
Restoration of Autophagy and Apoptosis in Myelodysplastic Syndromes: The Effect of Azacitidine in Disease Pathogenesis

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. The Study Group
2.2. Primary Cell Separation from MDS Patients
2.3. The MDS-L Cell Line Culture
2.4. Azacitidine Preparation
2.5. MDS-L Cell Viability After Azacitidine Treatment
2.6. RNA and Protein Extraction from the MDS-L Cells After Azacitidine Treatment
2.7. The Gene Expression Analysis Using Quantitative Real-Time PCR
2.8. The Protein Expression Analysis Using Western Blotting
2.9. The Phosphoprotein Expression Analysis Using a Multiplex ELISA Assay
2.10. Statistical Approach
3. Results
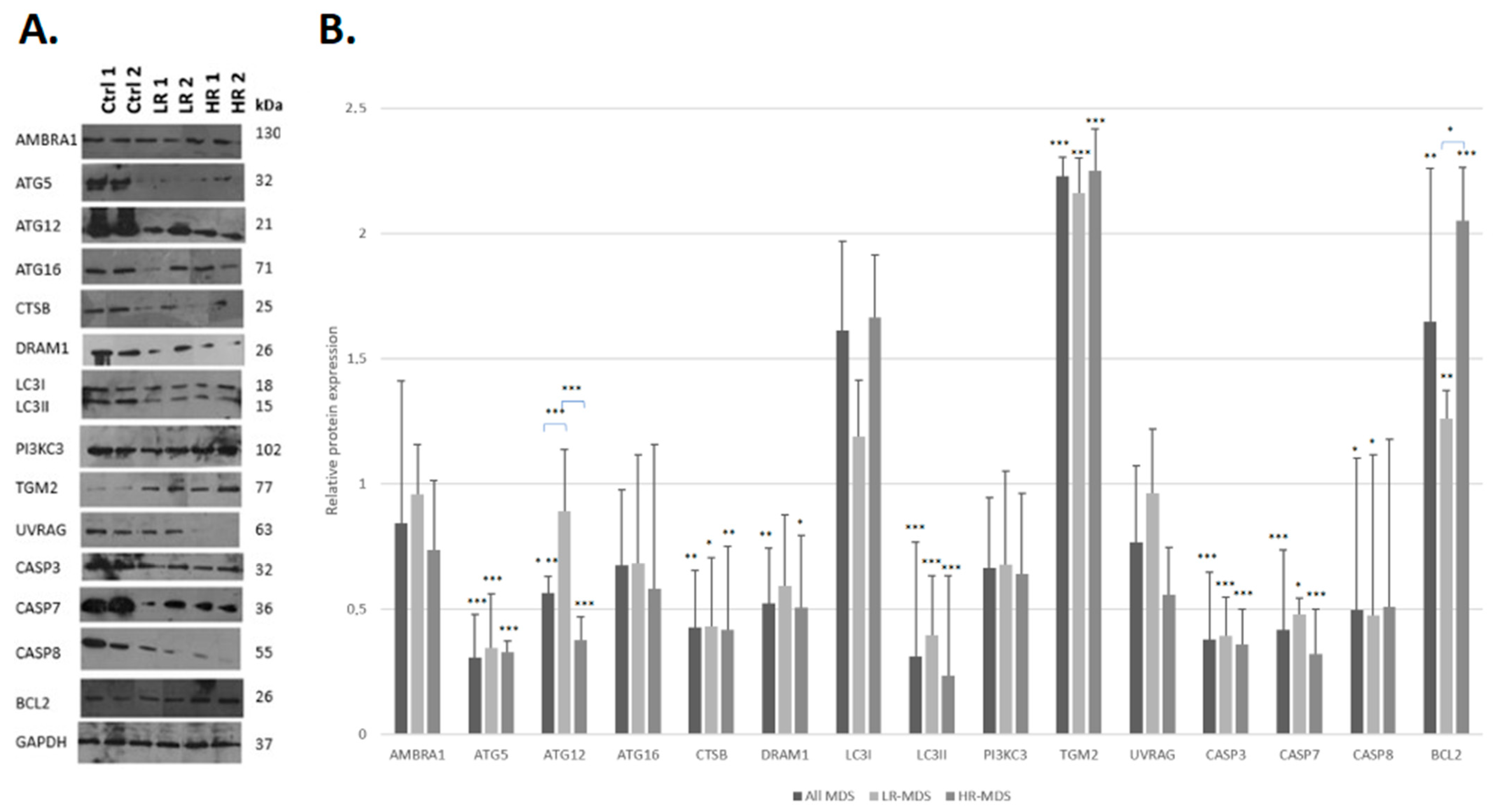
3.1. MDS Is Characterized by the Downregulation of Autophagy and Apoptosis Proteins
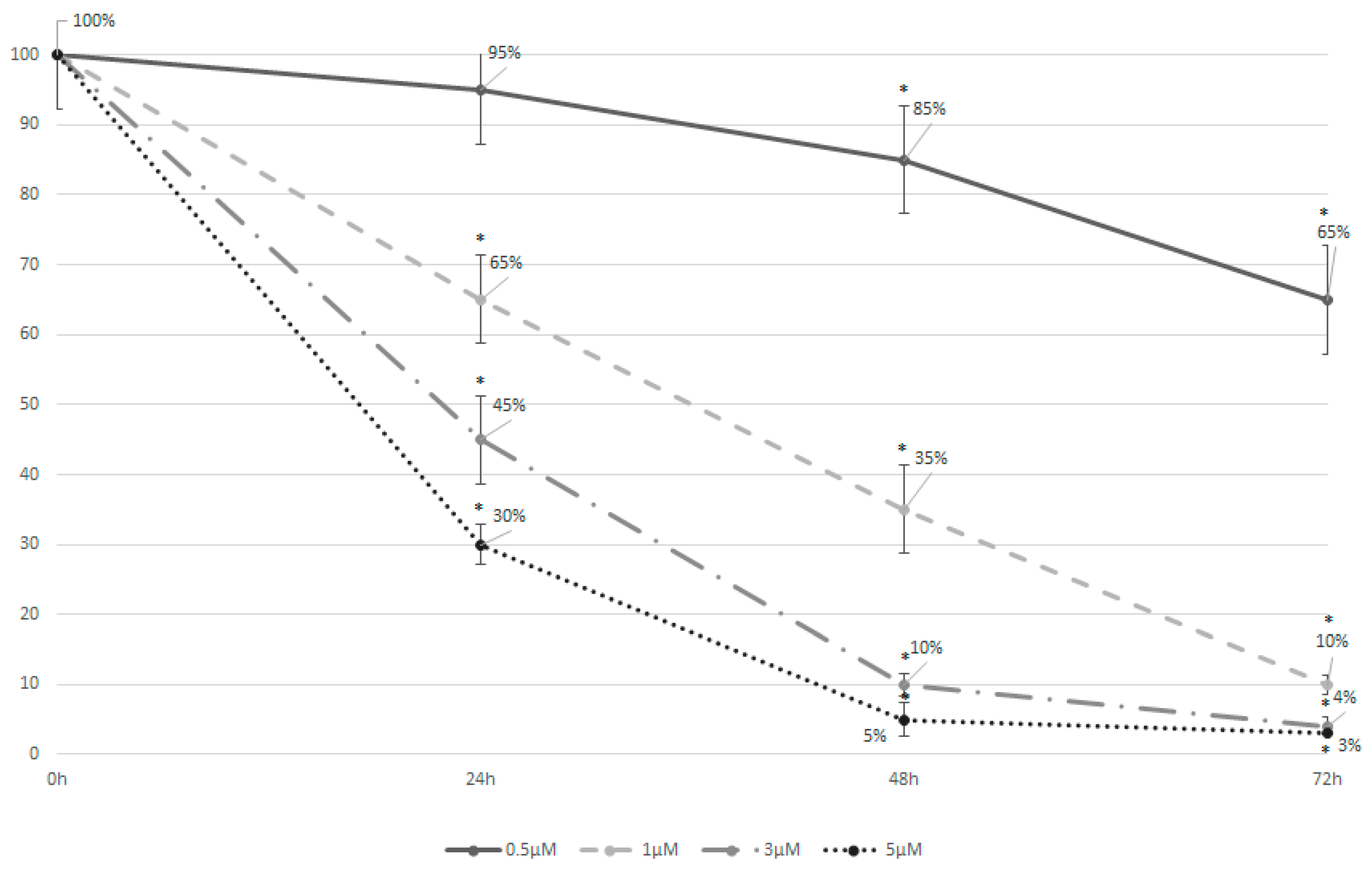
3.2. Azacytidine Induces Effective Cell Apoptosis in the MDS-L Cell Line
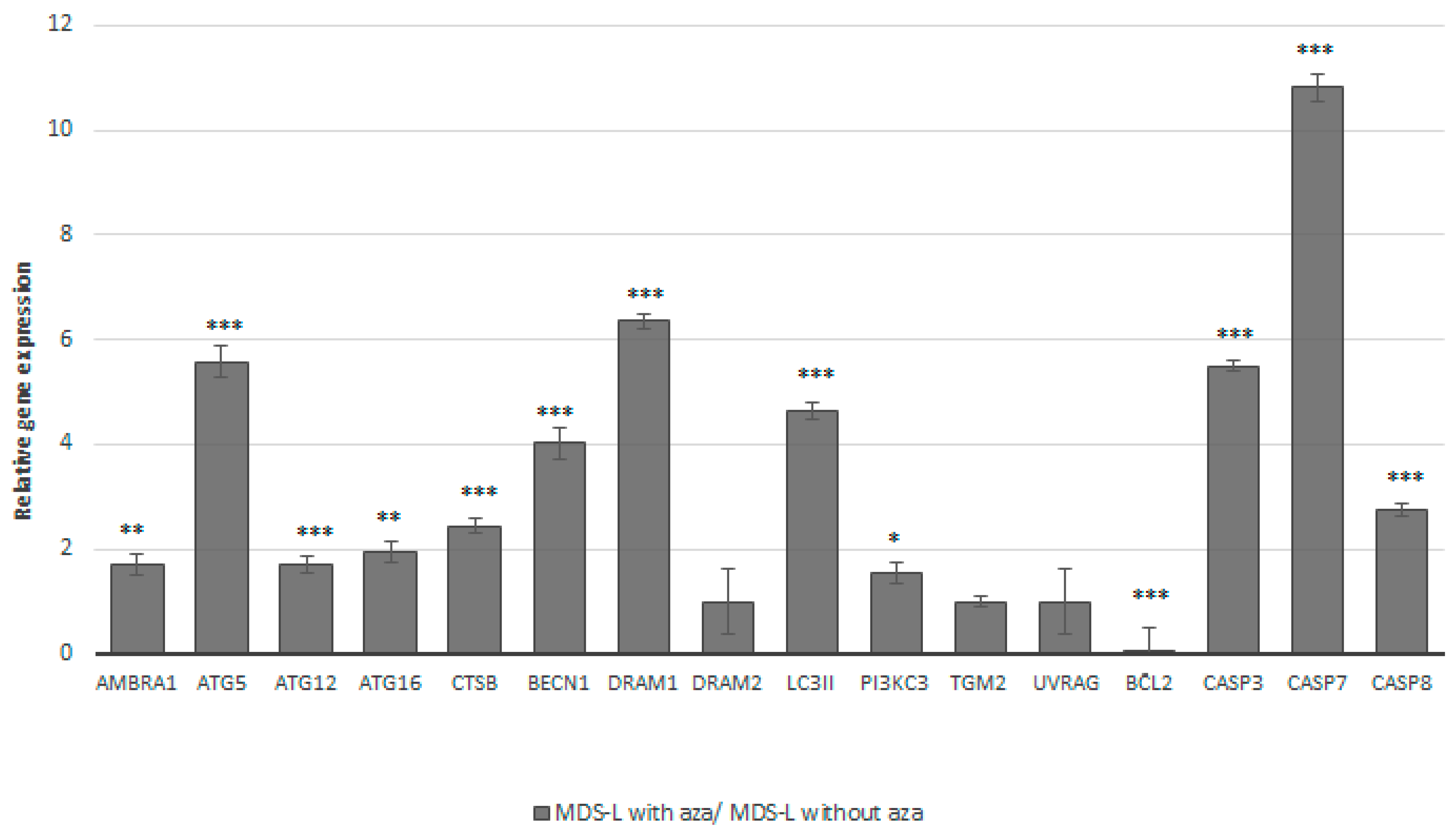
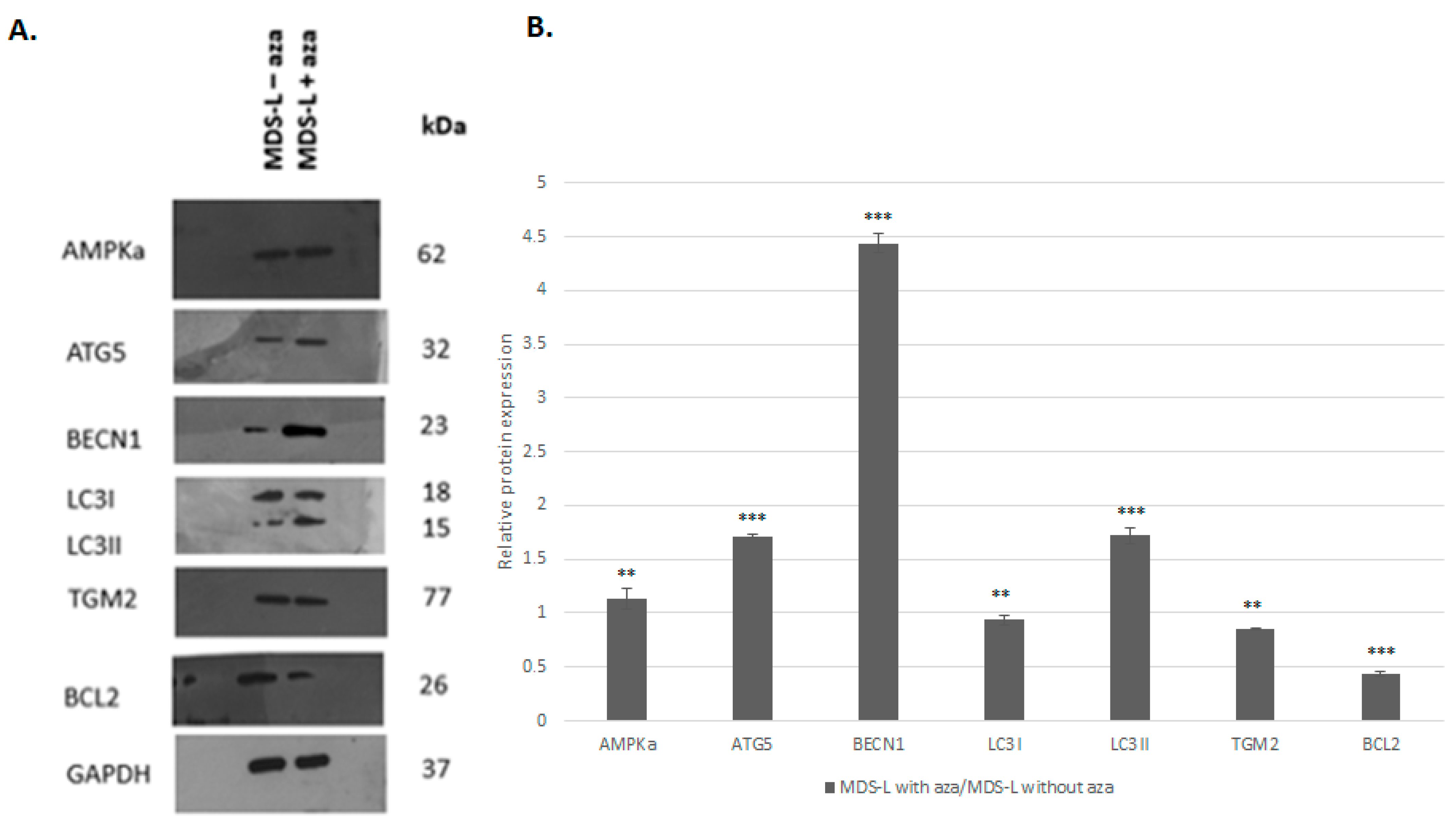
3.3. Treatment of the MDS-L Cells with Azacitidine Restored Autophagy and Apoptosis at the mRNA and Protein Levels
3.4. Azacitidine May Also Affect Phosphoproteins Related to Autophagy and Apoptosis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Garcia-Manero, G. Myelodysplastic syndromes: 2023 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2023, 98, 1307–1325. [Google Scholar] [CrossRef]
- McDonald, L.; McCarthy, P.; Khan, M.; Hogan, P.; Kelleher, E.C.; Murphy, P.; Quinn, J.; Desmond, R.; McHugh, J.; Strickland, M.; et al. Should Myelodysplastic Syndromes in Very Old Patients be More Actively Managed? Clinical Characteristics, Management and Outcomes for Patients 85 Years and Older. Blood 2018, 132 (Suppl. 1), 5515. [Google Scholar] [CrossRef]
- Watson, A.S.; Mortensen, M.; Simon, A.K. Autophagy in the pathogenesis of myelodysplastic syndrome and acute myeloid leukemia. Cell Cycle 2011, 10, 1719–1725. [Google Scholar] [CrossRef]
- Parker, J.E.; Mufti, G.J. The Role of Apoptosis in the Pathogenesis of the Myelodysplastic Syndromes. Int. J. Hematol. 2001, 73, 416–428. [Google Scholar] [CrossRef]
- Ames, K.; Gritsman, K. Unraveling the Link between Class 1A PI3-Kinase, Autophagy, and Myelodysplasia. Autophagy 2024, 20, 952–954. [Google Scholar] [CrossRef]
- Jiang, H.; Yang, L.; Guo, L.; Cui, N.; Zhang, G.; Liu, C.; Xing, L.; Shao, Z.; Wang, H. Impaired Mitophagy of Nucleated Erythroid Cells Leads to Anemia in Patients with Myelodysplastic Syndromes. Oxidative Med. Cell. Longev. 2018, 2018, 6328051. [Google Scholar] [CrossRef]
