_Kim.png)
Screening of Potential Drug Targets Based on the Genome-Scale Metabolic Network Model of Vibrio parahaemolyticus


Abstract
1. Introduction
2. Materials and Method
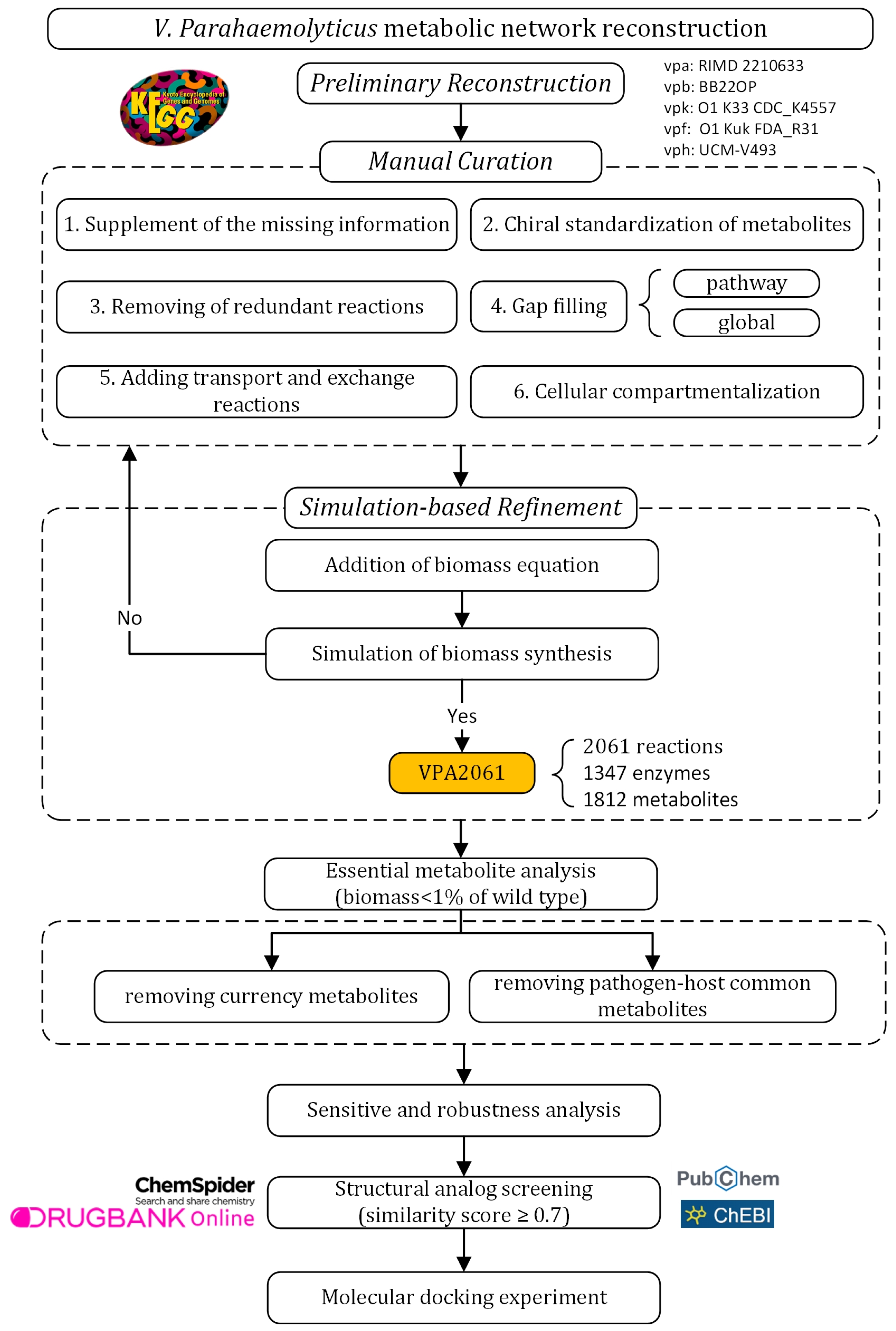
2.1. The Experimental Design
2.2. Reconstruction of the GSMN Model
2.2.1. Preliminary Reconstruction
2.2.2. Manual Refinement of the Model
Supplementing of the Missing Information
Chiral Standardization of Metabolites
Removal of Redundant Reactions
Gap Filling
Adding Transport and Exchange Reactions
Cellular Compartmentalization
2.2.3. Simulation-Based Refinement
Addition of Biomass Equation
Simulation of Biomass Synthesis
2.3. Essential Metabolite Analysis
2.4. Sensitivity and Robustness Analysis
2.5. Screening for Structural Analogs for Essential Metabolites
2.6. Molecular Docking Experiment
3. Results
3.1. Preliminary Reconstruction and Manual Curation of the GSMN for V. parahaemolyticus
3.1.1. Preliminary Reconstruction of the Model
3.1.2. Manual Curation of the Model
3.2. Simulation-Based Refinement of the Model
3.3. Essential Metabolite Analysis
3.4. Sensitivity and Robustness Analysis
3.5. Structural Analog Analysis and Drug Development Prediction
3.6. Molecular Docking of Essential Metabolites and Their Structure Analogs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GSMN | Genome-scale metabolic network model |
| KO | KEGG orthology |
| FBA | Flux balance analysis |
| WCC | Weakly connected component |
| FVA | Flux variability analysis |
| AMR | Anti-microbial resistance |
| ANI | Average nucleotide identity |
| isDDH | In silico DNA-DNA hybridization |
References
- Zhang, L.L.; Orth, K. Virulence determinants for Vibrio parahaemolyticus infection. Curr. Opin. Microbiol. 2013, 16, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Mala, W.; Alam, M.; Angkititrakul, S.; Wongwajana, S.; Lulitanond, V.; Huttayananont, S.; Kaewkes, W.; Faksri, K.; Chomvarin, C. Serogroup, virulence, and molecular traits of Vibrio parahaemolyticus isolated from clinical and cockle sources in northeastern Thailand. Infect. Genet. Evol. 2016, 39, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.C.; Liu, C.C. Vibrio parahaemolyticus: A concern of seafood safety. Food Microbiol. 2007, 24, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Raja, R.A.; Sridhar, R.; Balachandran, C.; Palanisammi, A.; Ramesh, S.; Nagarajan, K. Pathogenicity profile of Vibrio parahaemolyticus in farmed Pacific white shrimp, Penaeus vannamei. Fish Shellfish Immunol. 2017, 67, 368–381. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, J.B.; DePaola, A.; Bopp, C.A.; Martinek, K.A.; Napolilli, N.P.; Allison, C.G.; Murray, S.L.; Thompson, E.C.; Bird, M.M.; Middaugh, J.P. Outbreak of Vibrio parahaemolyticus gastroenteritis associated with Alaskan oysters. N. Engl. J. Med. 2005, 353, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Raszl, S.M.; Froelich, B.A.; Vieira, C.R.; Blackwood, A.D.; Noble, R.T. Vibrio parahaemolyticus and Vibrio vulnificus in South America: Water, seafood and human infections. J. Appl. Microbiol. 