
Inhibiting the Interaction Between Phospholipase A2 and Phospholipid Serine as a Potential Therapeutic Method for Pneumonia

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Animals
2.1.2. Cell Lines
2.1.3. Reagents
2.2. Methods
2.2.1. Construction of LPS Pulmonary Infection Model
2.2.2. Administration and Organ Collection in Mice
2.2.3. Western Blot
2.2.4. Reverse Transcription Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR)
2.2.5. Hematoxylin and Eosin (H&E) Staining
2.2.6. Immunofluorescence Staining
2.2.7. CCK-8
2.2.8. Flow Cytometry and Confocal Microscopy
2.3. Statistical Analysis
3. Results
3.1. The Impact of LPS and PLA2 on Cells
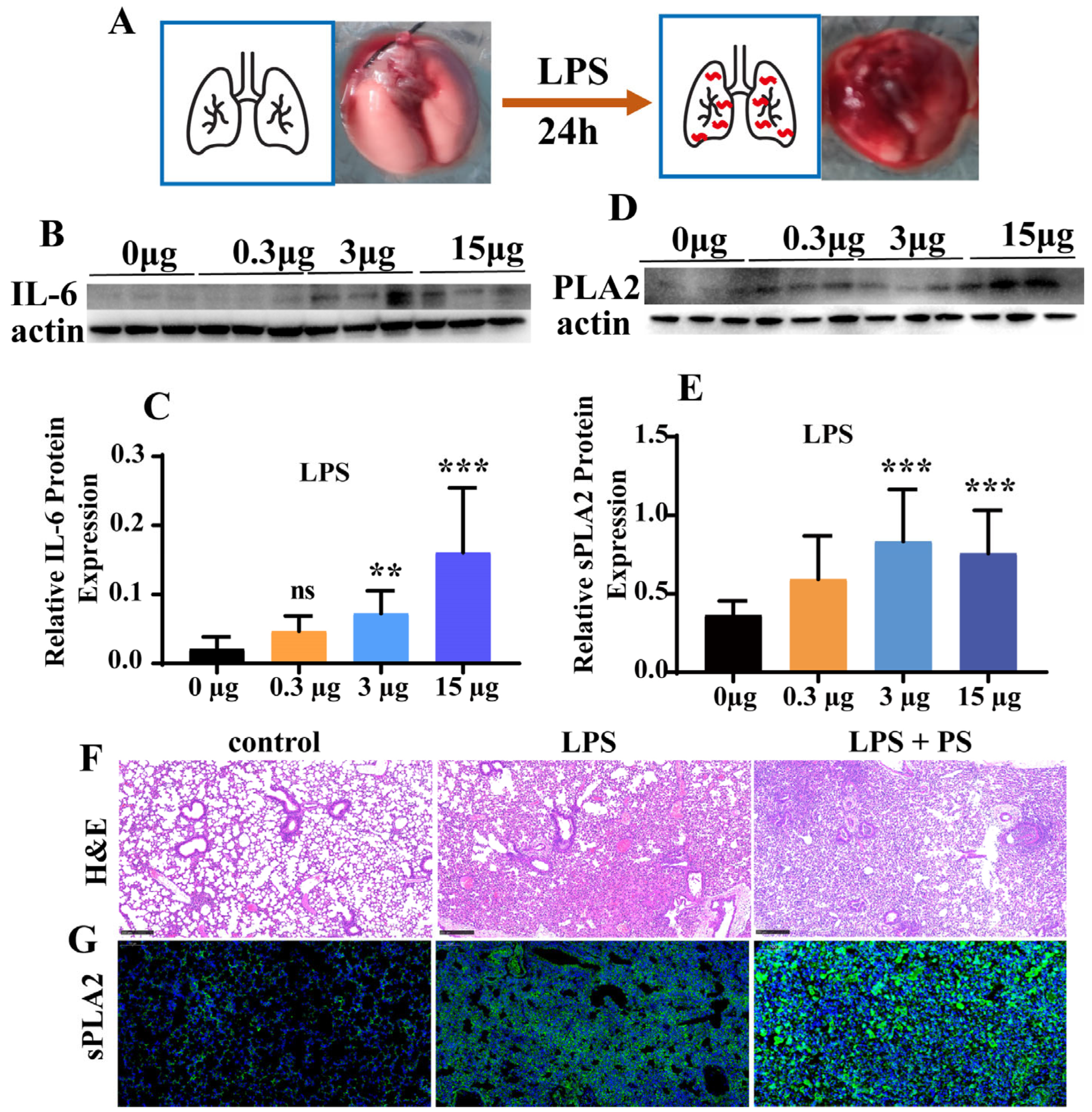
3.2. LPS and PLA2 Induce Inflammation In Vivo
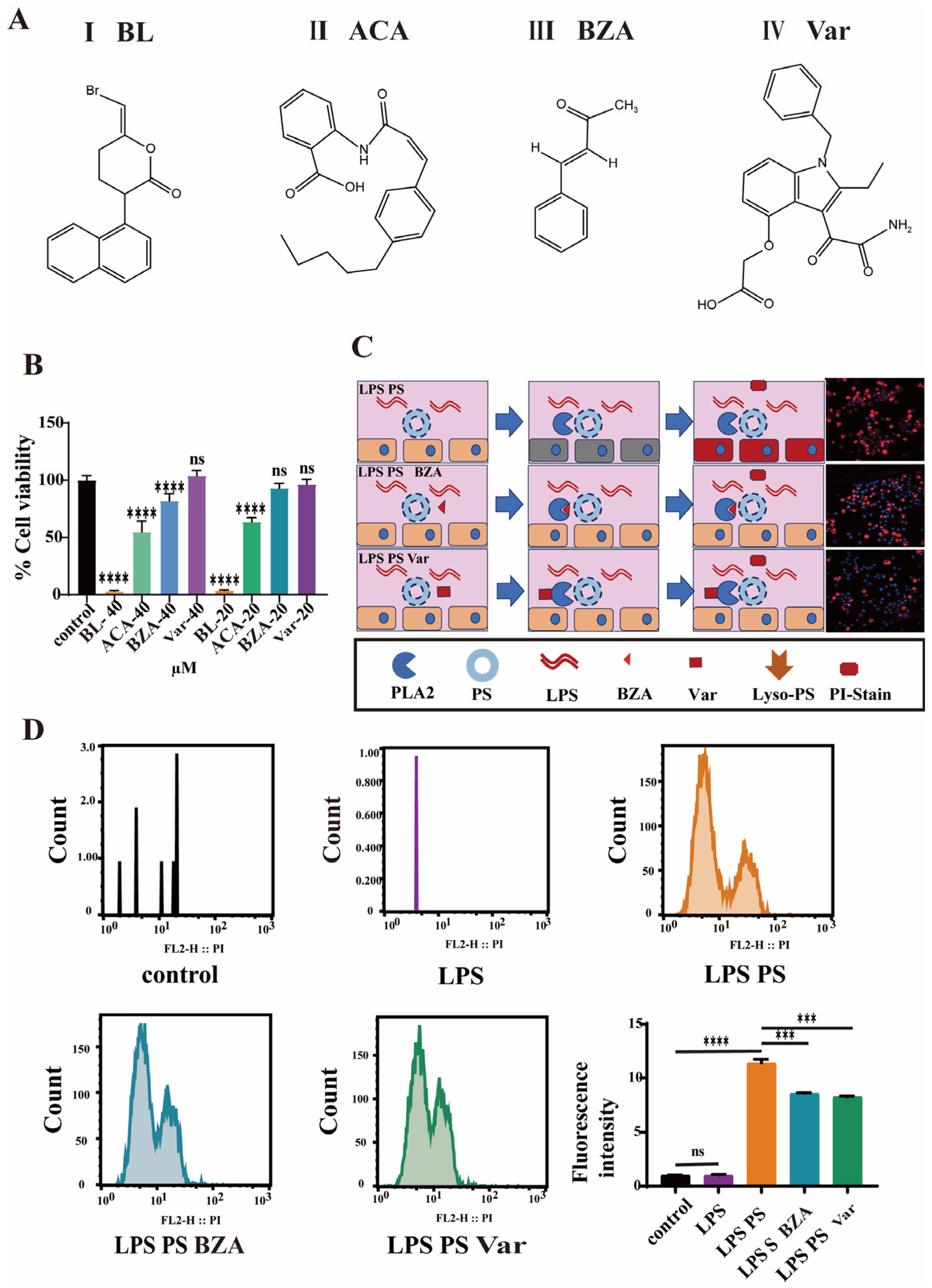
3.3. PLA2 Inhibitors Attenuate Cellular Damage In Vitro
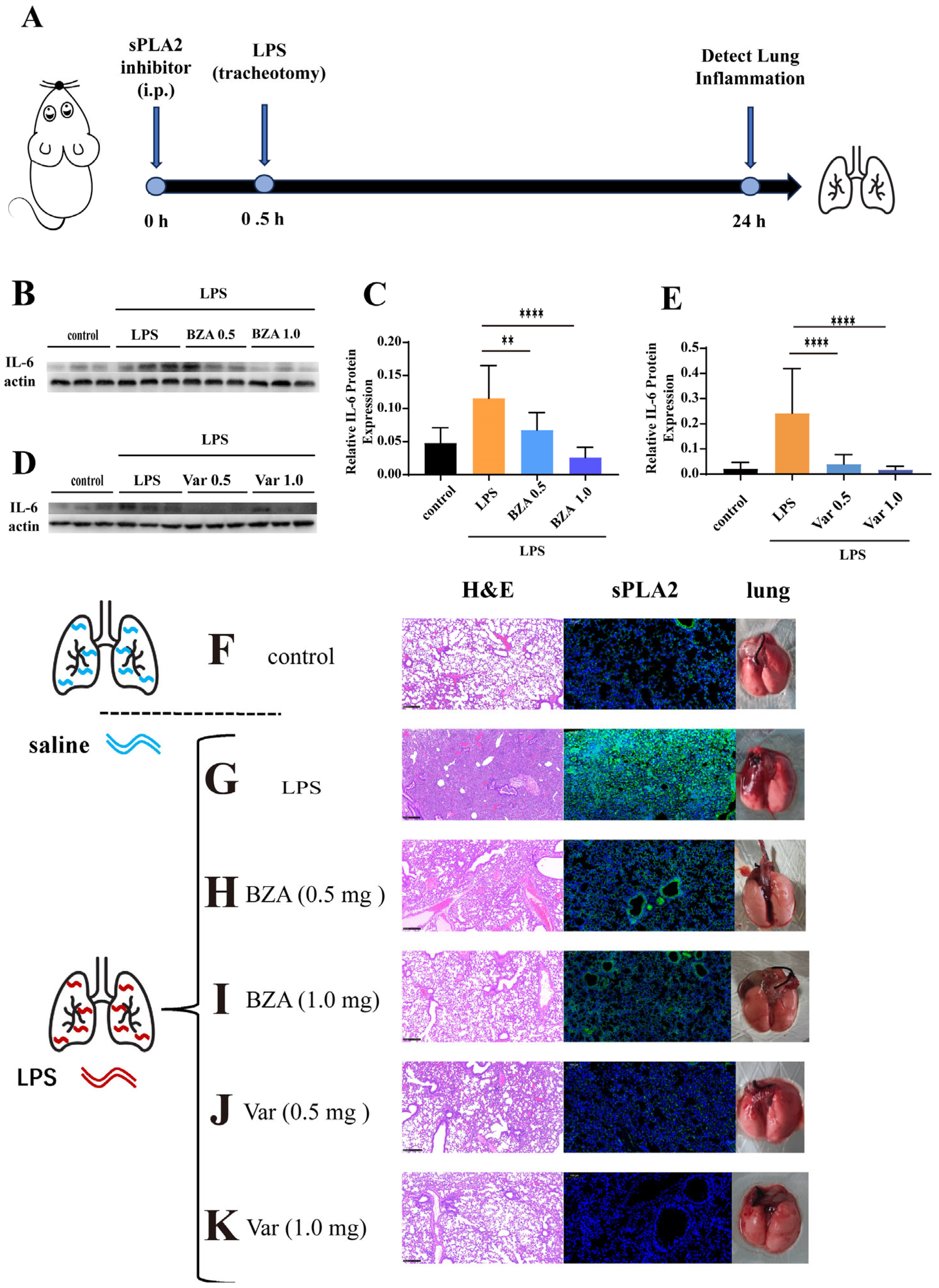
3.4. PLA2 Inhibitors Attenuate LPS Lung Injury in Mice
4. Discussion
4.1. LPS-Induced Lung Pneumonia Models
4.2. Non-Irritating Effects of LPS In Vitro
4.3. LPS Induces Injury In Vivo
4.4. PLA2 Inhibitors Attenuate Cellular Damage
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- English, M. Impact of bacterial pneumonias on world child health. Paediatr. Respir. Rev. 2000, 1, 21–25. [Google Scholar] [CrossRef]
- Kutter, J.S.; de Meulder, D.; Bestebroer, T.M.; Mulders, A.; Fouchier, R.A.M.; Herfst, S. Comparison of three air samplers for the collection of four nebulized respiratory viruses-Collection of respiratory viruses from air. Indoor Air 2021, 31, 1874–1885. [Google Scholar] [CrossRef]