- Wang, M.J.; Liu, W.Y.; Wang, X.Y.; Li, Y.M.; Xiao, H.Y.; Quan, R.C.; Huang, G.; Hu, X.M. Autophagy Gene Panel-Based Prognostic Model in Myelodysplastic Syndrome. Front. Oncol. 2021, 10, 606928. [Google Scholar] [CrossRef]
- Kerbauy, D.B.; Deeg, H.J. Apoptosis and antiapoptotic mechanisms in the progression of myelodysplastic syndrome. Exp. Hematol. 2007, 35, 1739–1746. [Google Scholar] [CrossRef]
- McBride, A.; Houtmann, S.; Wilde, L.; Vigil, C.; Eischen, C.M.; Kasner, M.; Palmisiano, N. The Role of Inhibition of Apoptosis in Acute Leukemias and Myelodysplastic Syndrome. Front. Oncol. 2019, 9, 192. [Google Scholar] [CrossRef]
- Garcia, J.S. Prospects for Venetoclax in Myelodysplastic Syndromes. Hematol. Oncol. Clin. N. Am. 2020, 34, 441–448. [Google Scholar] [CrossRef]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef]
- Tsekoura, G.; Agathangelidis, A.; Kontandreopoulou, C.-N.; Taliouraki, A.; Mporonikola, G.; Stavropoulou, M.; Diamantopoulos, P.T.; Viniou, N.-A.; Aleporou, V.; Papassideri, I.; et al. Deregulation of Autophagy and Apoptosis in Patients with Myelodysplastic Syndromes: Implications for Disease Development and Progression. Curr. Issues Mol. Biol. 2023, 45, 4135–4150. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Raj, K.; Mufti, G.J. Azacytidine (Vidaza®) in the treatment of myelodysplastic syndromes. Ther. Clin. Risk Manag. 2006, 2, 377–388. [Google Scholar] [CrossRef]
- Buckstein, R.; Yee, K.; Wells, R.A. 5-Azacytidine in myelodysplastic syndromes: A clinical practice guideline. Cancer Treat. Rev. 2011, 37, 160–167. [Google Scholar] [CrossRef]
- Campelo, M.D.; Delgado, R.G.; Molias, A.C.G.; Sanchez, J.F. Azacytidine for the treatment of myelodysplastic syndromes in the elderly. Adv. Ther. 2011, 28 (Suppl. 2), 10–15. [Google Scholar] [CrossRef]
- Zugasti, I.; Castaño-Díez, S.; Esteban, D.; Pita, A.A.; Pomares, H.; González, A.P.; Garcia-Avila, S.; Padilla-Conejo, I.; de la Fuente, C.; Martínez-Roca, A.; et al. Venetoclax and Azacytidine Treatment for High Risk Myelodysplastic Syndromes and Chronic Myelomonocytic Leukemia As a Bridge Therapy to Transplant. a GESMD Study. Blood 2023, 142 (Suppl. 1), 1858. [Google Scholar] [CrossRef]