2016, 121, 1201–1222. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.H.; Raghunath, P. Editorial: Vibrionaceae Diversity, Multidrug Resistance and Management. Front. Microbiol. 2018, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Ha, P.T.H.; Thi, Q.V.C.; Thuy, N.P.; Luan, N.T. Multi-antibiotics resistance phenotype of pathogenic Vibrio parahaemolyticus isolated from acute hepatopancreatic necrosis disease in Litopenaeus vannamei farmed in the Mekong Delta. J. World Aquacult. Soc. 2023, 54, 1070–1087. [Google Scholar] [CrossRef]
- Bakleh, M.Z.; Kohailan, M.; Marwan, M.; Alhaj Sulaiman, A. A Systematic Review and Comprehensive Analysis of mcr Gene Prevalence in Bacterial Isolates in Arab Countries. Antibiotics 2024, 13, 958. [Google Scholar] [CrossRef] [PubMed]
- Elmahdi, S.; DaSilva, L.V.; Parveen, S. Antibiotic resistance of Vibrio parahaemolyticus and Vibrio vulnificus in various countries: A review. Food Microbiol. 2016, 57, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.V.; Brabb, T.; Pekow, C.; Vasbinder, M.A. Administration of substances to laboratory animals: Routes of administration and factors to consider. J. Am. Assoc. Lab. Anim. Sci. JAALAS 2011, 50, 600–613. [Google Scholar] [PubMed]
- Kothari, D.; Patel, S.; Kim, S.K. Probiotic supplements might not be universally-effective and safe: A review. Biomed. Pharmacother.=Biomed. Pharmacother. 2019, 111, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Kah Sem, N.A.D.; Abd Gani, S.; Chong, C.M.; Natrah, I.; Shamsi, S. Management and Mitigation of Vibriosis in Aquaculture: Nanoparticles as Promising Alternatives. Int. J. Mol. Sci. 2023, 24, 12542. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Wei, X.; Shi, T.; Sun, X.; Xu, N.; Gao, C.; Zou, W. Genome-scale metabolic network models: From first-generation to next-generation. Appl. Microbiol. Biotechnol. 2022, 106, 4907–4920. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Ye, C. Development and applications of genome-scale metabolic network models. Adv. Appl. Microbiol. 2024, 126, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.E.; Tandon, K.; Verbruggen, H.; Nikoloski, Z. Comparative analysis of metabolic models of microbial communities reconstructed from automated tools and consensus approaches. NPJ Syst. Biol. Appl. 2024, 10, 54. [Google Scholar] [CrossRef] [PubMed]
- Seaver, S.M.D.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 2021, 49, D575–D588. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Martin-Sancho, L.; Pal, L.R.; Pu, Y.; Riva, L.; Yin, X.; Sinha, S.; Nair, N.U.; Chanda, S.K.; Ruppin, E. Genome-scale metabolic modeling reveals SARS-CoV-2-induced metabolic changes and antiviral targets. Mol. Syst. Biol. 2021, 17, e10260. [Google Scholar] [CrossRef] [PubMed]
