
TNF-α Promotes the Recovery of Dorsal Root Ganglion Neurons from Cisplatin-Induced Injury Through an NGF-Independent Mechanism
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Culture and Treatment of DRG Neurons in Adult Mice
2.3. Immunofluorescence Staining and Image Collection
2.4. Neurite Length Measurement
2.5. Intensity Analysis of Immunofluorescence Staining
2.6. RNA Interference
2.7. Real-Time Quantitative Polymerase Chain Reaction
2.8. Statistical Analysis
3. Results
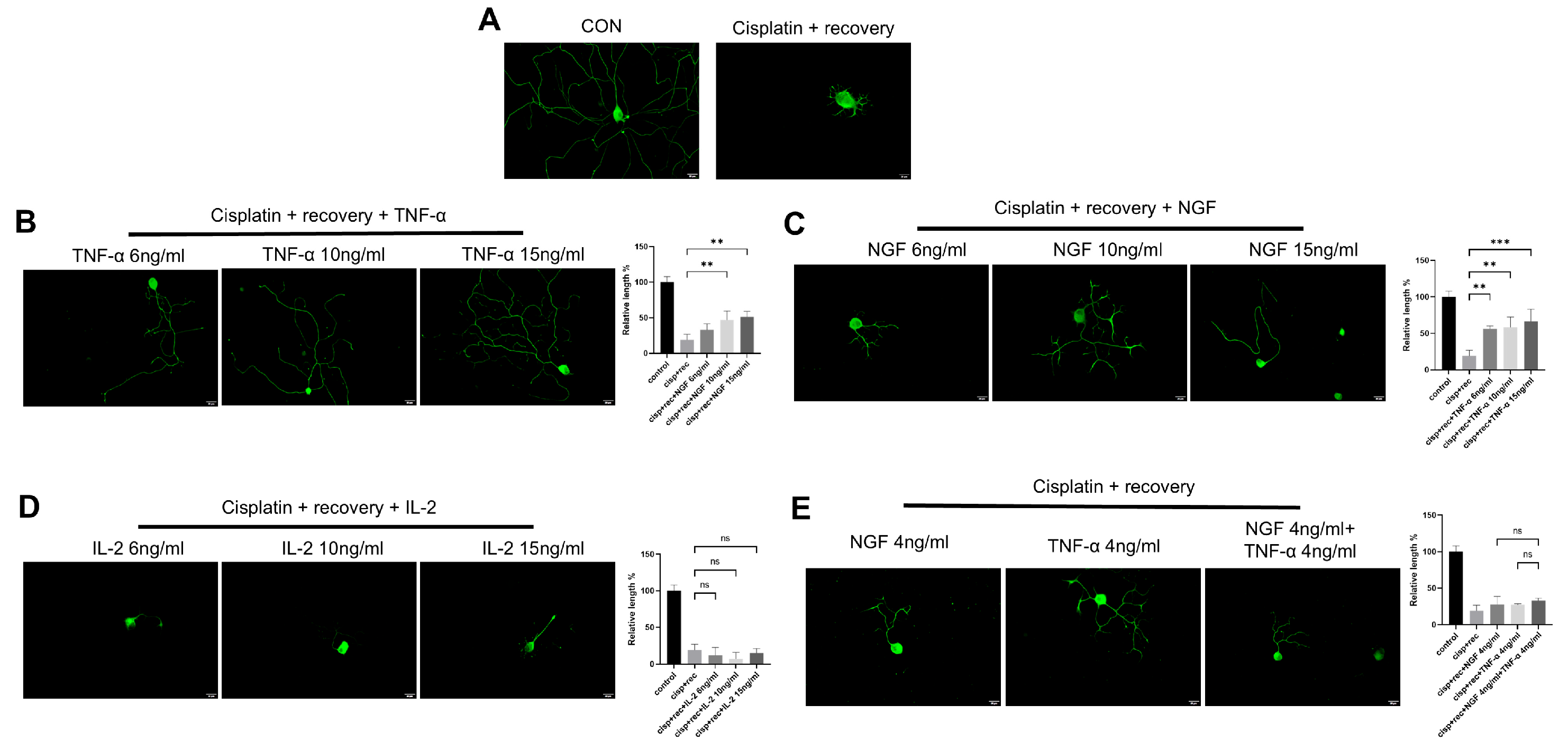
3.1. TNF-α Promotes Neurite Outgrowth in Cultured DRG Neurons In Vitro
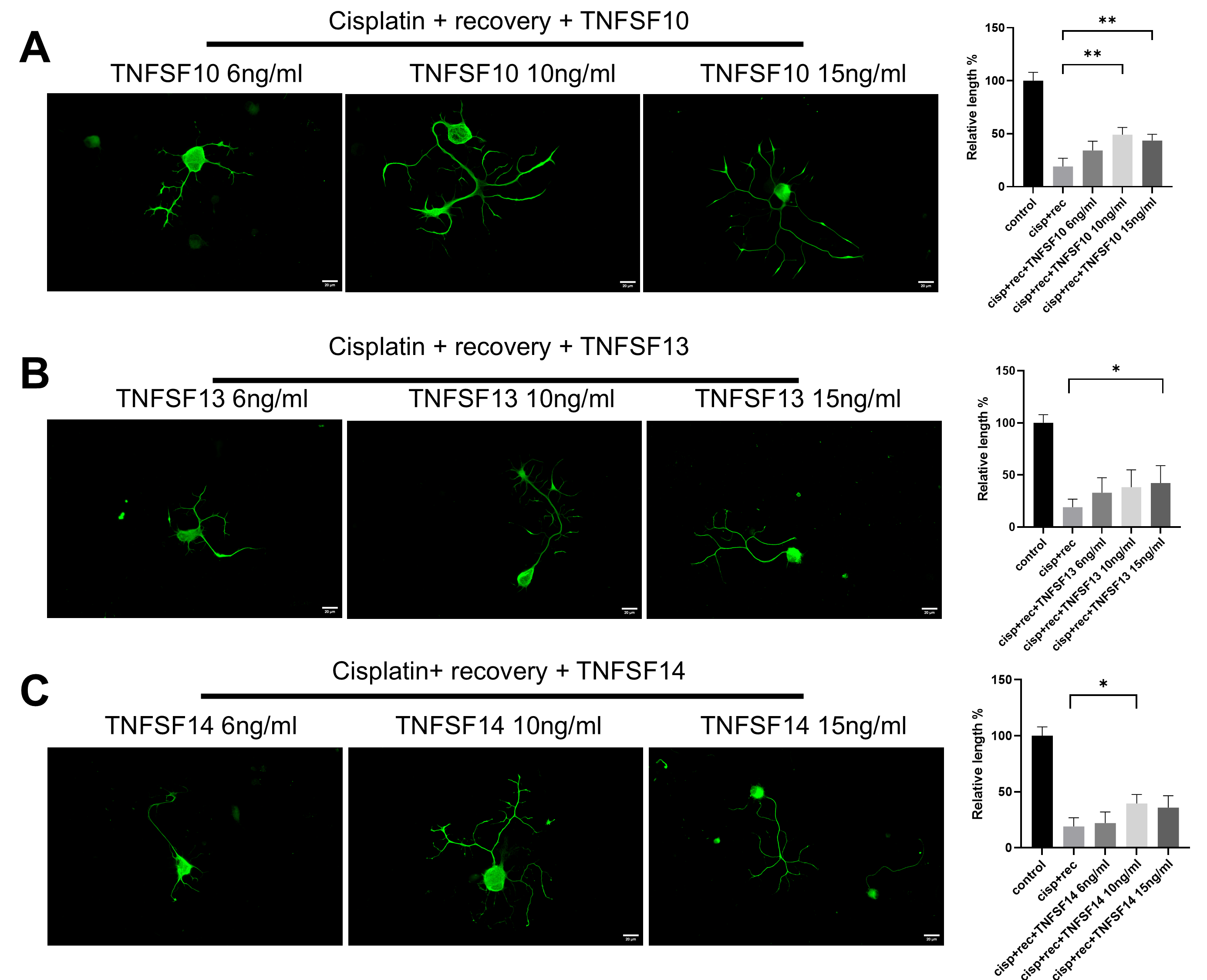
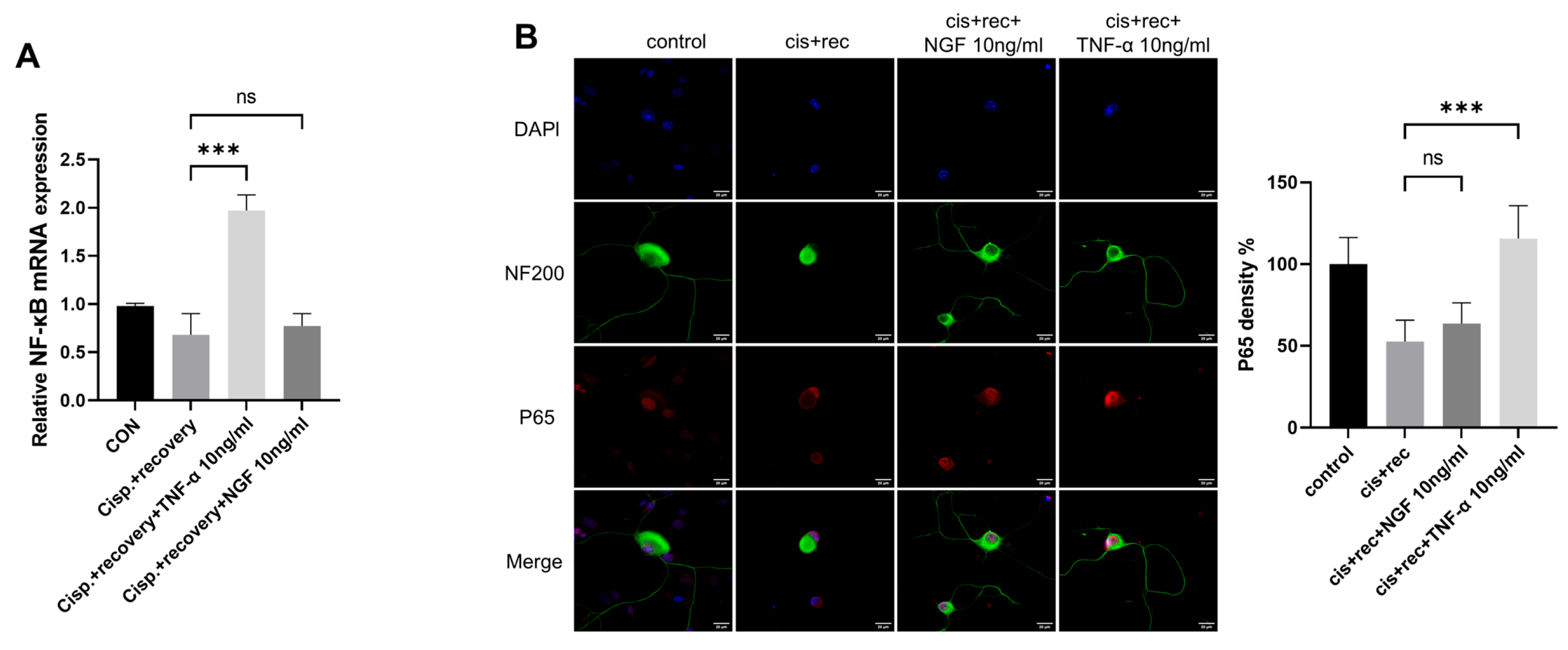
3.2. TNF-α Promotes Neurite Regeneration in DRG Neurons Damaged by Cisplatin
3.3. TNF-α Accelerates the Repairing Process of DNA Damage of DRG Neurons Induced by Cisplatin
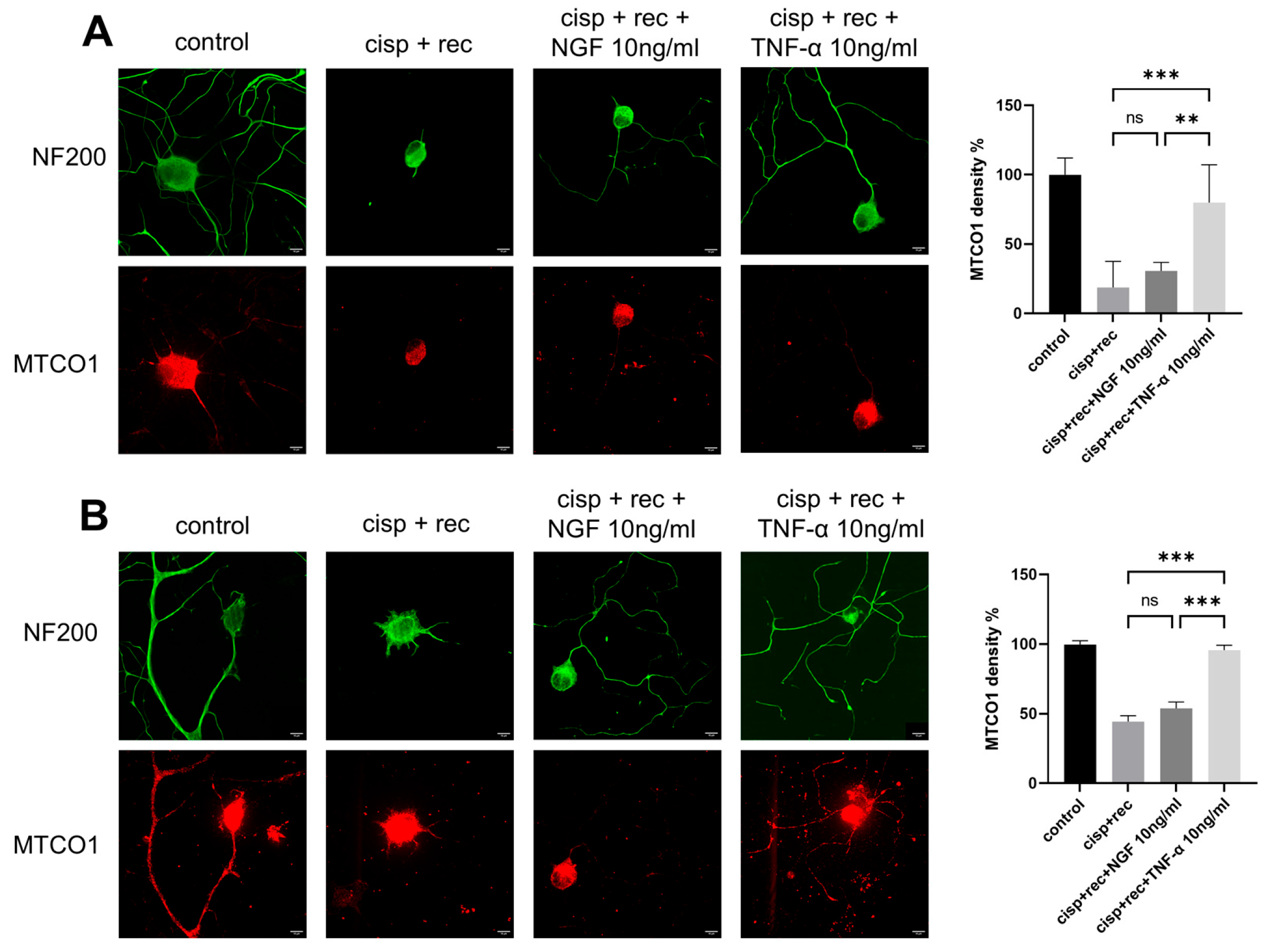
3.4. TNF-α Promotes Regeneration of Mitochondrial in DRG Neurons
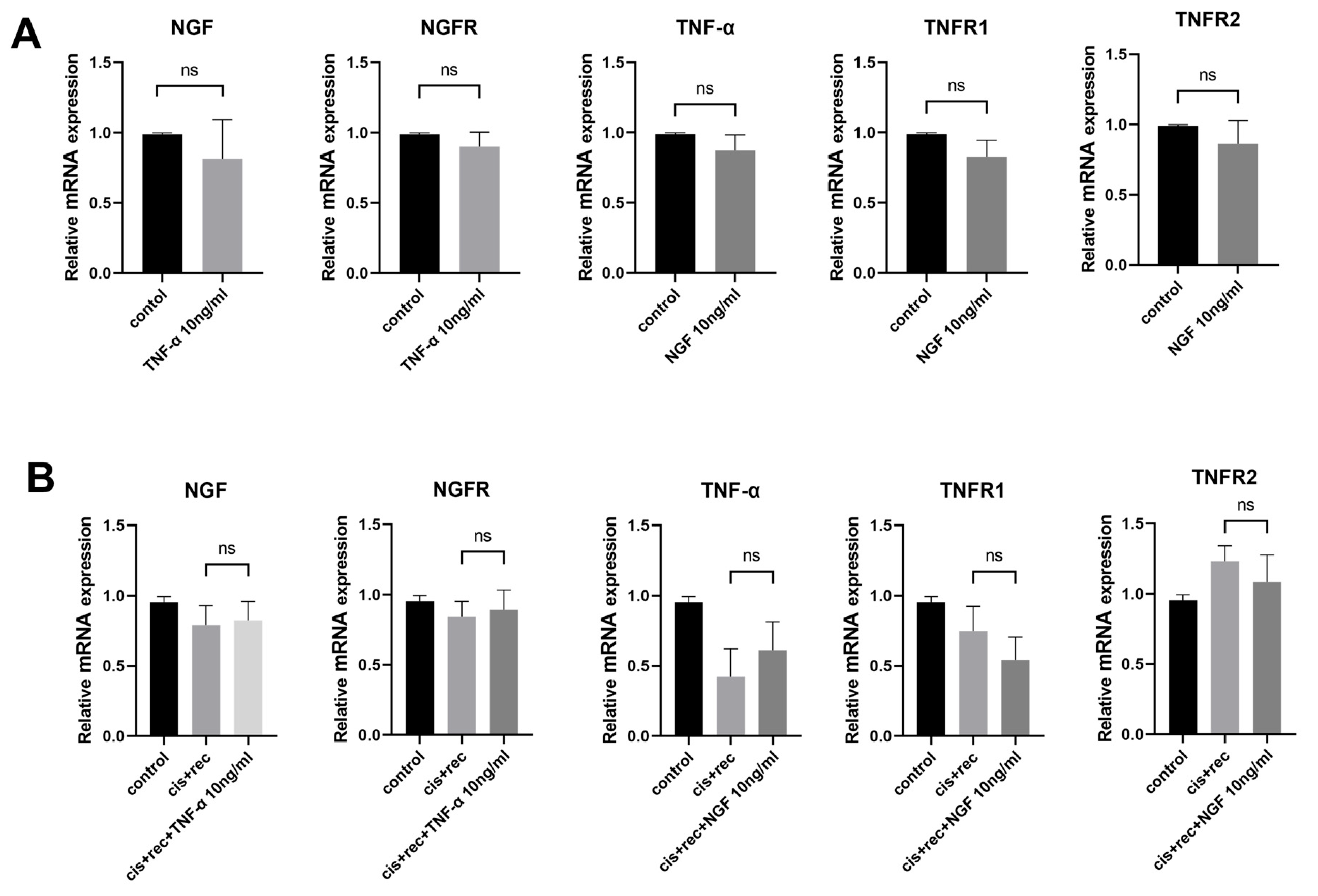
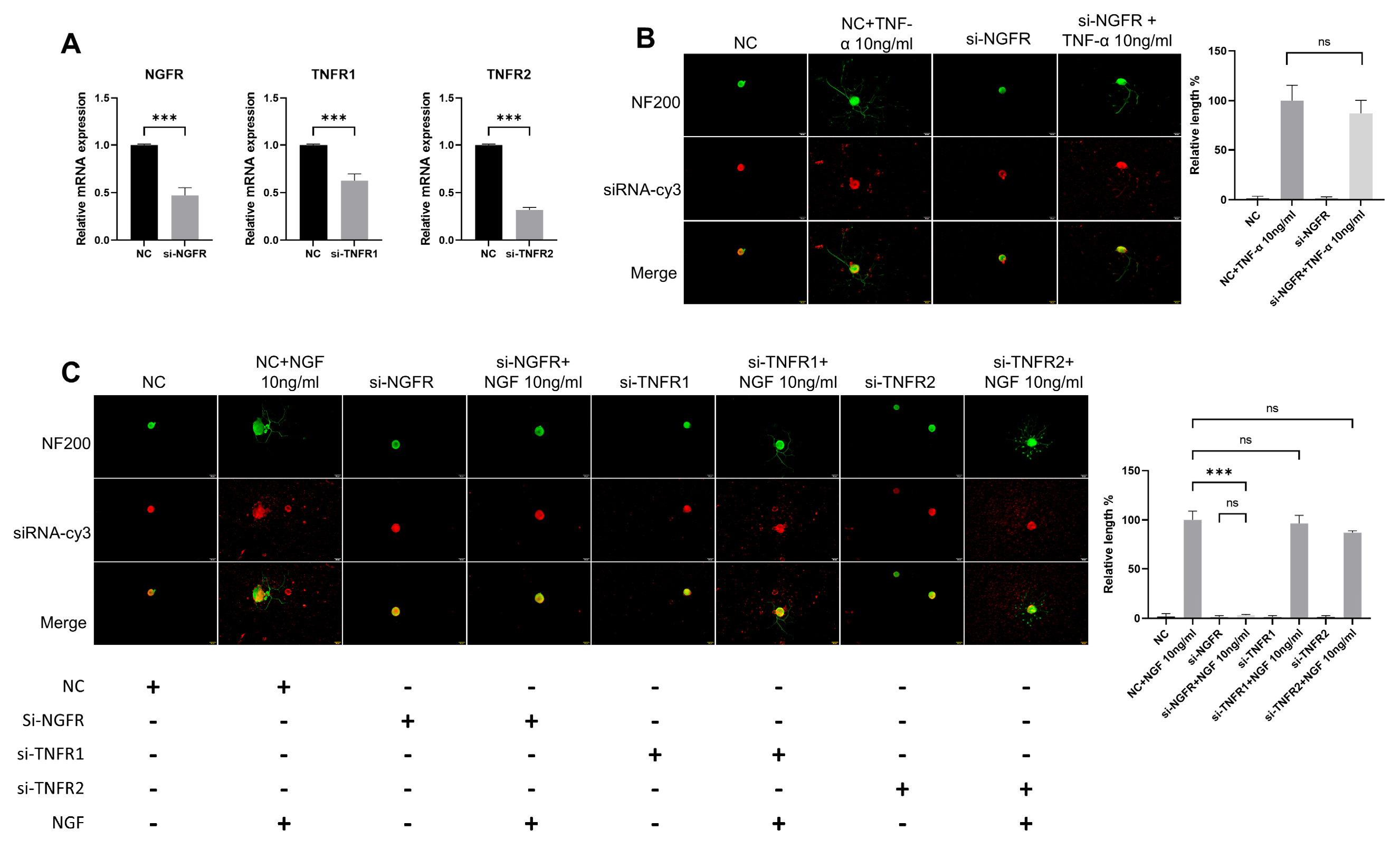
3.5. The Effect of TNF-α on DRG Neurons Is Independent of the NGF Signaling Pathway
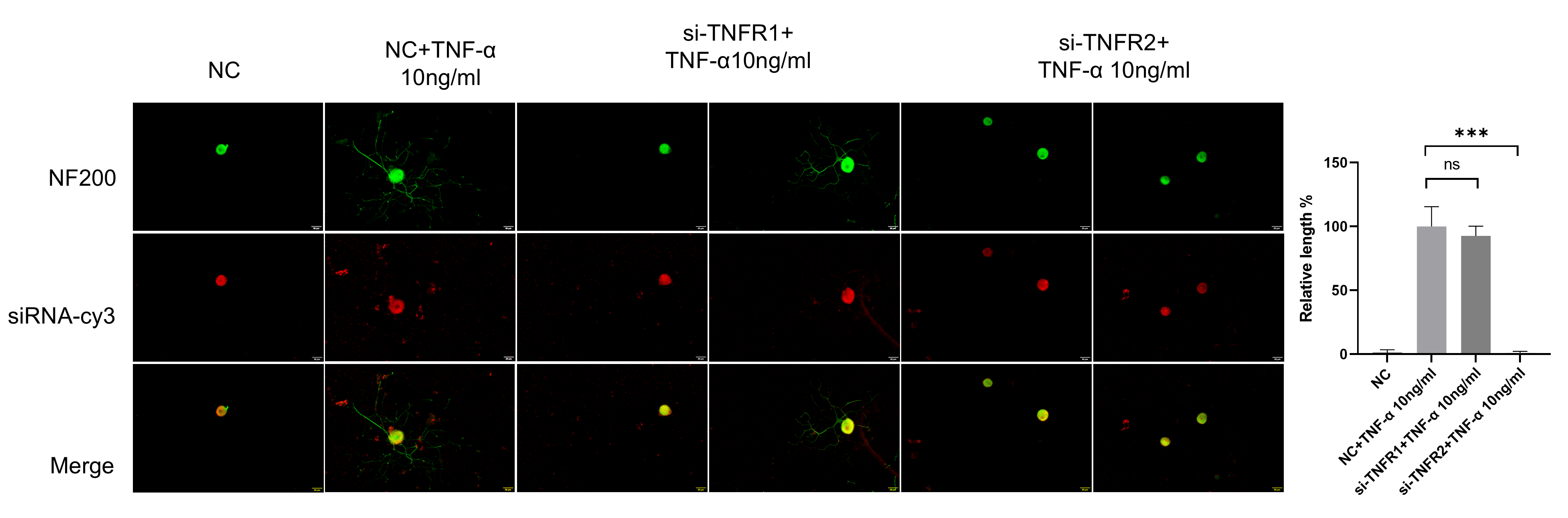
3.6. TNF-α Acts on DRG Neurons Depend on TNFR2-Mediated Signals
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Seretny, M.; Currie, G.L.; Sena, E.S.; Ramnarine, S.; Grant, R.; MacLeod, M.R.; Colvin, L.A.; Fallon, M. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: A systematic review and meta-analysis. Pain 2014, 155, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Staff, N.P.; Grisold, A.; Grisold, W.; Windebank, A.J. Chemotherapy-induced peripheral neuropathy: A current review. Ann. Neurol. 2017, 81, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Boyette-Davis, J.A.; Hou, S.; Abdi, S.; Dougherty, P.M. An Updated Understanding of the Mechanisms Involved in cHemotherapy-Induced Neuropathy. Pain Manag. 2018, 8, 363–375. [Google Scholar] [CrossRef]