- Kim, Y.J.; Jang, J.H.; Kwak, J.Y.; Lee, J.H.; Kim, H.J. Use of azacitidine for myelodysplastic syndromes: Controversial issues and practical recommendations. Blood Res. 2013, 48, 87–98. [Google Scholar] [CrossRef]
- Daw, S.; Law, S. Quercetin induces autophagy in myelodysplastic bone marrow including hematopoietic stem/progenitor compartment. Environ. Toxicol. 2021, 36, 149–167. [Google Scholar] [CrossRef]
- Ames, K.; Kaur, I.; Shi, Y.; Tong, M.M.; Sinclair, T.; Hemmati, S.; Glushakow-Smith, S.G.; Tein, E.; Gurska, L.; Steidl, U.; et al. PI3-kinase deletion promotes myelodysplasia by dysregulating autophagy in hematopoietic stem cells. Sci. Adv. 2023, 9, eade8222. [Google Scholar] [CrossRef]
- Im, H.; Rao, V.; Sridhar, K.; Bentley, J.; Mishra, T.; Chen, R.; Hall, J.; Graber, A.; Zhang, Y.; Li, X.; et al. Distinct transcriptomic and exomic abnormalities within myelodysplastic syndrome marrow cells. Leuk. Lymphoma 2018, 59, 2952–2962. [Google Scholar] [CrossRef]
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015, 17, 20–30. [Google Scholar] [CrossRef]
- Zang, D.Y.; Goodwin, R.G.; Loken, M.R.; Bryant, E.; Deeg, H.J. Expression of tumor necrosis factor–related apoptosis-inducing ligand, Apo2L, and its receptors in myelodysplastic syndrome: Effects on in vitro hemopoiesis. Blood 2001, 98, 3058–3065. [Google Scholar] [CrossRef]
- Jiang, M.; Qi, L.; Li, L.; Wu, Y.; Song, D.; Li, Y. Caspase-8: A key protein of cross-talk signal way in “PANoptosis” in cancer. Int. J. Cancer 2021, 149, 1408–1420. [Google Scholar] [CrossRef]
- Iriani, A.; Rachman, A.; Setiabudy, R.D.; Kresno, S.B.; Sudoyo, A.W.; Arief, M.; Harahap, A.R.; Fatina, M.K. TNFα induces Caspase-3 activity in hematopoietic progenitor cells CD34+, CD33+, and CD41 + of myelodysplastic syndromes. BMC Mol. Cell Biol. 2023, 24, 33. [Google Scholar] [CrossRef]
- Liu, S.; Joshi, K.; Zhang, L.; Li, W.; Mack, R.; Runde, A.; Hagen, P.A.; Barton, K.; Breslin, P.; Ji, H.-L.; et al. Caspase 8 deletion causes infection/inflammation-induced bone marrow failure and MDS-like disease in mice. Cell Death Dis. 2024, 15, 278. [Google Scholar] [CrossRef]
- Gillson, J.; El-Aziz, Y.S.A.; Leck, L.Y.W.; Jansson, P.J.; Pavlakis, N.; Samra, J.S.; Mittal, A.; Sahni, S. Autophagy: A Key Player in Pancreatic Cancer Progression and a Potential Drug Target. Cancers 2022, 14, 3528. [Google Scholar] [CrossRef]
- Wang, W.; Li, X.; Han, X.-Z.; Meng, F.-B.; Wang, Z.-X.; Zhai, Y.-Q.; Zhou, D.-S. Transglutaminase-2 is Involved in Cell Apoptosis of Osteosarcoma Cell Line U2OS Under Hypoxia Condition. Cell Biochem. Biophys. 2015, 72, 283–288. [Google Scholar] [CrossRef]