- Beste, D.J.; Hooper, T.; Stewart, G.; Bonde, B.; Avignone-Rossa, C.; Bushell, M.E.; Wheeler, P.; Klamt, S.; Kierzek, A.M.; McFadden, J. GSMN-TB: A web-based genome-scale network model of Mycobacterium tuberculosis metabolism. Genome Biol. 2007, 8, R89. [Google Scholar] [CrossRef] [PubMed]
- Rawls, K.; Dougherty, B.V.; Papin, J. Metabolic Network Reconstructions to Predict Drug Targets and Off-Target Effects. Methods Mol. Biol. 2020, 2088, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Raškevičius, V.; Mikalayeva, V.; Antanavičiūtė, I.; Ceslevičienė, I.; Skeberdis, V.A.; Kairys, V.; Bordel, S. Genome scale metabolic models as tools for drug design and personalized medicine. PLoS ONE 2018, 13, e0190636. [Google Scholar] [CrossRef] [PubMed]
- Cesur, M.F.; Siraj, B.; Uddin, R.; Durmuş, S.; Çakır, T. Network-based metabolism-centered screening of potential drug targets in Klebsiella pneumoniae at genome scale. Front. Cell. Infect. Microbiol. 2020, 9, 447. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.U.; Kim, T.Y.; Lee, S.Y. Genome-scale metabolic network analysis and drug targeting of multi-drug resistant pathogen Acinetobacter baumannii AYE. Mol. Biosyst. 2010, 6, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Palsson, B.O. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Matsuura, Y.; Ishiguro-Watanabe, M. KEGG: Biological systems database as a model of the real world. Nucleic Acids Res. 2025, 53, D672–D677. [Google Scholar] [CrossRef] [PubMed]
- Hao, T.; Ma, H.W.; Zhao, X.M.; Goryanin, I. Compartmentalization of the Edinburgh Human Metabolic Network. BMC Bioinform. 2010, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Hao, T.; Ma, H.W.; Zhao, X.M.; Goryanin, I. The reconstruction and analysis of tissue specific human metabolic networks. Mol. Biosyst. 2012, 8, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.U.; Kim, S.Y.; Jeong, H.; Kim, T.Y.; Kim, J.J.; Choy, H.E.; Yi, K.Y.; Rhee, J.H.; Lee, S.Y. Integrative genome-scale metabolic analysis of Vibrio vulnificus for drug targeting and discovery. Mol. Syst. Biol. 2011, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2022, 51, D1373–D1380. [Google Scholar] [CrossRef] [PubMed]
- Hastings, J.; Owen, G.; Dekker, A.; Ennis, M.; Kale, N.; Muthukrishnan, V.; Turner, S.; Swainston, N.; Mendes, P.; Steinbeck, C. ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic Acids Res. 2016, 44, D1214–D1219. [Google Scholar] [CrossRef] [PubMed]
- Editorial: ChemSpider—A tool for Natural Products research. Nat. Prod. Rep. 2015, 32, 1163–1164. [CrossRef] [PubMed]
- Knox, C.; Wilson, M.; Klinger, C.M.; Franklin, M.; Oler, E.; Wilson, A.; Pon, A.; Cox, J.; Chin, N.E.L.; Strawbridge, S.A.; et al. DrugBank 6.0: The DrugBank Knowledgebase for 2024. Nucleic Acids Res. 2024, 52, D1265–D1275. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Yang, F.; Kang, J.; Yang, X.; Lai, X.; Gao, Y. Multi-Layer Identification of Highly-Potent ABCA1 Up-Regulators Targeting LXRbeta Using Multiple QSAR Modeling, Structural Similarity Analysis, and Molecular Docking. Molecules 2016, 21, 1639. [Google Scholar] [CrossRef] [PubMed]
