodnitsky, E.; Jewell, B.M.; Hagopian, R.; Miya, J.; Wing, M.R.; Lyon, E.; Damodaran, S.; Bhatt, D.; Reeser, J.W.; Datta, J.; et al. Evaluation of Hybridization Capture Versus Amplicon-Based Methods for Whole-Exome Sequencing. Hum. Mutat. 2015, 36, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Q.; Li, J.; Salemi, R.; Sheppard, K.E.; Do, H.; Tothill, R.W.; McArthur, G.A.; Dobrovic, A. Targeted-capture massively-parallel sequencing enables robust detection of clinically informative mutations from formalin-fixed tumours. Sci. Rep. 2013, 3, 3494. [Google Scholar] [CrossRef]
- Wang, J.; Yu, H.; Zhang, V.W.; Tian, X.; Feng, Y.; Wang, G.; Gorman, E.; Wang, H.; Lutz, R.E.; Schmitt, E.S.; et al. Capture-based high-coverage NGS: A powerful tool to uncover a wide spectrum of mutation types. Genet. Med. 2016, 18, 513–521. [Google Scholar] [CrossRef]
- Bogdan, L.; Saleh, R.R.; Avery, L.; Del Rossi, S.; Yu, C.; Bedard, P.L. Clinical Utility of Tumor Next-Generation Sequencing Panel Testing to Inform Treatment Decisions for Patients with Advanced Solid Tumors in a Tertiary Care Center. JCO Precis. Oncol. 2024, 8, e2400092. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Variants Previously Found (CGL) | Variants Found in This Study |
|---|---|---|
| CC001 | TSC2 c.5185C>T TP53 c.524G>A - | TSC2 c.5185C>T TP53 c.524G>A ARID1A c.2959G>A |
| CC002 | KRAS c.34G>T | KRAS c.34G>T |
| CC003 | KRAS c.35G>T TP53 c.524G>A | KRAS c.35G>T TP53 c.524G>A |
| CC004 | KRAS c.183A>C PIK3CA c.1634A>G | KRAS c.183A>C PIK3CA c.1634A>G |
| CC005 | TP53 c.455C>T | TP53 c.455C>T |
| CC006 | KRAS c.35G>A | KRAS c.35G>A |
| CC008 | KRAS c.346A>C TP53 c.524G>A PIK3CA c.3140A>G BRAF c.1741A>T | KRAS c.346A>C TP53 c.524G>A PIK3CA c.3140A>G BRAF c.1741A>T |
| CC009 | BRCA2 c.2588delA ARID1A c.3977delC PIK3CA c.3140A>G | BRCA2 c.2588delA ARID1A c.3977delC PIK3CA c.3140A>G |
| CC011 | KRAS c.183A>T TSC2 c.4352G>A - - | KRAS c.183A>T TSC2 c.4352G>A BRCA2 c.3586T>A TSC2 c.4759T>C |
| CC012 | KRAS c.35G>A TP53 c.839G>A | KRAS c.35G>A TP53 c.839G>A |
| CC014 | TP53 c.818G>A - - - | TP53 c.818G>A BRCA2 c.7573G>A BRCA1 c.2905A>C PDGFRA c.1676G>T |
| CC015 | BRCA1 c.2521C>T TP53 c.524G>A - - - | BRCA1 c.2521C>T TP53 c.524G>A TSC1 c.1692A>T PTCH1 c.2042C>A PTCH1 c.29C>T |
| CC016 | KRAS c.35G>A TP53 c.833C>T - - | KRAS c.35G>A TP53 c.833C>T BRCA2 c.3961G>A PIK3CA c.1070G>T |
| CC017 | KRAS c.351A>C TP53 c.844C>T | KRAS c.351A>C TP53 c.844C>T |
| CC019 | KRAS c.35G>A TP53 c.778_799del - - | KRAS c.35G>A TP53 c.778_779del BRCA1 c.2188G>A PIK3CA c.2983G>T |
| CC020 | KRAS c.182A>T BRCA2 c.9440C>A BRCA1 c.4039A>G TP53c.745A>G | KRAS c.182A>T BRCA2 c.9440C>A BRCA1 c.4039A>G TP53c.745A>G |
| CC021 | KRAS c.34G>T ARID1A c.1833_1836del | KRAS c.34G>T ARID1A c.1833_1836del |
| CC022 | TP53 c.659A>G ARID1A c.2169G>C - | TP53 c.659A>G ARID1A c.2169G>C BRCA2 c.5125G>A |