- Rossin, F.; D’Eletto, M.; Macdonald, D.; Farrace, M.G.; Piacentini, M. TG2 transamidating activity acts as a reostat controlling the interplay between apoptosis and autophagy. Amino Acids 2012, 42, 1793–1802. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: Where do they meet? Apoptosis 2014, 19, 555–566. [Google Scholar] [CrossRef]
- Mortensen, M.; Ferguson, D.; Edelmann, M.; Kessler, B.; Morten, K.; Komatsu, M.; Simon, A. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 832–837. [Google Scholar] [CrossRef]
- Watson, A.; Riffelmacher, T.; Stranks, A.; Williams, O.; De Boer, J.; Cain, K.; MacFarlane, M.; McGouran, J.; Kessler, B.; Khandwala, S.; et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 2015, 1, 15008. [Google Scholar] [CrossRef]
- Liu, F.; Lee, J.Y.; Wei, H.; Tanabe, O.; Engel, J.D.; Morrison, S.J.; Guan, J.-L. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010, 116, 4806–4814. [Google Scholar] [CrossRef]
- Rai, R.; Patel, F.; Feld, J.; Melana, S.; Navada, S.C.; Odchimar-Reissig, R.; Demakos, E.P.; Reddy, E.P.; Silverman, L.R. Combination of Ras Modulator and Azacitidine Impacts Innate Immune Signaling Pathway in MDS-L Cell Line. Blood 2021, 138 (Suppl. 1), 4325. [Google Scholar] [CrossRef]
- Romano, A.; Giallongo, C.; La Cava, P.; Parrinello, N.L.; Chiechi, A.; Vetro, C.; Tibullo, D.; Di Raimondo, F.; Liotta, L.A.; Espina, V.; et al. Proteomic Analysis Reveals Autophagy as Pro-Survival Pathway Elicited by Long-Term Exposure with 5-Azacitidine in High-Risk Myelodysplasia. Front. Pharmacol. 2017, 8, 204. [Google Scholar] [CrossRef]
- Wu, N.; Zhu, Y.; Xu, X.; Zhu, Y.; Song, Y.; Pang, L.; Chen, Z. The anti-tumor effects of dual PI3K/mTOR inhibitor BEZ235 and histone deacetylase inhibitor Trichostatin A on inducing autophagy in esophageal squamous cell carcinoma. J. Cancer 2018, 9, 987–997. [Google Scholar] [CrossRef]
- Liang, S.; Zhou, X.; Cai, D.; Rodrigues-Lima, F.; Chi, J.; Wang, L. Network Pharmacology and Experimental Validation Reveal the Effects of Chidamide Combined With Aspirin on Acute Myeloid Leukemia-Myelodysplastic Syndrome Cells Through PI3K/AKT Pathway. Front. Cell Dev. Biol. 2021, 9, 685954. [Google Scholar] [CrossRef]
