
Effect of a Novel Antidepressant and Anticancer Nuc01 on Depression in Cancer Survivors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Molecular Docking
2.3. In Vitro LSD1 Inhibitor Experiment
2.4. LSD1 Inhibitor in MDA-MB-231 Cells
2.5. Tail Suspension Test (TST) in Mice
3. Results
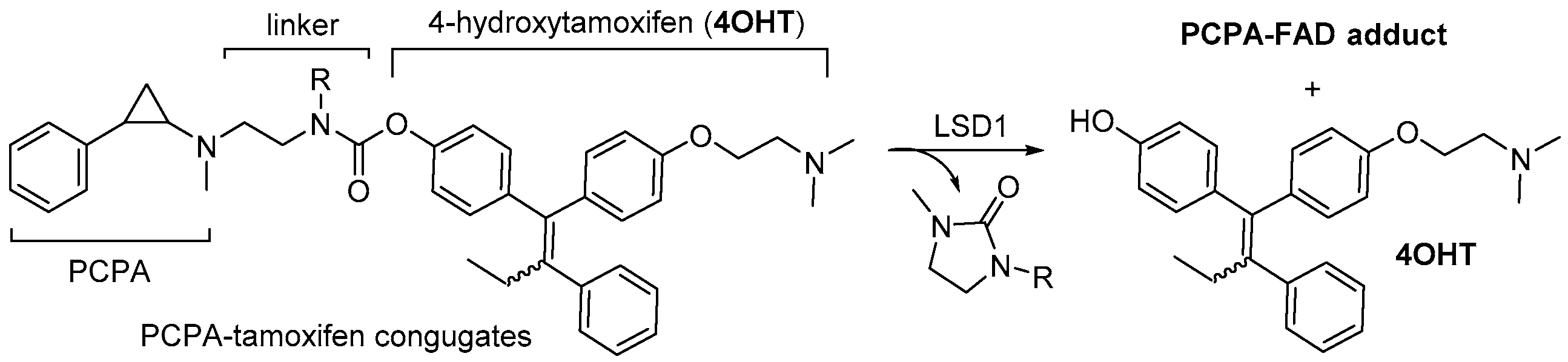
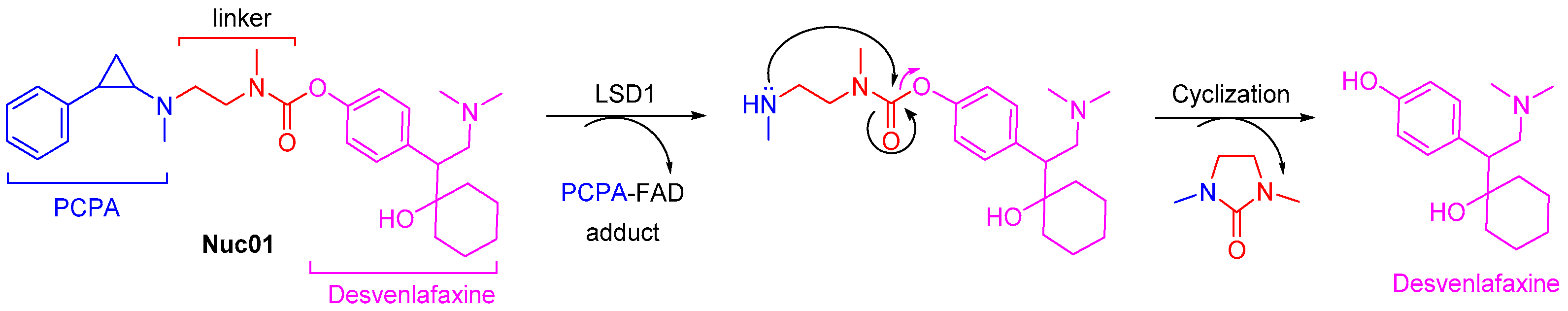
3.1. Design Concept of Synergistic Antidepressant and Anticancer Therapy Through the Combination of Two Drugs
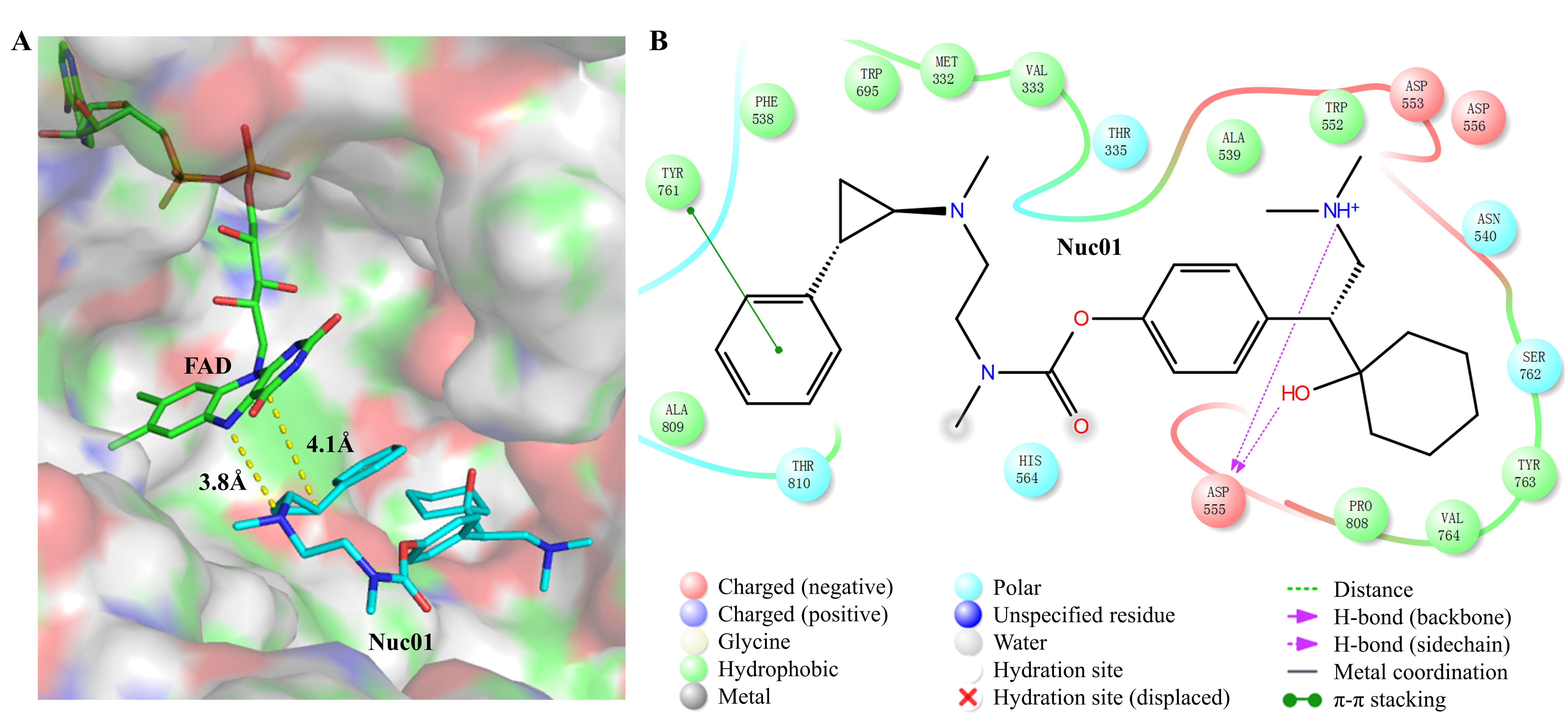
3.2. Analysis of the Binding Mode Between Nuc01 and Its Target LSD1
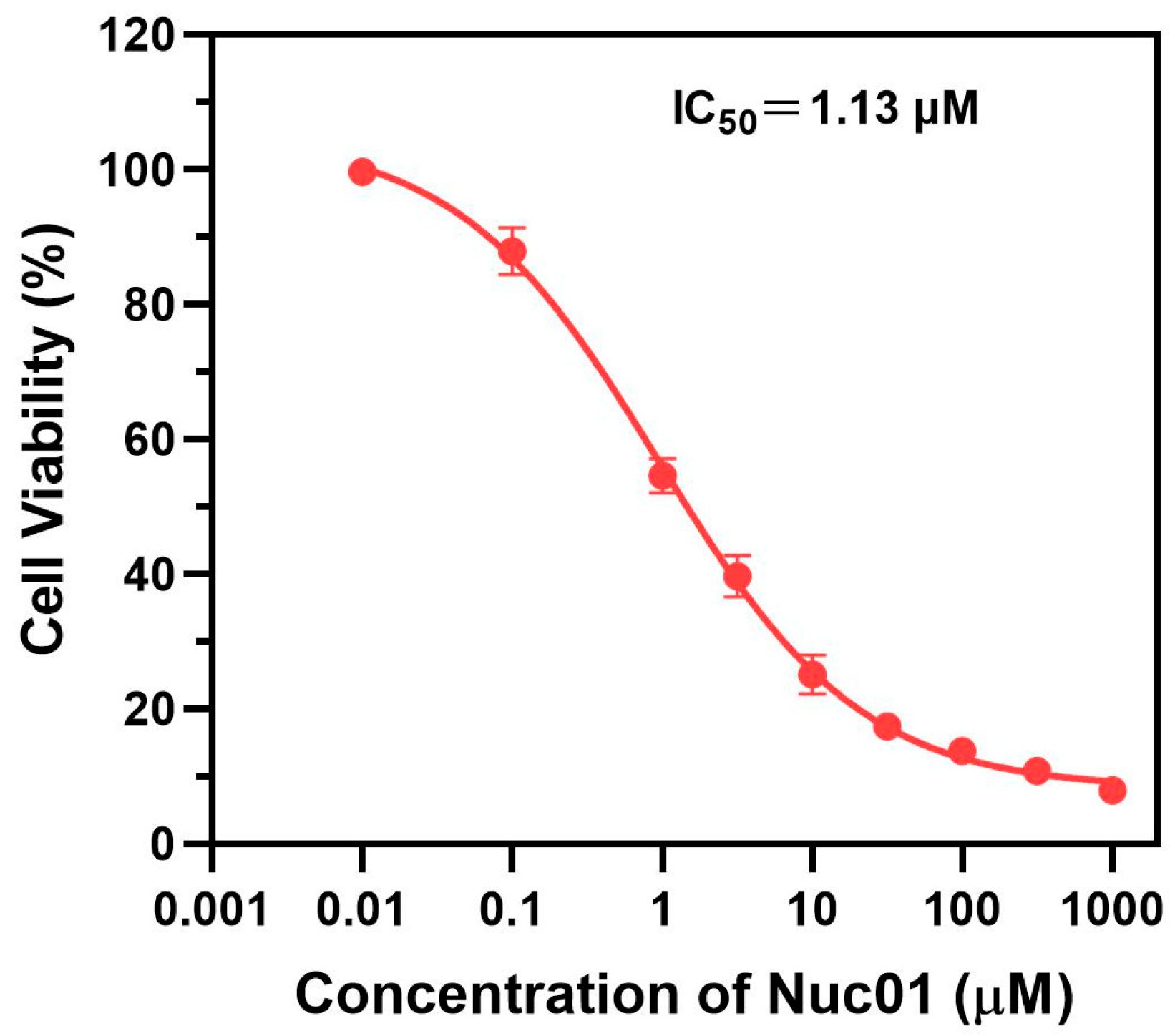
3.3. Anti-Proliferative Effects of Nuc01 on MDA-MB-231 Cells
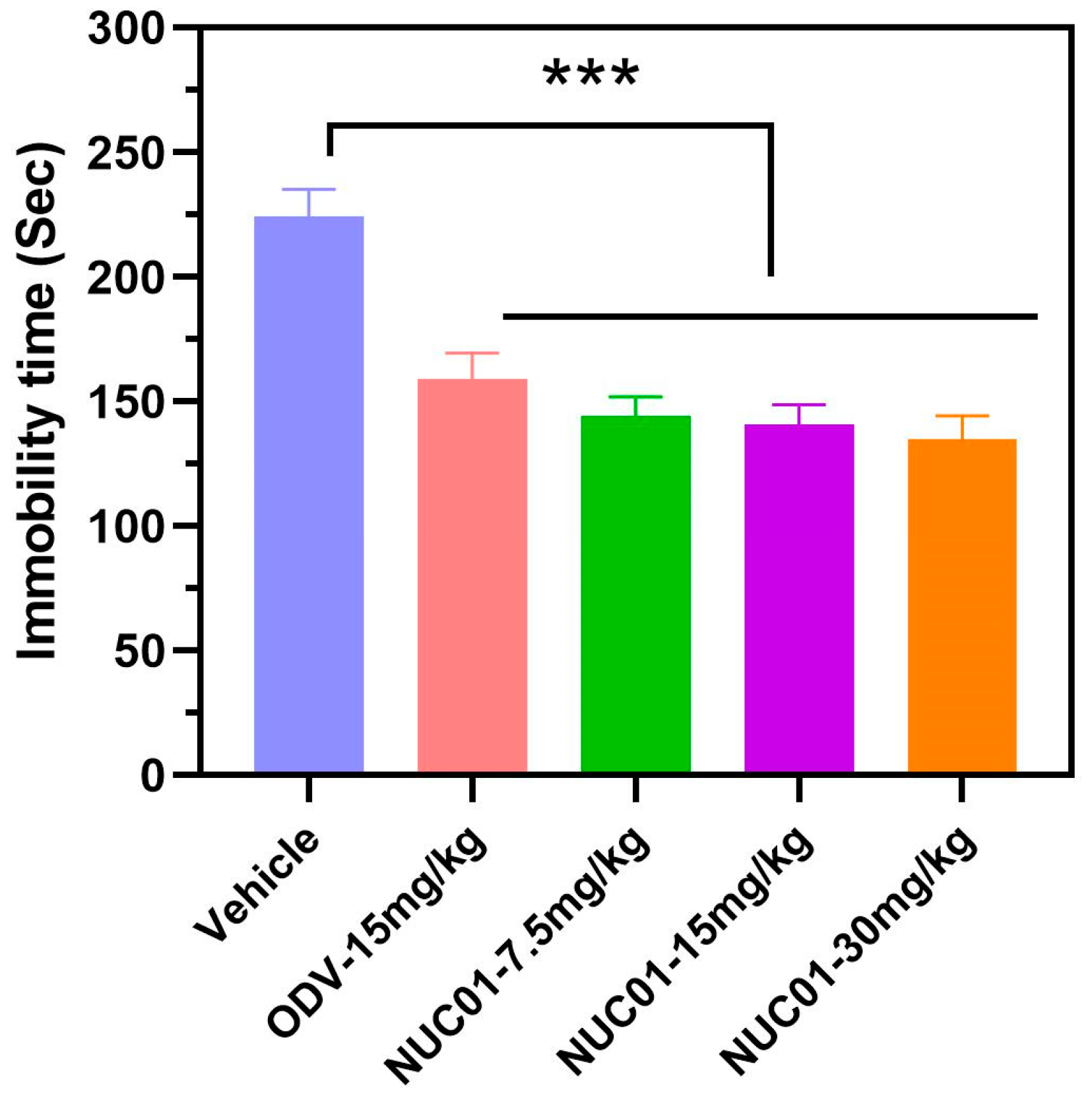
3.4. Effect of Nuc01 on the Tail Suspension Test (TST) in Mouse
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SNRIs | Serotonin and norepinephrine reuptake inhibitors |
| LSD1 | Lysine-specific demethylase 1 |
| PCPA | Trans-2-phenylcyclopropylamine |
| FAD | Flavin adenine dinucleotide |
| ODV | Desmethylvenlafaxine |
| TST | Tail suspension test |
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Cuijpers, P.; Smit, F. Excess mortality in depression: A meta-analysis of community studies. J. Affect. Disord. 2002, 72, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Mausbach, B.T.; Decastro, G.; Schwab, R.B.; Tiamson-Kassab, M.; Irwin, S.A. Healthcare use and costs in adult cancer patients with anxiety and depression. Depress. Anxiety 2020, 37, 908–915. [Google Scholar] [CrossRef] [PubMed]
- McFarland, D.C.; Walsh, L.; Napolitano, S.; Morita, J.; Jaiswal, R. Suicide in patients with cancer: Identifying the risk factors. Oncology 2019, 33, 221–226. [Google Scholar] [PubMed]