- Liu, Q.; Su, Q.; Zhang, F.; Tun, H.M.; Mak, J.W.Y.; Lui, G.C.-Y.; Ng, S.S.S.; Ching, J.Y.L.; Li, A.; Lu, W.; et al. Multi-kingdom gut microbiota analyses define COVID-19 severity and post-acute COVID-19 syndrome. Nat. Commun. 2022, 13, 6806. [Google Scholar] [CrossRef]
- Cheng, L.; Ren, Y.; Lin, D.; Peng, S.a.; Zhong, B.; Ma, Z. The Anti-Inflammatory Properties of Citrus wilsonii Tanaka Extract in LPS-Induced RAW 264.7 and Primary Mouse Bone Marrow-Derived Dendritic Cells. Molecules 2017, 22, 1213. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, X.; Ling, P.; Liao, J.; Zhao, M.; Mei, L.; Shao, H.; Jiang, P.; Song, Z.; Chen, Q.; et al. Immunomodulatory effects of xanthan gum in LPS-stimulated RAW 264.7 macrophages. Carbohydr. Polym. 2017, 169, 65–74. [Google Scholar] [CrossRef]
- Hite, R.D.; Grier, B.L.; Waite, B.M.; Veldhuizen, R.A.; Possmayer, F.; Yao, L.-J.; Seeds, M.C. Surfactant protein B inhibits secretory phospholipase A2 hydrolysis of surfactant phospholipids. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 302, L257–L265. [Google Scholar] [CrossRef]
- Kim, W.; Son, B.; Lee, S.; Do, H.; Youn, B. Targeting the enzymes involved in arachidonic acid metabolism to improve radiotherapy. Cancer Metastasis Rev. 2018, 37, 213–225. [Google Scholar] [CrossRef]