- Zeng, W.; Dai, H.; Yan, M.; Cai, X.; Luo, H.; Ke, M.; Liu, Z. Decitabine-Induced Changes in Human Myelodysplastic Syndrome Cell Line SKM-1 Are Mediated by FOXO3A Activation. J. Immunol. Res. 2017, 2017, 4302320. [Google Scholar] [CrossRef]
- Li, L.; Liu, W.; Sun, Q.; Zhu, H.; Hong, M.; Qian, S. Decitabine Downregulates TIGAR to Induce Apoptosis and Autophagy in Myeloid Leukemia Cells. Oxidative Med. Cell. Longev. 2021, 2021, 8877460. [Google Scholar] [CrossRef]
- Saba, H.I. Decitabine in the treatment of myelodysplastic syndromes. Ther. Clin. Risk Manag. 2007, 3, 807–817. [Google Scholar]
- Khan, R.; Schmidt-Mende, J.; Karimi, M.; Gogvadze, V.; Hassan, M.; Ekström, T.J.; Zhivotovsky, B.; Hellström-Lindberg, E. Hypomethylation and apoptosis in 5-azacytidine–treated myeloid cells. Exp. Hematol. 2008, 36, 149–157. [Google Scholar] [CrossRef]
- Galante, J.M.; Mortenson, M.M.; Schlieman, M.G.; Virudachalam, S.; Bold, R.J. Targeting NF-kB/BCL-2 pathway increases apoptotic susceptibility to chemotherapy in pancreatic cancer. J. Surg. Res. 2004, 121, 306–307. [Google Scholar] [CrossRef]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The Role of the Transcription Factor CREB in Immune Function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef]
- Cluzeau, T.; Robert, G.; Mounier, N.; Karsenti, J.M.; Dufies, M.; Puissant, A.; Jacquel, A.; Renneville, A.; Preudhomme, C.; Cassuto, J.-P.; et al. BCL2L10 is a predictive factor for resistance to Azacitidine in MDS and AML patients. Oncotarget 2012, 3, 490–501. [Google Scholar] [CrossRef]
- Du, Y.; Li, C.; Yan, J. The efficacy and safety of venetoclax and azacytidine combination treatment in patients with acute myeloid leukemia and myelodysplastic syndrome: Systematic review and meta-analysis. Hematology 2023, 28, 2198098. [Google Scholar] [CrossRef]
- Abuetabh, Y.; Wu, H.H.; Chai, C.; Al Yousef, H.; Persad, S.; Sergi, C.M.; Leng, R. DNA damage response revisited: The p53 family and its regulators provide endless cancer therapy opportunities. Exp. Mol. Med. 2022, 54, 1658–1669. [Google Scholar] [CrossRef]
- Hirao, A.; Kong, Y.-Y.; Matsuoka, S.; Wakeham, A.; Ruland, J.; Yoshida, H.; Liu, D.; Elledge, S.J.; Mak, T.W. DNA Damage-Induced Activation of p53 by the Checkpoint Kinase Chk2. Science 2000, 287, 1824–1827. [Google Scholar] [CrossRef]