- Partridge, A.H.; Jacobsen, P.B.; Andersen, B.L. Challenges to standardizing the care for adult cancer survivors: Highlighting ASCO’s fatigue and anxiety and depression guidelines. Am. Soc. Clin. Oncol. Educ. Book 2015, 35, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Tu, S.; Sheng, J.; Shao, A. Depression in sleep disturbance: A review on a bidirectional relationship, mechanisms and treatment. J. Cell. Mol. Med. 2019, 23, 2324–2332. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Fiorentino, L.; Natarajan, L.; Parker, B.A.; Mills, P.J.; Sadler, G.R.; Dimsdale, J.E.; Rissling, M.; He, F.; Ancoli-Israel, S. Pre-treatment symptom cluster in breast cancer patients is associated with worse sleep, fatigue and depression during chemotherapy. Psycho-Oncology 2009, 18, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Lan, A.; Li, H.; Shen, M.; Li, D.; Shu, D.; Liu, Y.; Tang, H.; Li, K.; Peng, Y.; Liu, S. Association of depressive symptoms and sleep disturbances with survival among US adult cancer survivors. BMC Med. 2024, 22, 225. [Google Scholar] [CrossRef] [PubMed]
- Pinquart, M.; Duberstein, P.R. Depression and cancer mortality: A meta-analysis. Psychol. Med. 2010, 40, 1797–1810. [Google Scholar] [CrossRef] [PubMed]
- Satin, J.R.; Linden, W.; Phillips, M.J. Depression as a predictor of disease progression and mortality in cancer patients: A meta-analysis. Cancer 2009, 115, 5349–5361. [Google Scholar] [CrossRef] [PubMed]
- Bortolato, B.; Hyphantis, T.N.; Valpione, S.; Perini, G.; Maes, M.; Morris, G.; Kubera, M.; Köhler, C.A.; Fernandes, B.S.; Stubbs, B.; et al. Depression in cancer: The many biobehavioral pathways driving tumor progression. Cancer Treat. Rev. 2017, 52, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Pitman, A.; Suleman, S.; Hyde, N.; Hodgkiss, A. Depression and anxiety in patients with cancer. BMJ 2018, 361, k1415. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.M.; Rodriguez, A.I.; Kohn, B.; Ball, R.M.; Pegg, J.; Pocock, J.R.; Nunez, R.; Peterson, E.; Jakary, S.; Levine, R.A. Acupuncture versus venlafaxine for the management of vasomotor symptoms in patients with hormone receptor-positive breast cancer: A randomized controlled trial. J. Clin. Oncol. 