- Whitsett, J.A.; Wert, S.E.; Weaver, T.E. Alveolar Surfactant Homeostasis and the Pathogenesis of Pulmonary Disease. Annu. Rev. Med. 2010, 61, 105–119. [Google Scholar] [CrossRef]
- Hurley Bryan, P.; McCormick Beth, A. Multiple Roles of Phospholipase A2 during Lung Infection and Inflammation. Infect. Immun. 2008, 76, 2259–2272. [Google Scholar] [CrossRef]
- Neidlinger, N.A.; Larkin, S.K.; Bhagat, A.; Victorino, G.P.; Kuypers, F.A. Hydrolysis of Phosphatidylserine-exposing Red Blood Cells by Secretory Phospholipase A2 Generates Lysophosphatidic Acid and Results in Vascular Dysfunction *. J. Biol. Chem. 2006, 281, 775–781. [Google Scholar] [CrossRef]
- Iaria, A.; Schwarz, C.; Waldinger, F. Frontier Knowledge and Scientific Production: Evidence from the Collapse of International Science*. Q. J. Econ. 2018, 133, 927–991. [Google Scholar] [CrossRef]
- Cochet, F.; Peri, F. The Role of Carbohydrates in the Lipopolysaccharide (LPS)/Toll-Like Receptor 4 (TLR4) Signalling. Int. J. Mol. Sci. 2017, 18, 2318. [Google Scholar] [CrossRef]
- Du, Z.-A.; Sun, M.-N.; Hu, Z.-S. Saikosaponin a Ameliorates LPS-Induced Acute Lung Injury in Mice. Inflammation 2018, 41, 193–198. [Google Scholar] [CrossRef]
- Shan, M.R.; Zhou, S.N.; Fu, C.N.; Song, J.W.; Wang, X.Q.; Bai, W.W.; Li, P.; Song, P.; Zhu, M.L.; Ma, Z.M.; et al. Vitamin B6 inhibits macrophage activation to prevent lipopolysaccharide-induced acute pneumonia in mice. J. Cell. Mol. Med. 2020, 24, 3139–3148. [Google Scholar] [CrossRef]