- Bossy-Wetzel, E.; Bakiri, L.; Yaniv, M. Induction of apoptosis by the transcription factor c-Jun. EMBO J. 1997, 16, 1695–1709. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Ye, Y.-C.; Shi, Q.-F.; Chai, K.; Tashiro, S.-I.; Onodera, S.; Ikejima, T. Activation of ERK–p53 and ERK-Mediated Phosphorylation of Bcl-2 Are Involved in Autophagic Cell Death Induced by the c-Met Inhibitor SU11274 in Human Lung Cancer A549 Cells. J. Pharmacol. Sci. 2012, 118, 423–432. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J. ERK and cell death: Mechanisms of ERK-induced cell death—Apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, L.; Yang, Y.; Liu, H.; Kang, X.; Nie, Y.; Fan, D. DNMT1 expression partially dictates 5-Azacytidine sensitivity and correlates with RAS/MEK/ERK activity in gastric cancer cells. Epigenetics 2023, 18, 2254976. [Google Scholar] [CrossRef]
- Carew, J.S.; Nawrocki, S.T.; Kahue, C.N.; Zhang, H.; Yang, C.; Chung, L.; Houghton, J.A.; Huang, P.; Giles, F.J.; Cleveland, J.L. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl–mediated drug resistance. Blood 2007, 110, 313–322. [Google Scholar] [CrossRef]
- Parks, M.; Tillhon, M.; Donà, F.; Prosperi, E.; Scovassi, A.I. 2-Methoxyestradiol: New perspectives in colon carcinoma treatment. Mol. Cell Endocrinol. 2011, 331, 119–128. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Harari, P.M.; Allen, G.W.; Bonner, J.A. Biology of Interactions: Antiepidermal Growth Factor Receptor Agents. J. Clin. Oncol. 2007, 25, 4057–4065. [Google Scholar] [CrossRef]
- El Amri, M.; Fitzgerald, U.; Schlosser, G. MARCKS and MARCKS-like proteins in development and regeneration. J. Biomed. Sci. 2018, 25, 43. [Google Scholar] [CrossRef]
- Daw, S.; Law, A.; Law, S. Myelodysplastic Syndrome related alterations of MAPK signaling in the bone marrow of experimental mice including stem/progenitor compartment. Acta Histochem. 2019, 121, 330–343. [Google Scholar] [CrossRef]
- Ziemba, B.P.; Burke, J.E.; Masson, G.; Williams, R.L.; Falke, J.J. Regulation of PI3K by PKC and MARCKS: Single-Molecule Analysis of a Reconstituted Signaling Pathway. Biophys. J. 2016, 110, 1811–1825. [Google Scholar] [CrossRef]
- Sandoval, S.; Pigazzi, M.; Sakamoto, K.M. CREB: A Key Regulator of Normal and Neoplastic Hematopoiesis. Adv. Hematol. 2009, 2009, 634292. [Google Scholar] [CrossRef]
- Cheng, J.C.; Kinjo, K.; Judelson, D.R.; Chang, J.; Wu, W.S.; Schmid, I.; Shankar, D.B.; Kasahara, N.; Stripecke, R.; Bhatia, R.; et al. CREB is a critical regulator of normal hematopoiesis and leukemogenesis. Blood 2008, 111, 1182–1192. [Google Scholar] [CrossRef]