2010, 28, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Okajima, Y.; Fujii, T.; Natori, A.; Kobayashi, D. The efficacy of nonestrogenic therapy to hot flashes in cancer patients under hormone manipulation therapy: A systematic review and meta-analysis. J. Cancer Res. Clin. Oncol. 2013, 139, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Hickey, M.; Saunders, C.M.; Stuckey, B.G.A. Management of menopausal symptoms in patients with breast cancer: An evidence-based approach. Lancet Oncol. 2005, 6, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Ota, Y.; Itoh, Y.; Kaise, A.; Ohta, K.; Endo, Y.; Masuda, M.; Sowa, Y.; Sakai, T.; Suzuki, T. Targeting cancer with PCPA-drug conjugates: LSD1 inhibition-triggered release of 4-hydroxytamoxifen. Angew. Chem. Int. Ed. 2016, 55, 16115–16118. [Google Scholar] [CrossRef] [PubMed]
- Ota, Y.; Itoh, Y.; Takada, Y.; Yamashita, Y.; Hu, C.; Horinaka, M.; Sowa, Y.; Masuda, M.; Sakai, T.; Suzuki, T. Design, synthesis, and biological evaluation of phenylcyclopropylamine-entinostat conjugates that selectively target cancer cells. Bioorg. Med. Chem. 2024, 100, 117632. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Janzer, A.; Becker, A.; Zimmer, A.; Schüle, R.; Buettner, R.; Kirfel, J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010, 31, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H.; Chen, Y.; Sun, Y.; Yang, F.; Yu, W.; Liang, J.; Sun, L.; Yang, X.; Shi, L.; et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 2009, 138, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.H.; Lim, S.; Schramm, A.; Friedrichs, N.; Koster, J.; Versteeg, R.; Ora, I.; Pajtler, K.; Klein-Hitpass, L.; Kuhfittig-Kulle, S.; et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: Implications for therapy. Cancer Res. 2009, 69, 2065–2071. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Takezawa, S.; Schüle, R.; Kitagawa, H.; Kato, S. Transrepressive function of TLX requires the histone demethylase LSD1. Mol. Cell. Biol. 2008, 28, 3995–4003. [Google Scholar] [CrossRef] [PubMed]