- Ma, J.; Kavelaars, A.; Dougherty, P.M.; Heijnen, C.J. Beyond symptomatic relief for chemotherapy-induced peripheral neuropathy: Targeting the source. Cancer 2018, 124, 2289–2298. [Google Scholar] [CrossRef]
- Burakgazi, A.Z.; Messersmith, W.; Vaidya, D.; Hauer, P.; Hoke, A.; Polydefkis, M. Longitudinal Assessment of Oxaliplatin-Induced Neuropathy. Neurology 2011, 77, 980–986. [Google Scholar]
- Pachman, D.R.; Barton, D.L.; Watson, J.C.; Loprinzi, C.L. Chemotherapy-Induced Peripheral Neuropathy: Prevention and Treatment. Clin. Pharmacol. Ther. 2011, 90, 377–387. [Google Scholar] [CrossRef]
- Windebank, A.J.; Grisold, W. Chemotherapy-induced neuropathy. J. Peripher. Nerv. Syst. 2008, 13, 27–46. [Google Scholar] [CrossRef] [PubMed]
- Podratz, J.L.; Knight, A.M.; Ta, L.E.; Staff, N.P.; Gass, J.M.; Genelin, K.; Schlattau, A.; Lathroum, L.; Windebank, A.J. Cisplatin induced Mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol. Dis. 2011, 41, 661–668. [Google Scholar] [CrossRef]
- Lykissas, M.; Batistatou, A.; Charalabopoulos, K.; Beris, A. The Role of Neurotrophins in Axonal Growth, Guidance, and Regeneration. Curr. Neurovascular Res. 2007, 4, 143–151. [Google Scholar] [CrossRef]
- Khan, N.; Smith, M. Neurotrophins and Neuropathic Pain: Role in Pathobiology. Molecules 2015, 20, 10657–10688. [Google Scholar] [CrossRef]
- Donnerer, J. Regeneration of Primary Sensory Neurons. Pharmacology 2003, 67, 169–181. [Google Scholar] [CrossRef]
- Webber, C.A.; Xu, Y.; Vanneste, K.J.; Martinez, J.A.; Verge, V.M.K.; Zochodne, D.W. Guiding Adult Mammalian Sensory Axons During Regeneration. J. Neuropathol. Exp. Neurol. 2008, 67, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Piao, X.; Bonaldo, P. Role of macrophages in Wallerian degeneration and axonal regeneration after peripheral nerve injury. Acta Neuropathol. 2015, 130, 605–618. [Google Scholar] [CrossRef]