- Kinjo, K.; Sandoval, S.; Sakamoto, K.M.; Shankar, D.B. The Role of CREB as a Proto-oncogene in Hematopoiesis. Cell Cycle 2005, 4, 1134–1135. [Google Scholar] [CrossRef]
- LeBlanc, F.; Bennett, J.; Choi, K.; Starczynowski, D.T. Targeting CREB-Binding Protein (CREBBP) Overcomes Resistance to Azacitidine and Venetoclax Therapy in Acute Myeloid Leukemia (AML). Blood 2023, 142 (Suppl. 1), 5765. [Google Scholar] [CrossRef]
- Navas, T.A.; Mohindru, M.; Estes, M.; Ma, J.Y.; Sokol, L.; Pahanish, P.; Parmar, S.; Haghnazari, E.; Zhou, L.; Collins, R.; et al. Inhibition of overactivated p38 MAPK can restore hematopoiesis in myelodysplastic syndrome progenitors. Blood 2006, 108, 4170–4177. [Google Scholar] [CrossRef]
- Zhang, T.; Ma, C.; Zhang, Z.; Zhang, H.; Hu, H. NF-κB signaling in inflammation and cancer. MedComm 2021, 2, 618–653. [Google Scholar] [CrossRef]
- Park, M.; Hong, J. Roles of NF-κB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef]
- Saluja, S.; Bansal, I.; Bhardwaj, R.; Beg, M.S.; Palanichamy, J.K. Inflammation as a driver of hematological malignancies. Front. Oncol. 2024, 14, 1347402. [Google Scholar] [CrossRef]
- Shastri, A.; Choudhary, G.; Teixeira, M.; Gordon-Mitchell, S.; Ramachandra, N.; Bernard, L.; Bhattacharyya, S.; Lopez, R.; Pradhan, K.; Giricz, O.; et al. Antisense STAT3 inhibitor decreases viability of myelodysplastic and leukemic stem cells. J. Clin. Investig. 2018, 128, 5479–5488. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All MDS | p-Value | LR-MDS | p-Value | HR-MDS | p-Value | |
|---|---|---|---|---|---|---|
| AMBRA1 | −1.190165 | −1.042547 | −1.361363 | |||
| ATG5 | −3.274832 | <0.001 | −2.911969 | <0.001 | −3.066072 | <0.001 |
| ATG12 | −1.774992 | <0.001 | −1.126027 | −2.659288 | <0.001 | |
| ATG16 | −1.483484 | −1.470517 | −1.720712 | |||
| CTSB | −2.335516 | <0.02 | −2.318447 | <0.05 | −2.393512 | <0.02 |
| DRAM1 | −1.916758 | <0.02 | −1.692008 | −1.984616 | <0.05 | |
| LC3I | 1.612848 | 1.188060 | 1.664416 | |||
| LC3II | −3.231055 | <0.001 | −2.523108 | <0.001 | −4.274320 | <0.001 |
| PI3KC3 | −1.509030 | −1.478196 | −1.566015 | |||
| TGM2 | 2.229709 | <0.001 | 2.162048 | <0.001 | 2.249299 | <0.001 |
| UVRAG | −1.307206 | −1.042216 | −1.797932 | |||
| CASP3 | −2.650488 | <0.001 | −2.542133 | <0.001 | −2.799367 | <0.001 |
| CASP7 | −2.410310 | <0.001 | −2.095823 | <0.05 | −3.116006 | <0.001 |
| CASP8 | −2.013422 | <0.05 | −2.109531 | <0.05 | −1.961725 | |