| CC023 | KRAS c.176C>G KRAS c.35G>A TP53 c.641A>G | KRAS c.176C>G KRAS c.35G>A TP53 c.641A>G |
| CC024 | TSC2 c.5383C>T NRAS c.182A>G ARID1A c.3211delA | - NRAS c.182A>G ARID1A c.3216delA |
| CC025 | TP53 c.817C>T | TP53 c.817C>T |
| CC026 | TP53 c.151G>T ARID1A c.1113delG | TP53 c.151G>T ARID1A c.1113delG |
| CC027 | TP53 c.742C>T PIK3CA c.1633G>A | TP53 c.742C>T PIK3CA c.1633G>A |
| CC028 | PTEN c.445C>T KRAS c.38G>A TSC2 c.5137C>T BRCA1 c.3083G>A TP53 c.817C>T TP53 c.473G>A ARID1A c.4003C>T PTCH1 c.1946G>A | PTEN c.445C>T KRAS c.38G>A TSC2 c.5137C>T BRCA1 c.3083G>A TP53 c.817C>T TP53 c.473G>A ARID1A c.4003C>T PTCH1 c.1946G>A |
| CC029 | BRCA2 c.9004G>A TP53 c.527G>A | BRCA2 c.9004G>A TP53 c.527G>A |
| CC030 | TP53 c.584T>C | TP53 c.584T>C |
| CC031 | TP53 c.524G>A ARID1A c.5693C>T - | TP53 c.524G>A ARID1A c.5693C>T KRAS c.35G>T |
| CC032 | NRAS c.35G>A PTCH1 c.3727G>A | NRAS c.35G>A PTCH1 c.3727G>A |
| Total # of variants | 63 | 77 |
| Sample | Capture-Based | Amplicon-Based |
|---|---|---|
| CC001 | TP53 c.524G>A | TP53 c.524G>A |
| CC002 | KRAS c.34G>T | KRAS c.34G>T |
| CC003 | KRAS c.35G>T TP53 c.524G>A | KRAS c.35G>T TP53 c.524G>A |
| CC004 | PIK3CA c.1634A>G KRAS c.183A>C | PIK3CA c.1634A>G KRAS c.183A>C |
| CC005 | TP53 c.455C>T | TP53 c.455C>T |
| CC006 | KRAS c.35G>A | KRAS c.35G>A |
| CC007 | No variants detected | |
| CC009 | No variants detected | KRAS c.182A>T TP53 c.745A>G |
| CC011 | KRAS c.183A>T | KRAS c.183A>T |
| CC012 | KRAS c.35G>A TP53 c.839G>A | KRAS c.35G>A TP53 c.839G>A |
| CC014 | TP53 c.818G>A PDGFRA c.1676G>T | No variants detected |
| CC016 | KRAS c.35G>A TP53 c.833C>T PIK3CA c.1070G>T | KRAS c.35G>A TP53 c.833C>T - |
| CC017 | KRAS c.315A>C TP53 c.844C>T | KRAS c.315A>C TP53 c.844C>T |
| CC018 | KRAS c.37G>T PTEN c.607A>G | KRAS c.37G>T - |
| CC019 | KRAS c.35G>A TP53 c.778_779delTC PIK3CA c.2983G>T | KRAS c.35G>A - - |
| CC021 | KRAS c.34G>T | KRAS c.34G>T |
| CC022 | TP53 c.659A>G | TP53 c.659A>G |
| CC023 | KRAS c.35G>A KRAS c.176C>G TP53 c.641A>G | KRAS c.35G>A KRAS c.176C>G TP53 c.641A>G |
| CC024 | NRAS c.182A>G | NRAS c.182A>G |
| CC025 | TP53 c.817C>T | TP53 c.817C>T |
| CC026 | TP53 c.151G>T | No variants detected |
| CC027 | PIK3CA c.1633G>A TP53 c.742C>T | PIK3CA c.1633G>A TP53 c.742C>T |
| CC028 | KRAS c.38G>A TP53 c.473G>A TP53 c.817C>T PTEN c.445C>T | KRAS c.38G>A TP53 c.473G>A TP53 c.817C>T - |
| CC029 | TP53 c.527G>A | TP53 c.527G>A |
| CC030 | TP53 c.584T>C | TP53 c.584T>C |
| CC031 | KRAS c.35G>T TP53 c.524G>A | KRAS c.35G>T TP53 c.524G>A |
| CC032 | NRAS c.35G>A - | NRAS c.35G>A TP53 c.376-1G>A |
| 11438 | TP53 c.472C>G | TP53 c.472C>G |
| 11442 | No variants detected | |
| 11545 | PIK3CA c.1633G>A KRAS c.38G>A KRAS c.68T>G TP53 c.481G>A PTEN c.1-519T>C TP53 c.911C>T TP53 c.53C>T EGFR c.844G>A EGFR c.2264C>T | PIK3CA c.1633G>A KRAS c.38G>A KRAS c.68T>G TP53 c.481G>A - - - - - |