- Zhao, Q.; Jiang, C.; Zhao, L.; Dai, X.; Yi, S. Unleashing Axonal Regeneration Capacities: Neuronal and Non-neuronal Changes After Injuries to Dorsal Root Ganglion Neuron Central and Peripheral Axonal Branches. Mol. Neurobiol. 2024, 61, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Cuellar, M.J.; Montesano, X.P.; Carstens, E. Role of TNF-alpha in sensitization of nociceptive dorsal horn neurons induced by application of nucleus pulposus to L5 dorsal root ganglion in rats. Pain 2004, 110, 578–587. [Google Scholar] [CrossRef]
- Zelenka, M.; Schäfers, M.; Sommer, C. Intraneural injection of interleukin-1β and tumor necrosis factor-alpha into rat sciatic nerve at physiological doses induces signs of neuropathic pain. Pain 2005, 116, 257–263. [Google Scholar] [CrossRef]
- Nadeau, S.; Filali, M.; Zhang, J.; Kerr, B.J.; Rivest, S.; Soulet, D.; Iwakura, Y.; De Rivero Vaccari, J.P.; Keane, R.W.; Lacroix, S. Functional Recovery after Peripheral Nerve Injury is Dependent on the Pro-Inflammatory Cytokines IL-1β and TNF: Implications for Neuropathic Pain. J. Neurosci. 2011, 31, 12533–12542. [Google Scholar] [CrossRef] [PubMed]
- Takano, S.; Uchida, K.; Miyagi, M.; Inoue, G.; Fujimaki, H.; Aikawa, J.; Iwase, D.; Minatani, A.; Iwabuchi, K.; Takaso, M. Nerve Growth Factor Regulation by TNF- α and IL-1 β in Synovial Macrophages and Fibroblasts in Osteoarthritic Mice. J. Immunol. Res. 2016, 2016, 5706359. [Google Scholar] [CrossRef]