- Szilagyi, K.; Flachner, B.; Hajdu, I.; Szaszko, M.; Dobi, K.; Lorincz, Z.; Cseh, S.; Dorman, G. Rapid Identification of Potential Drug Candidates from Multi-Million Compounds’ Repositories. Combination of 2D Similarity Search with 3D Ligand/Structure Based Methods and In Vitro Screening. Molecules 2021, 26, 5593. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Santos, A.; von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef] [PubMed]
- UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [CrossRef] [PubMed]
- Bugnon, M.; Rohrig, U.F.; Goullieux, M.; Perez, M.A.S.; Daina, A.; Michielin, O.; Zoete, V. SwissDock 2024: Major enhancements for small-molecule docking with Attracting Cavities and AutoDock Vina. Nucleic Acids Res. 2024, 52, W324–W332. [Google Scholar] [CrossRef] [PubMed]
- Makino, K.; Oshima, K.; Kurokawa, K.; Yokoyama, K.; Uda, T.; Tagomori, K.; Iijima, Y.; Najima, M.; Nakano, M.; Yamashita, A.; et al. Genome sequence of Vibrio parahaemolyticus: A pathogenic mechanism distinct from that of V. cholerae. Lancet 2003, 361, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.V.; Depasquale, S.M.; Harbolick, E.A.; Hong, T.; Kernell, A.L.; Kruchko, D.H.; Modise, T.; Smith, C.E.; McCarter, L.L.; Stevens, A.M. Complete Genome Sequence of Prepandemic Vibrio parahaemolyticus BB22OP. Genome Announc. 2013, 1, e00002-12. [Google Scholar] [CrossRef] [PubMed]
- Kalburge, S.S.; Polson, S.W.; Boyd Crotty, K.; Katz, L.; Turnsek, M.; Tarr, C.L.; Martinez-Urtaza, J.; Boyd, E.F. Complete Genome Sequence of Vibrio parahaemolyticus Environmental Strain UCM-V493. Genome Announc. 2014, 2, e00159-14. [Google Scholar] [CrossRef] [PubMed]
- Ludeke, C.H.M.; Kong, N.; Weimer, B.C.; Fischer, M.; Jones, J.L. Complete Genome Sequences of a Clinical Isolate and an Environmental Isolate of Vibrio parahaemolyticus. Genome Announc. 2015, 3, e00216-15. [Google Scholar] [CrossRef] [PubMed]
- Ashok Kumar, J.; Vinaya Kumar, K.; Avunje, S.; Akhil, V.; Ashok, S.; Kumar, S.; Sivamani, B.; Grover, M.; Rai, A.; Alavandi, S.V.; et al. Phylogenetic Relationship Among Brackishwater Vibrio Species. Evol. Bioinform. Online 2020, 16, 1176934320903288. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gou, Y.; Yang, J.; Zhao, L.; Wang, B.; Hao, T.; Sun, J. Genome-scale metabolic network model of Eriocheir sinensis icrab4665 and nutritional requirement analysis. BMC Genom. 2022, 23, 475. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Yang, J.; Hao, T.; Li, J.; Sun, J. Reconstruction of Litopenaeus vannamei Genome-Scale Metabolic Network Model and Nutritional Requirements Analysis of Different Shrimp Commercial Varieties. Front. Genet. 2021, 12, 658109. [Google Scholar] [CrossRef] [PubMed]