- Willmann, D.; Lim, S.; Wetzel, S.; Metzger, E.; Jandausch, A.; Wilk, W.; Jung, M.; Forne, I.; Imhof, A.; Janzer, A.; et al. Impairment of prostate cancer cell growth by a selective and reversible lysine-specific demethylase 1 inhibitor. Int. J. Cancer 2012, 131, 2704–2709. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, V.; Ahmad, S.; Nilsson, E.M.; Helczynski, L.; Kenna, S.; Persson, J.L.; Gudas, L.J.; Mongan, N.P. The lysine specific demethylase-1 (LSD1/KDM1A) regulates VEGF-A expression in prostate cancer. Mol. Oncol. 2013, 7, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Ketscher, A.; Jilg, C.A.; Willmann, D.; Hummel, B.; Imhof, A.; Rüsseler, V.; Hölz, S.; Metzger, E.; Müller, J.M.; Schüle, R. LSD1 controls metastasis of androgen-independent prostate cancer cells through PXN and LPAR6. Oncogenesis 2014, 3, e120. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Miyata, N. Lysine demethylases inhibitors. J. Med. Chem. 2011, 54, 8236–8250. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, D.; Itoh, Y.; Tsumoto, H.; Kakizawa, T.; Mino, K.; Fukuhara, K.; Nakagawa, H.; Hasegawa, M.; Sasaki, R.; Mizukami, T.; et al. Lysine-specific demethylase 1-selective inactivators: Protein-targeted drug delivery mechanism. Angew. Chem. Int. Ed. Engl. 2013, 52, 8620–8624. [Google Scholar] [CrossRef] [PubMed]
- McAllister, T.E.; England, K.S.; Hopkinson, R.J.; Brennan, P.E.; Kawamura, A.; Schofield, C.J. Recent progress in histone demethylase inhibitors. J. Med. Chem. 2016, 59, 1308–1329. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.M.Z. Trans-2-phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 2007, 46, 4408–4416. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Culhane, J.C.; Szewczuk, L.M.; Jalili, P.; Ball, H.L.; Machius, M.; Cole, P.A.; Yu, H. Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry 2007, 46, 8058–8065. [Google Scholar] [CrossRef]