- Ohta, M.; Chosa, N.; Kyakumoto, S.; Yokota, S.; Okubo, N.; Nemoto, A.; Kamo, M.; Joh, S.; Satoh, K.; Ishisaki, A. IL-1β and TNF-α suppress TGF-β-promoted NGF expression in periodontal ligament-derived fibroblasts through inactivation of TGF-β-induced Smad2/3- and p38 MAPK-mediated signals. Int. J. Mol. Med. 2018, 42, 1484–1494. [Google Scholar] [CrossRef]
- Nagura, N.; Kenmoku, T.; Uchida, K.; Nakawaki, M.; Inoue, G.; Takaso, M. Nerve growth factor continuously elevates in a rat rotator cuff tear model. J. Shoulder Elb. Surg. 2019, 28, 143–148. [Google Scholar] [CrossRef]
- Meyers, C.A.; Lee, S.; Sono, T.; Xu, J.; Negri, S.; Tian, Y.; Wang, Y.; Li, Z.; Miller, S.; Chang, L.; et al. A Neurotrophic Mechanism Directs Sensory Nerve Transit in Cranial Bone. Cell Rep. 2020, 31, 107696. [Google Scholar] [CrossRef]
- Yang, L.; Lindholm, K.; Konishi, Y.; Li, R.; Shen, Y. Target Depletion of Distinct Tumor Necrosis Factor Receptor Subtypes Reveals Hippocampal Neuron Death and Survival through Different Signal Transduction Pathways. J. Neurosci. 2002, 22, 3025–3032. [Google Scholar] [CrossRef] [PubMed]
- Sleigh, J.N.; Weir, G.A.; Schiavo, G. A simple, step-by-step dissection protocol for the rapid isolation of mouse dorsal root ganglia. BMC Res. Notes 2016, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Sleigh, J.N.; West, S.J.; Schiavo, G. A video protocol for rapid dissection of mouse dorsal root ganglia from defined spinal levels. BMC Res. Notes 2020, 13, 302. [Google Scholar] [CrossRef]
- Gorgun, M.F.; Zhuo, M.; Englander, E.W. Cisplatin Toxicity in Dorsal Root Ganglion Neurons Is Relieved by Meclizine via Diminution of Mitochondrial Compromise and Improved Clearance of DNA Damage. Mol. Neurobiol. 2017, 54, 7883–7895. [Google Scholar] [CrossRef] [PubMed]