- Hao, T.; Han, B.; Ma, H.; Fu, J.; Wang, H.; Wang, Z.; Tang, B.; Chen, T.; Zhao, X. In silico metabolic engineering of Bacillus subtilis for improved production of riboflavin, Egl-237, (R,R)-2,3-butanediol and isobutanol. Mol. Biosyst. 2013, 9, 2034–2044. [Google Scholar] [CrossRef] [PubMed]
- Dobson, P.D.; Patel, Y.; Kell, D.B. ‘Metabolite-likeness’ as a criterion in the design and selection of pharmaceutical drug libraries. Drug Discov. Today 2009, 14, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa-Gerasmio, I.R.; Tanaka, T.; Sumi, A.; Kondo, H.; Hirono, I.J.M.B. Effects of 5-Aminolevulinic acid on gene expression, immunity, and ATP levels in Pacific white shrimp, Litopenaeus vannamei. Mar. Biotechnol. 2018, 20, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, T.; Lin, G.; Li, M.; Zhang, Y.; Mai, K. The Assessment of Dietary Organic Zinc on Zinc Homeostasis, Antioxidant Capacity, Immune Response, Glycolysis and Intestinal Microbiota in White Shrimp (Litopenaeus vannamei Boone, 1931). Antioxidants 2022, 11, 1492. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Z.; Lin, G.; Li, M.; Zhang, Y.; Mai, K. Organic copper promoted copper accumulation and transport, enhanced low temperature tolerance and physiological health of white shrimp (Litopenaeus vannamei Boone, 1931). Fish Shellfish Immunol. 2023, 132, 108459. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, W.; Blanot, D.; De Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Nugraha, M.A.R.; Lin, Y.R.; Dewi, N.R.; Huang, H.T.; Nan, F.H.; Hu, Y.F. Effects of Taiwanese indigenous cinnamon (Cinnamomum osmophloeum) leaf hot-water extract on nonspecific immune responses, resistance against Vibrio parahaemolyticus, nonviable cells, and haemocyte subpopulations in white shrimp (Penaeusvannamei). Fish Shellfish Immunol. 2024, 151, 109680. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Strain ID | Strain Name | Number of KOs | References |
|---|---|---|---|
| vpa | RIMD 2210633 | 2187 | [39] |
| vpb | BB22OP | 2197 | [40] |
| vpk | O1 K33 CDC_K4557 | 2261 | [41] |
| vpf | O1 Kuk FDA_R31 | 2275 | [41] |
| vph | UCM-V493 | 2265 | [42] |
| Items | Number |
|---|---|
| Reaction | 2061 |
| Enzyme | 1132 |
| Metabolite | 1812 |
| Pathway | 150 |
| Subsystem | 13 |
| Item | Name |
|---|---|
| C00002 | ATP |
| C00003 | NAD |
| C00005 | NADPH |
| C00006 | NADP |
| C00008 | ADP |
| C00009 | PI |
| C00010 | CoA |
| C00014 | NH3 |
| C00015 | UDP |
| C00016 | FAD |
| C00041 | ALA |
| C00022 | Pyruvate |
| C00063 | CTP |
| C00075 | UTP |
| C00101 | Tetrahydrofolate |
| C00105 | UMP |
| KEGG ID | Name | Matched Structural Analogs |
|---|---|---|
| ADPHEP | ADP-L-glycero-D-manno-heptose | DB00171, DB00131, DB01992, DB00157, DB03147 |
| C00133 | D-Alanine | DB00160, DB01786, DB02952, DB00133, DB03929, DB19057, DB03320, DB04454 |
| C00498 | ADP-glucose | DB00171, DB00131, DB01992, DB00157, DB03147 |