- Akechi, T. Psychotherapy for depression among patients with advanced cancer. Jpn. J. Clin. Oncol. 2012, 42, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, H.; Smitheman, K.; van Aller, G.; Cusan, M.; Kamat, S.; Liu, Y.; Johnson, N.; Hann, C.; Armstrong, S.; Kruger, R. 212 Novel anti-tumor activity of targeted LSD1 inhibition by GSK2879552. Eur. J. Cancer 2014, 50, 72. [Google Scholar] [CrossRef]
- Lee, S.H.; Liu, X.M.; Diamond, M.; Dostalik, V.; Favata, M.; He, C.; Wu, L.; Wynn, R.; Yao, W.; Hollis, G.; et al. Abstract 4704: The evaluation of INCB059872, an FAD-directed inhibitor of LSD1, in preclinical models of human small cell lung cancer. Cancer Res. 2016, 76, 4704. [Google Scholar] [CrossRef]
- Lee, S.H.; Stubbs, M.; Liu, X.M.; Diamond, M.; Dostalik, V.; Ye, M.; Lo, Y.; Favata, M.; Yang, G.; Gallagher, K.; et al. Abstract 4712: Discovery of INCB059872, a novel FAD-directed LSD1 inhibitor that is effective in preclinical models of human and murine AML. Cancer Res. 2016, 76, 4712. [Google Scholar] [CrossRef]
- Romussi, A.; Cappa, A.; Vianello, P.; Brambillasca, S.; Cera, M.R.; Dal Zuffo, R.; Fagà, G.; Fattori, R.; Moretti, L.; Trifirò, P.; et al. Discovery of reversible inhibitors of KDM1A efficacious in acute myeloid leukemia models. ACS Med. Chem. Lett. 2020, 11, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Liu, H.-M. Pharmacoepigenetics of LSD1 inhibitors in cancer. In Pharmacoepigenetics; Elsevier: Amsterdam, The Netherlands, 2019; Volume 10, pp. 523–530. [Google Scholar]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Liao, G.; Yu, B. LSD1/KDM1A inhibitors in clinical trials: Advances and prospects. J. Hematol. Oncol. 