- Venegas, V.; Wang, J.; Dimmock, D.; Wong, L.J. Real-Time Quantitative PCR Analysis of Mitochondrial DNA Content. Curr. Protoc. Hum. Genet. 2011, 68, 19.7.1–19.7.12. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Wang, J.; Yin, L.; Zhang, J.; Zhang, Y.; Zhang, X.; Ding, D.; Gao, Y.; Li, Q.; Chen, H. The profiles of gamma-H2AX along with ATM/DNA-PKcs activation in the lymphocytes and granulocytes of rat and human blood exposed to gamma rays. Radiat. Environ. Biophys. 2016, 55, 359–370. [Google Scholar] [CrossRef]
- Zhuo, M.; Gorgun, M.F.; Englander, E.W. Translesion Synthesis DNA Polymerase Kappa Is Indispensable for DNA Repair Synthesis in Cisplatin Exposed Dorsal Root Ganglion Neurons. Mol. Neurobiol. 2018, 55, 2506–2515. [Google Scholar] [CrossRef]
- Dolga, A.M.; Granic, I.; Blank, T.; Knaus, H.G.; Spiess, J.; Luiten, P.G.M.; Eisel, U.L.M.; Nijholt, I.M. TNF-α-mediates neuroprotection against glutamate-induced excitotoxicity via NF-κB-dependent up-regulation of Kca 2.2 channels. J. Neurochem. 2008, 107, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, T.; Fairless, R.; Pichi, K.; Fischer, R.; Richter, F.; Kontermann, R.E.; Pfizenmaier, K.; Diem, R.; Williams, S.K. Co-modulation of TNFR1 and TNFR2 in an animal model of multiple sclerosis. J. Neuroinflammation 2023, 20, 100. [Google Scholar] [CrossRef]
- Petratos, S.; Butzkueven, H.; Shipham, K.; Cooper, H.; Bucci, T.; Reid, K.; Lopes, E.; Emery, B.; Cheema, S.S.; Kilpatrick, T.J. Schwann Cell Apoptosis in the Postnatal Axotomized Sciatic Nerve Is Mediated Via NGF through the Low-Affinity Neurotrophin Receptor. J. Neuropathol. Exp. Neurol. 2003, 62, 398–411. [Google Scholar] [CrossRef]
- Barker, P.A.; Mantyh, P.; Arendt-Nielsen, L.; Viktrup, L.; Tive, L. Nerve Growth Factor Signaling and Its Contribution to Pain. J. Pain Res. 2020, 13, 1223–1241. [Google Scholar] [CrossRef]