- Xi, X.; Yao, Y.; Liu, N.; Li, P. MiR-297 alleviates LPS-induced A549 cell and mice lung injury via targeting cyclin dependent kinase 8. Int. Immunopharmacol. 2020, 80, 106197. [Google Scholar] [CrossRef]
- Wang, J.; Cao, Y.; Liu, Y.; Zhang, X.; Ji, F.; Li, J.; Zou, Y. PIM1 inhibitor SMI-4a attenuated lipopolysaccharide-induced acute lung injury through suppressing macrophage inflammatory responses via modulating p65 phosphorylation. Int. Immunopharmacol. 2019, 73, 568–574. [Google Scholar] [CrossRef]
- Kay, J.G.; Grinstein, S. Sensing Phosphatidylserine in Cellular Membranes. Sensors 2011, 11, 1744–1755. [Google Scholar] [CrossRef]
- Gao, J.; Teng, L.; Yang, S.; Huang, S.; Li, L.; Zhou, L.; Liu, G.; Tang, H. MNK as a potential pharmacological target for suppressing LPS-induced acute lung injury in mice. Biochem. Pharmacol. 2021, 186, 114499. [Google Scholar] [CrossRef]
- Tang, X.; Weng, R.; Guo, G.; Wei, J.; Wu, X.; Chen, B.; Liu, S.; Zhong, Z.; Chen, X. USP10 regulates macrophage inflammation responses via stabilizing NEMO in LPS-induced sepsis. Inflamm. Res. 2023, 72, 1621–1632. [Google Scholar] [CrossRef]
- Lu, J.; Zhu, B.; Zhou, F.; Ding, X.; Qian, C.; Ding, Z.; Ye, X. Polysaccharides From the Aerial Parts of Tetrastigma Hemsleyanum Diels et Gilg Induce Bidirectional Immunity and Ameliorate LPS-Induced Acute Respiratory Distress Syndrome in Mice. Front. Pharmacol. 2022, 13, 838873. [Google Scholar] [CrossRef]
- Abdulkhaleq, L.A.; Assi, M.A.; Abdullah, R.; Zamri-Saad, M.; Taufiq-Yap, Y.H.; Hezmee, M.N.M. The crucial roles of inflammatory mediators in inflammation: A review. Vet. World 2018, 11, 627–635. [Google Scholar] [CrossRef]