| 11546 | TP53 c.743G>A | TP53 c.743G>A |
| 11548 | BRAF c.1799T>A PTEN c.515G>A KRAS c.118T>A | BRAF c.1799T>A PTEN c.515G>A - |
| 11557 | KRAS c.34G>T | KRAS c.34G>T |
| 11676 | TP53 c.843_862dup PIK3CA c.2309G>A | TP53 c.843_862dup - |
| 11678 | KRAS c.35G>A PIK3CA c.328_330del EGFR c.2314C>T | KRAS c.35G>A - - |
| 11681 | KRAS c.436G>A BRAF c.1781A>G | KRAS c.436G>A BRAF c.1781A>G |
| 11685 | - KRAS c.38G>A TP53 c.584T>A | PIK3CA c.1624G>A KRAS c.38G>A TP53 c.584T>A |
| 11690 | PIK3CA c.1258T>C PTEN c.800del | PIK3CA c.1258T>C PTEN c.800del |
| 11691 | TP53 c.722C>G | TP53 c.722C>G |
| 11694 | No variants detected | |
| 117107 | KRAS c.35G>T PIK3CA c.337_339del | KRAS c.35G>T PIK3CA c.337_339del |
| 117118 | KRAS c.35G>T | KRAS c.35G>T |
| 117125 | KRAS c.35G>A TP53 c.916C>T MET c.2962C>T | KRAS c.35G>A TP53 c.916C>T MET c.2962C>T |
| 117134 | No variants detected | |
| 118142 | NRAS c.182A>G PIK3CA c.40C>A PIK3CA c.42C>G PIK3CA c.44T>G | NRAS c.182A>G - - - |
| 118144 | TP53 c.614A>G KRAS c.187G>A BRAF c.1857G>C | TP53 c.614A>G - - |
| 118159 | TP53 c.818G>C | TP53 c.818G>C |
| Sample | Variant | TumorSecTM | CLC/ Franklin | LRM/ Franklin |
|---|---|---|---|---|
| 11442 | KRAS c.35G>T | ✔ | ✔ | ✔ |
| 11545 | PIK3CA c.1633G>A | ✔ | ✔ | ✔ |
| KRAS c.38G>A | ✔ | ✔ | ✔ | |
| KRAS c.68T>G | ✔ | ✔ | ✔ | |
| TP53 c.743G>A | ✔ | ✔ | ✔ | |
| 11548 | BRAF c.1799T>A | ✔ | ✔ | ✔ |
| PTEN c.515G>A | ✔ | ✔ | ✔ | |
| 11557 | KRAS c.34G>T | ✔ | ✔ | ✔ |
| 11676 | TP53 c.843_862dup | ✔ | ✔ | ✔ |
| 11678 | KRAS c.35G>A | ✔ | ✔ | ✔ |
| PIK3CA c.328_330del | ✕ | ✔ | ✔ | |
| 11681 | KRAS c.436G>A | ✔ | ✔ | ✔ |
| BRAF c.1781A>G | ✔ | ✔ | ✔ | |
| 11685 | PIK3CA c.1624G>A | ✔ | ✔ | ✔ |
| KRAS c.38G>A | ✔ | ✔ | ✔ | |
| TP53 c.584T>A | ✔ | ✔ | ✔ | |
| 11690 | PIK3CA c.1258T>C | ✔ | ✔ | ✔ |
| PTEN c.800del | ✔ | ✔ | ✔ | |
| 11691 | TP53 c.722C>G | ✔ | ✔ | ✔ |
| 11694 | No variants detected | |||
| 117107 | KRAS c.35G>T | ✔ | ✔ | ✔ |
| PIK3CA c.337_339del | ✔ | ✔ | ✔ | |
| 117118 | KRAS c.35G>T | ✔ | ✔ | ✔ |
| 117125 | KRAS c.35G>A | ✔ | ✔ | ✔ |
| TP53 c.916C>T | ✔ | ✔ | ✔ | |
| 117134 | No variants detected | |||
| 118142 | NRAS c.182A>G | ✔ | ✔ | ✔ |
| TP53 c.614A>G | ✔ | ✔ | ✔ | |
| 118159 | No variants detected | |||
| CC001 | TP53 c.524G>A | ✔ | ✔ | ✔ |
| CC002 | KRAS c.34G>C | ✔ | ✔ | ✔ |
| CC003 | KRAS c.35G>T | ✔ | ✔ | ✔ |
| TP53 c.524G>A | ✔ | ✔ | ✔ | |
| CC004 | KRAS c.183A>C | ✔ | ✔ | ✔ |
| PIK3CA c.1634A>G | ✔ | ✔ | ✔ | |
| CC005 | TP53 c.455C>T | ✔ | ✔ | ✔ |
| CC006 | KRAS c.35G>A | ✔ | ✔ | ✔ |
| CC007 | No variants detected | |||
| CC009 | PIK3CA c.3140A>G | ✕ | ✕ | ✕ |
| CC011 | KRAS c.183A>T | ✔ | ✔ | ✔ |
| CC012 | KRAS c.35G>A | ✔ | ✔ | ✔ |
| TP53 c.839G>A | ✔ | ✔ | ✔ | |