- Ji, R.-R.; Nackley, A.; Huh, Y.; Terrando, N.; Maixner, W. Neuroinflammation and Central Sensitization in Chronic and Widespread Pain. Anesthesiology 2018, 129, 343–366. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.G. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Honjoh, K.; Watanabe, S.; Kubota, A.; Matsumine, A. Distribution and polarization of microglia and macrophages at injured sites and the lumbar enlargement after spinal cord injury. Neurosci. Lett. 2020, 737, 135152. [Google Scholar] [CrossRef]
- Li, X.-C.; Luo, S.-J.; Fan, W.; Zhou, T.-L.; Tan, D.-Q.; Tan, R.-X.; Xian, Q.-Z.; Li, J.; Huang, C.-M.; Wang, M.-S. Macrophage polarization regulates intervertebral disc degeneration by modulating cell proliferation, inflammation mediator secretion, and extracellular matrix metabolism. Front. Immunol. 2022, 13, 922173. [Google Scholar] [CrossRef]
- Geisler, S.; Doan, R.A.; Strickland, A.; Huang, X.; Milbrandt, J.; DiAntonio, A. Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain 2016, 139, 3092–3108. [Google Scholar] [CrossRef]
- Alé, A.; Bruna, J.; Navarro, X.; Udina, E. Neurotoxicity induced by antineoplastic proteasome inhibitors. NeuroToxicology 2014, 43, 28–35. [Google Scholar] [CrossRef]
- Snavely, A.R.; Heo, K.; Petrova, V.; Ho, T.S.-Y.; Huang, X.; Hermawan, C.; Kagan, R.; Deng, T.; Singeç, I.; Chen, L.; et al. Bortezomib-induced neurotoxicity in human neurons is the consequence of nicotinamide adenine dinucleotide depletion. Dis. Models Mech. 2022, 15, dmm049358. [Google Scholar] [CrossRef] [PubMed]
- Jaggi, A.S.; Singh, N. Mechanisms in cancer-chemotherapeutic drugs-induced peripheral neuropathy. Toxicology 2012, 291, 1–9. [Google Scholar] [CrossRef]
- Fontaine, V.; Mohand-Said, S.; Hanoteau, N.; Fuchs, C.; Pfizenmaier, K.; Eisel, U. Neurodegenerative and Neuroprotective Effects of Tumor Necrosis Factor (TNF) in Retinal Ischemia: Opposite Roles of TNF Receptor 1 and TNF Receptor 2. J. Neurosci. 2002, 22, RC216. [Google Scholar] [CrossRef]