| C00692 | Uridine-5’-Diphosphate-N-Acetylmuramoyl-L-Alanine-D-Glutamate | DB02314, DB01673, DB02196, DB03397, DB04147, DB03041, DB01861, DB02421, DB03501, DB01713, DB03723, DB03751, DB04097 |
| C00993 | D-Alanyl-D-alanine | DB07939, DB02518 |
| C01050 | UDP-N-acetylmuramate | DB01673, DB02196, DB03397, DB04174, DB02314, DB03041 DB01861, DB02421, DB03501, DB01713, DB03723, DB03751 DB04097, DB03161, DB02976, DB03488, DB02554, DB02485 |
| C01212 | UDP-N-acetylmuramoyl-L-alanine | DB01673, DB02196, DB03397, DB02314, DB04174, DB03041 DB01861, DB02421, DB03501, DB01713, DB03723, DB03751 DB04097, DB03161, DB02976, DB03488, DB02554 |
| C04576 | Pentagalloylglucose | DB03208, DB09372 |
| C04877 | UDP-N-acetylmuramoyl-L-alanyl-gamma-D-glutamyl-meso-2,6-diaminopimelate | DB02314, DB01673, DB04174, DB02196, DB03397, DB03041 DB01861, DB02421, DB03501, DB01713 |
| C17556 | di-trans,poly-cis-Undecaprenyl phosphate | DB07780, DB07841, DB02552 |
| Category | DrugBank ID |
|---|---|
| Energy and Redox Coenzymes | DB00171, DB00131, DB01992, DB00157, DB03147 |
| Amino Acid Analogs | DB00160, DB01786, DB00133, DB03929, DB19057, DB03320, DB04454, DB02518, DB07939, DB02952 |
| Nucleotide Sugar/Peptidoglycan Precursors | DB02314, DB01673, DB02196, DB3397, DB04147, DB03041, DB01861, DB02421, DB03501, DB01713, DB03723, DB03751, DB04097, DB03161, DB02976, DB03488, DB02554, DB02485, DB04174 |
| Isoprenoid Diphosphates | DB07780, DB07841, DB02552 |
| Plant-Derived Polyphenols | DB03208, DB09372 |
| Essential Metabolite | Structure Analog | Target Protein | ΔG1 1 (kcal/mol) | ΔG2 2 (kcal/mol) |
|---|---|---|---|---|
| ADP-heptose | ATP | HldE | −8.2 | −7.7 |
| D-Alanine | D-Alanine | HemE | −5.1 | −5.1 |
| UDP-MurNAc | UDP-MurNAc-L-Ala | MurE | −9.1 | −8.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Wang, B.; Zhang, R.; He, Z.; Zhang, M.; Hao, T.; Sun, J. Screening of Potential Drug Targets Based on the Genome-Scale Metabolic Network Model of Vibrio parahaemolyticus. Curr. Issues Mol. Biol. 2025, 47, 575. https://doi.org/10.3390/cimb47070575
Zhang L, Wang B, Zhang R, He Z, Zhang M, Hao T, Sun J. Screening of Potential Drug Targets Based on the Genome-Scale Metabolic Network Model of Vibrio parahaemolyticus. Current Issues in Molecular Biology. 2025; 47(7):575. https://doi.org/10.3390/cimb47070575
Chicago/Turabian StyleZhang, Lingrui, Bin Wang, Ruiqi Zhang, Zhen He, Mingzhi Zhang, Tong Hao, and Jinsheng Sun. 2025. "Screening of Potential Drug Targets Based on the Genome-Scale Metabolic Network Model of Vibrio parahaemolyticus" Current Issues in Molecular Biology 47, no. 7: 575. https://doi.org/10.3390/cimb47070575
APA StyleZhang, L., Wang, B., Zhang, R., He, Z., Zhang, M., Hao, T., & Sun, J. (2025). Screening of Potential Drug Targets Based on the Genome-Scale Metabolic Network Model of Vibrio parahaemolyticus. Current Issues in Molecular Biology, 47(7), 575. https://doi.org/10.3390/cimb47070575