- Vazquez-Medina, J.P.; Tao, J.Q.; Patel, P.; Bannitz-Fernandes, R.; Dodia, C.; Sorokina, E.; Feinstein, S.I.; Chatterjee, S.; Fisher, A. 318-Genetic Inactivation of the Phospholipase A2 Activity of Prdx6 Ameliorates Sepsis-induced Acute Lung Injury. Free Radical. Biol. Med. 2017, 112, 208–209. [Google Scholar] [CrossRef]
- Fensome-Green, A.; Stannard, N.; Li, M.; Bolsover, S.; Cockcroft, S. Bromoenol lactone, an inhibitor of Group V1A calcium-independent phospholipase A2 inhibits antigen-stimulated mast cell exocytosis without blocking Ca2+ influx. Cell Calcium 2007, 41, 145–153. [Google Scholar] [CrossRef]
- Kraft, R.; Grimm, C.; Frenzel, H.; Harteneck, C. Inhibition of TRPM2 cation channels by N-(p-amylcinnamoyl)anthranilic acid. Br. J. Pharmacol. 2006, 148, 264–273. [Google Scholar] [CrossRef]
- Kwon, B.; Kim, Y. Benzylideneacetone, an Immunosuppressant, Enhances Virulence of Bacillus thuringiensis Against Beet Armyworm (Lepidoptera: Noctuidae). J. Econ. Entomol. 2008, 101, 36–41. [Google Scholar] [CrossRef]
- Hori, Y.; Spurr-Michaud, S.J.; Russo, C.L.; Argüeso, P.; Gipson, I.K. Effect of Retinoic Acid on Gene Expression in Human Conjunctival Epithelium: Secretory Phospholipase A2 Mediates Retinoic Acid Induction of MUC16. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4050–4061. [Google Scholar] [CrossRef]