| CC014 | TP53 c.422G>A | ✕ | ✔ | ✔ |
| PDGFRA c.1676G>T | ✕ | ✕ | ✕ | |
| CC016 | KRAS c.35G>A | ✔ | ✔ | ✔ |
| TP53 c.833C>T | ✔ | ✔ | ✔ | |
| PIK3CA c.1070G>T | ✕ | ✕ | ✕ | |
| CC017 | KRAS c.351A>C | ✔ | ✔ | ✔ |
| TP53 c.844C>T | ✔ | ✔ | ✔ | |
| CC018 | PTEN c.607A>G | ✕ | ✕ | ✕ |
| KRAS c.37G>T | ✔ | ✔ | ✔ | |
| CC019 | KRAS c.35G>A | ✔ | ✔ | ✔ |
| TP53 c.382_383del | ✕ | ✕ | ✕ | |
| PIK3CA c.2983G>T | ✕ | ✕ | ✕ | |
| CC021 | KRAS c.34G>T | ✔ | ✔ | ✔ |
| CC022 | TP53 c.659A>G | ✔ | ✔ | ✔ |
| CC023 | KRAS c.176C>G | ✔ | ✔ | ✔ |
| KRAS c.35G>A | ✔ | ✔ | ✔ | |
| TP53 c.245A>G | ✔ | ✔ | ✔ | |
| CC024 | NRAS c.182A>G | ✔ | ✔ | ✔ |
| CC025 | TP53 c.817C>T | ✔ | ✔ | ✔ |
| CC026 | TP53 c.34G>T | ✕ | ✕ | ✕ |
| CC027 | TP53 c.742C>T | ✔ | ✔ | ✔ |
| PIK3CA c.1633G>A | ✔ | ✔ | ✔ | |
| CC028 | PTEN c.445C>T | ✕ | ✕ | ✕ |
| KRAS c.38G>A | ✔ | ✔ | ✔ | |
| TP53 c.817C>T | ✔ | ✔ | ✔ | |
| TP53 c.473G>A | ✔ | ✔ | ✔ | |
| CC029 | TP53 c.527G>A | ✔ | ✔ | ✔ |
| CC030 | TP53 c.584T>C | ✔ | ✔ | ✔ |
| CC031 | KRAS c.35G>T | ✔ | ✔ | ✔ |
| TP53 c.524G>A | ✔ | ✔ | ✔ | |
| CC032 | NRAS c.35G>A | ✔ | ✔ | ✔ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Durán-Jara, E.; Ponce, I.; Rojas-Herrera, M.; Toro, J.; Covarrubias, P.; González, E.; Santis-Alay, N.T.; Soto-Marchant, M.E.; Marcelain, K.; Parra, B.; et al. Towards Personalized Precision Oncology: A Feasibility Study of NGS-Based Variant Analysis of FFPE CRC Samples in a Chilean Public Health System Laboratory. Curr. Issues Mol. Biol. 2025, 47, 599. https://doi.org/10.3390/cimb47080599
Durán-Jara E, Ponce I, Rojas-Herrera M, Toro J, Covarrubias P, González E, Santis-Alay NT, Soto-Marchant ME, Marcelain K, Parra B, et al. Towards Personalized Precision Oncology: A Feasibility Study of NGS-Based Variant Analysis of FFPE CRC Samples in a Chilean Public Health System Laboratory. Current Issues in Molecular Biology. 2025; 47(8):599. https://doi.org/10.3390/cimb47080599
Chicago/Turabian StyleDurán-Jara, Eduardo, Iván Ponce, Marcelo Rojas-Herrera, Jessica Toro, Paulo Covarrubias, Evelin González, Natalia T. Santis-Alay, Mario E. Soto-Marchant, Katherine Marcelain, Bárbara Parra, and et al. 2025. "Towards Personalized Precision Oncology: A Feasibility Study of NGS-Based Variant Analysis of FFPE CRC Samples in a Chilean Public Health System Laboratory" Current Issues in Molecular Biology 47, no. 8: 599. https://doi.org/10.3390/cimb47080599
APA StyleDurán-Jara, E., Ponce, I., Rojas-Herrera, M., Toro, J., Covarrubias, P., González, E., Santis-Alay, N. T., Soto-Marchant, M. E., Marcelain, K., Parra, B., & Fernández, J. (2025). Towards Personalized Precision Oncology: A Feasibility Study of NGS-Based Variant Analysis of FFPE CRC Samples in a Chilean Public Health System Laboratory. Current Issues in Molecular Biology, 47(8), 599. https://doi.org/10.3390/cimb47080599