- Shen, H.-M.; Pervaiz, S. TNF receptor superfamily-induced cell death: Redox-dependent execution. FASEB J. 2006, 20, 1589–1598. [Google Scholar] [CrossRef]
- Wang, M.; Tsai, B.M.; Crisostomo, P.R.; Meldrum, D.R. Tumor Necrosis Factor Receptor 1 Signaling Resistance in the Female Myocardium During Ischemia. Circulation 2006, 114, I282–I289. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Kontermann, R.E.; Pfizenmaier, K. Selective Targeting of TNF Receptors as a Novel Therapeutic Approach. Front. Cell Dev. Biol. 2020, 8, 401. [Google Scholar] [CrossRef]
- Marchetti, L.; Klein, M.; Schlett, K.; Pfizenmaier, K.; Eisel, U.L.M. Tumor Necrosis Factor (TNF)-mediated Neuroprotection against Glutamate-induced Excitotoxicity Is Enhanced by N-Methyl-D-aspartate Receptor Activation. J. Biol. Chem. 2004, 279, 32869–32881. [Google Scholar] [CrossRef]
- Rauert, H.; Wicovsky, A.; Müller, N.; Siegmund, D.; Spindler, V.; Waschke, J.; Kneitz, C.; Wajant, H. Membrane Tumor Necrosis Factor (TNF) Induces p100 Processing via TNF Receptor-2 (TNFR2). J. Biol. Chem. 2010, 285, 7394–7404. [Google Scholar] [CrossRef] [PubMed]
- Naudé, P.J.W.; Den Boer, J.A.; Luiten, P.G.M.; Eisel, U.L.M. Tumor necrosis factor receptor cross-talk. FEBS J. 2011, 278, 888–898. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| siRNA Name | Sequence (5′-3′) |
|---|---|
| si-NC | sense: 5′-UUCUCCGAACGUGUCACGUTT-3′ |
| anti-sense: 5′-ACGUGACACGUUCGGAGAATT-3′ | |
| si-NGFR | sense: 5′-GGGCCUUGUGGCCUAUAUUTT-3′ |
| anti-sense: 5′-AAUAUAGGCCACAAGGCCCTT-3′ | |
| si-TNFR1 | sense: 5′-UACGGCUUCCCAGAAUUACTT-3′ |
| anti-sense: 5′-GUAAUUCUGGGAAGCCGUATT-3′ | |
| si-TNFR2 | sense: 5′-CUGAUGAAAUCCCAGGATT-3′ |
| anti-sense: 5′-UCCUGGGAUUUCUCAUCAGTT-3′ |
| Primer Name | Sequence (5′-3′) |
|---|---|