| BCL2 | 1.647007 | <0.02 | 1.257670 | <0.02 | 2.051875 | <0.001 |
| Gene/Protein | Gene Expression | Protein Expression |
|---|---|---|
| AMPKα | - | 1.32 * |
| ATG5 | 5.58 * | 1.72 * |
| BECN1 | 4.03 * | 4.44 * |
| LC3II | 4.63 * | 1.72 * |
| TGM2 | 1 | −1.19 * |
| BCL2 | −18.54 * | −2.28 * |
| Phosphoprotein | Fold Change |
|---|---|
| AKT | −2 * |
| AKT1S1 | −1.25 |
| CHK2 | 1.17 * |
| c-JUN | 1.13 * |
| CREB1 | −2.2 * |
| EGFR | −1.62 * |
| ERK1 | 1.76 * |
| FAK1 | 1.1 |
| GSK3a/β | −1.15 |
| HSPB1 | −1.21 |
| IKBA | 1.18 |
| MARCKS | −1.65 * |
| MEK1 | 1.2 |
| MTOR | −1.2 * |
| NFKB | −1.97 * |
| P38 MAPK | −1.12 |
| P53 | 2.3 * |
| PTN11 | −1.21 |
| RSK1 | −1.16 * |
| SMAD3 | −1.12 |
| STAT3 | −1.23 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsekoura, G.; Agathangelidis, A.; Kontandreopoulou, C.-N.; Fasouli, E.S.; Katsantoni, E.; Pliaka, V.; Alexopoulos, L.; Katana, E.; Papaioannou, M.; Taktikou, G.; et al. Restoration of Autophagy and Apoptosis in Myelodysplastic Syndromes: The Effect of Azacitidine in Disease Pathogenesis. Curr. Issues Mol. Biol. 2025, 47, 520. https://doi.org/10.3390/cimb47070520
Tsekoura G, Agathangelidis A, Kontandreopoulou C-N, Fasouli ES, Katsantoni E, Pliaka V, Alexopoulos L, Katana E, Papaioannou M, Taktikou G, et al. Restoration of Autophagy and Apoptosis in Myelodysplastic Syndromes: The Effect of Azacitidine in Disease Pathogenesis. Current Issues in Molecular Biology. 2025; 47(7):520. https://doi.org/10.3390/cimb47070520
Chicago/Turabian StyleTsekoura, Georgia, Andreas Agathangelidis, Christina-Nefeli Kontandreopoulou, Eirini Sofia Fasouli, Eleni Katsantoni, Vaia Pliaka, Leonidas Alexopoulos, Eleni Katana, Myrto Papaioannou, Georgia Taktikou, and et al. 2025. "Restoration of Autophagy and Apoptosis in Myelodysplastic Syndromes: The Effect of Azacitidine in Disease Pathogenesis" Current Issues in Molecular Biology 47, no. 7: 520. https://doi.org/10.3390/cimb47070520
APA StyleTsekoura, G., Agathangelidis, A., Kontandreopoulou, C.-N., Fasouli, E. S., Katsantoni, E., Pliaka, V., Alexopoulos, L., Katana, E., Papaioannou, M., Taktikou, G., Strataki, M. E., Taliouraki, A., Mantzourani, M., Viniou, N.-A., Diamantopoulos, P. T., & Kollia, P. (2025). Restoration of Autophagy and Apoptosis in Myelodysplastic Syndromes: The Effect of Azacitidine in Disease Pathogenesis. Current Issues in Molecular Biology, 47(7), 520. https://doi.org/10.3390/cimb47070520