2019, 12, 129. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.L.; Zhang, T.; Suo, F.Z.; Chang, J.; Wan, X.B.; Feng, X.J.; Zheng, Y.C.; Liu, H.M. Lysine-specific demethylase 1 activation by vitamin B2 attenuates efficacy of apatinib for proliferation and migration of gastric cancer cell MGC-803. J. Cell. Biochem. 2018, 119, 4957–4966. [Google Scholar] [CrossRef] [PubMed]
- Kaniskan, H.Ü.; Martini, M.L.; Jin, J. Inhibitors of protein methyltransferases and demethylases. Chem. Rev. 2018, 118, 989–1068. [Google Scholar] [CrossRef] [PubMed]
- Niwa, H.; Umehara, T. Structural insight into inhibitors of flavin adenine dinucleotide-dependent lysine demethylases. Epigenetics 2017, 12, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.C.; Chang, J.; Zhang, T.; Suo, F.Z.; Chen, X.B.; Liu, Y.; Zhao, B.; Yu, B.; Liu, H.M. An overview on screening methods for lysine specific demethylase 1 (LSD1) Inhibitors. Curr. Med. Chem. 2017, 24, 2496–2504. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.C.; Ma, J.L.; Liu, Y.; Liu, H.M. Writers and erasers of histone lysine methylation with clinically applied modulators: Promising target for cancer therapy. Curr. Pharm. Des. 2016, 22, 5943–5947. [Google Scholar] [CrossRef] [PubMed]
- Maes, T.; Mascaró, C.; Tirapu, I.; Estiarte, A.; Ciceri, F.; Lunardi, S.; Guibourt, N.; Perdones, A.; Lufino, M.M.P.; Somervaille, T.C.P.; et al. ORY-1001, a potent and selective covalent KDM1A Inhibitor, for the treatment of acute leukemia. Cancer Cell 2018, 33, 495–511.e12. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, H.P.; Smitheman, K.N.; Kamat, C.D.; Soong, D.; Federowicz, K.E.; Van Aller, G.S.; Schneck, J.L.; Carson, J.D.; Liu, Y.; Butticello, M.; et al. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell 2015, 28, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.C.; Yu, B.; Jiang, G.Z.; Feng, X.J.; He, P.X.; Chu, X.Y.; Zhao, W.; Liu, H.M. Irreversible LSD1 inhibitors: Application of tranylcypromine and its derivatives in cancer treatment. Curr. Top. Med. Chem. 2016, 16, 2179–2188. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.S.; Nakajima, S.T. Hormonal and nonhormonal treatment of vasomotor symptoms. Obstet. Gynecol. Clin. N. Am. 2015, 5, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Boekhout, A.H.; Vincent, A.D.; Dalesio, O.B.; van den Bosch, J.; Foekema-Töns, J.H.; Adriaansz, S.; Sprangers, S.; Nuijen, B.; Beijnen, J.H.; Schellens, J.H.M. Management of hot flashes in patients who have breast cancer with venlafaxine and clonidine: A randomized, double-blind, placebo-controlled trial. J. Clin. Oncol. 2011, 29, 3862–3868. [Google Scholar] [CrossRef] [PubMed]
- Barton, D.; Loprinzi, C.; Wahner-Roedler, D. Hot flashes: Aetiology and management. Drugs Aging 2001, 18, 597–606. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, C.; Shi, X.; Wang, Z.; Li, Y.; Ma, W.; Fu, K. Effect of a Novel Antidepressant and Anticancer Nuc01 on Depression in Cancer Survivors. Curr. Issues Mol. Biol. 2025, 47, 587. https://doi.org/10.3390/cimb47080587
Yuan C, Shi X, Wang Z, Li Y, Ma W, Fu K. Effect of a Novel Antidepressant and Anticancer Nuc01 on Depression in Cancer Survivors. Current Issues in Molecular Biology. 2025; 47(8):587. https://doi.org/10.3390/cimb47080587
Chicago/Turabian StyleYuan, Changchun, Xudong Shi, Zhiqiang Wang, Yuqiang Li, Wenbing Ma, and Kai Fu. 2025. "Effect of a Novel Antidepressant and Anticancer Nuc01 on Depression in Cancer Survivors" Current Issues in Molecular Biology 47, no. 8: 587. https://doi.org/10.3390/cimb47080587
APA StyleYuan, C., Shi, X., Wang, Z., Li, Y., Ma, W., & Fu, K. (2025). Effect of a Novel Antidepressant and Anticancer Nuc01 on Depression in Cancer Survivors. Current Issues in Molecular Biology, 47(8), 587. https://doi.org/10.3390/cimb47080587