| B2M | Forward: 5′-ATCCAAATGCTGAAGAACGG-3′ |
| Reverse: 5′-ATCAGTCTCAGTGGGGGTGA-3′ | |
| NF-κB | Forward: 5′-TGAAACACTGGAAGCACGGATGAC-3′ |
| Reverse: 5′-TCTCCTCCGCCTTCTGCTTGTAG-3′ | |
| NGF | Forward: 5′-AAGTGCCGAGCCTCCAATCC-3′ |
| Reverse: 5′-TTCTCATCTGTTGTCAACGCCTTG-3′ | |
| NGFR | Forward: 5′-CACAGCGACAGCGGCATCTC-3′ |
| Reverse: 5′-AGCAGCTTCTCGACCTCCTCAC-3′ | |
| TNF-α | Forward: 5′-GCCACCACGCTCTTCTGTCTAC-3′ |
| Reverse: 5′-GGTTTGTGAGTGTGAGGGTCTGG-3′ | |
| TNFR1 | Forward: 5′-AGTTCCAACGCTACCTGAGTGAG-3′ |
| Reverse: 5′-ACGGTGTTCTGAGTCTCCTTACAG-3′ | |
| TNFR2 | Forward: 5′-CCTGCCTACAAAGAGATGCCAAG-3′ |
| Reverse: 5′-GAACTGGGTGCTGTGGTCAAC-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Y.; Xu, X.; Wu, P.; Chen, X.; Mo, Q.; Zhuo, M. TNF-α Promotes the Recovery of Dorsal Root Ganglion Neurons from Cisplatin-Induced Injury Through an NGF-Independent Mechanism. Curr. Issues Mol. Biol. 2025, 47, 482. https://doi.org/10.3390/cimb47070482
Wei Y, Xu X, Wu P, Chen X, Mo Q, Zhuo M. TNF-α Promotes the Recovery of Dorsal Root Ganglion Neurons from Cisplatin-Induced Injury Through an NGF-Independent Mechanism. Current Issues in Molecular Biology. 2025; 47(7):482. https://doi.org/10.3390/cimb47070482
Chicago/Turabian StyleWei, Yiling, Xianlin Xu, Pan Wu, Xiang Chen, Qingmei Mo, and Ming Zhuo. 2025. "TNF-α Promotes the Recovery of Dorsal Root Ganglion Neurons from Cisplatin-Induced Injury Through an NGF-Independent Mechanism" Current Issues in Molecular Biology 47, no. 7: 482. https://doi.org/10.3390/cimb47070482
APA StyleWei, Y., Xu, X., Wu, P., Chen, X., Mo, Q., & Zhuo, M. (2025). TNF-α Promotes the Recovery of Dorsal Root Ganglion Neurons from Cisplatin-Induced Injury Through an NGF-Independent Mechanism. Current Issues in Molecular Biology, 47(7), 482. https://doi.org/10.3390/cimb47070482