- Lewin, M.R.; Carter, R.W.; Matteo, I.A.; Samuel, S.P.; Rao, S.; Fry, B.G.; Bickler, P.E. Varespladib in the Treatment of Snakebite Envenoming: Development History and Preclinical Evidence Supporting Advancement to Clinical Trials in Patients Bitten by Venomous Snakes. Toxins 2022, 14, 783. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Xing, H.; Wang, L.; Xu, Z.; Sui, X.; Luo, Y.; Yang, J.; Wang, Y. Inhibiting the Interaction Between Phospholipase A2 and Phospholipid Serine as a Potential Therapeutic Method for Pneumonia. Curr. Issues Mol. Biol. 2025, 47, 516. https://doi.org/10.3390/cimb47070516
Wang J, Xing H, Wang L, Xu Z, Sui X, Luo Y, Yang J, Wang Y. Inhibiting the Interaction Between Phospholipase A2 and Phospholipid Serine as a Potential Therapeutic Method for Pneumonia. Current Issues in Molecular Biology. 2025; 47(7):516. https://doi.org/10.3390/cimb47070516
Chicago/Turabian StyleWang, Jianyu, Huanchun Xing, Lin Wang, Zhongxing Xu, Xin Sui, Yuan Luo, Jun Yang, and Yongan Wang. 2025. "Inhibiting the Interaction Between Phospholipase A2 and Phospholipid Serine as a Potential Therapeutic Method for Pneumonia" Current Issues in Molecular Biology 47, no. 7: 516. https://doi.org/10.3390/cimb47070516
APA StyleWang, J., Xing, H., Wang, L., Xu, Z., Sui, X., Luo, Y., Yang, J., & Wang, Y. (2025). Inhibiting the Interaction Between Phospholipase A2 and Phospholipid Serine as a Potential Therapeutic Method for Pneumonia. Current Issues in Molecular Biology, 47(7), 516. https://doi.org/10.3390/cimb47070516