
Microarray Analysis Reveals Sepsis Is a Syndrome with Hyperactivity of TH17 Immunity, with Over-Presentation of the Treg Cell Cytokine TGF-β †


Abstract
1. Introduction
2. Materials and Methods
2.1. Microarray Dataset
2.2. Statistical Analysis
2.3. RT-PCR Confirmation
3. Results
3.1. RMA Analysis of Whole Blood
3.2. Toll-like Signaling and Heat Shock Protein Expression in Patients with Sepsis
3.3. Antigen Processing and Antigen Presentation Genes in Sepsis
3.4. TH17-like Innate Immune Transcription Factors in Sepsis
3.5. TH17-like and Treg-Related Cytokines Are Upregulated During Sepsis
3.6. Upregulation of Th17-like Innate Immunity-Related Effector Molecules During Sepsis
3.7. Dysregulation of Coagulation-, Glycolysis-, Acidosis-, and Vasodilation-Related Genes in Sepsis
3.8. Failure of T-Lymphocyte Adaptive Immunity During Sepsis
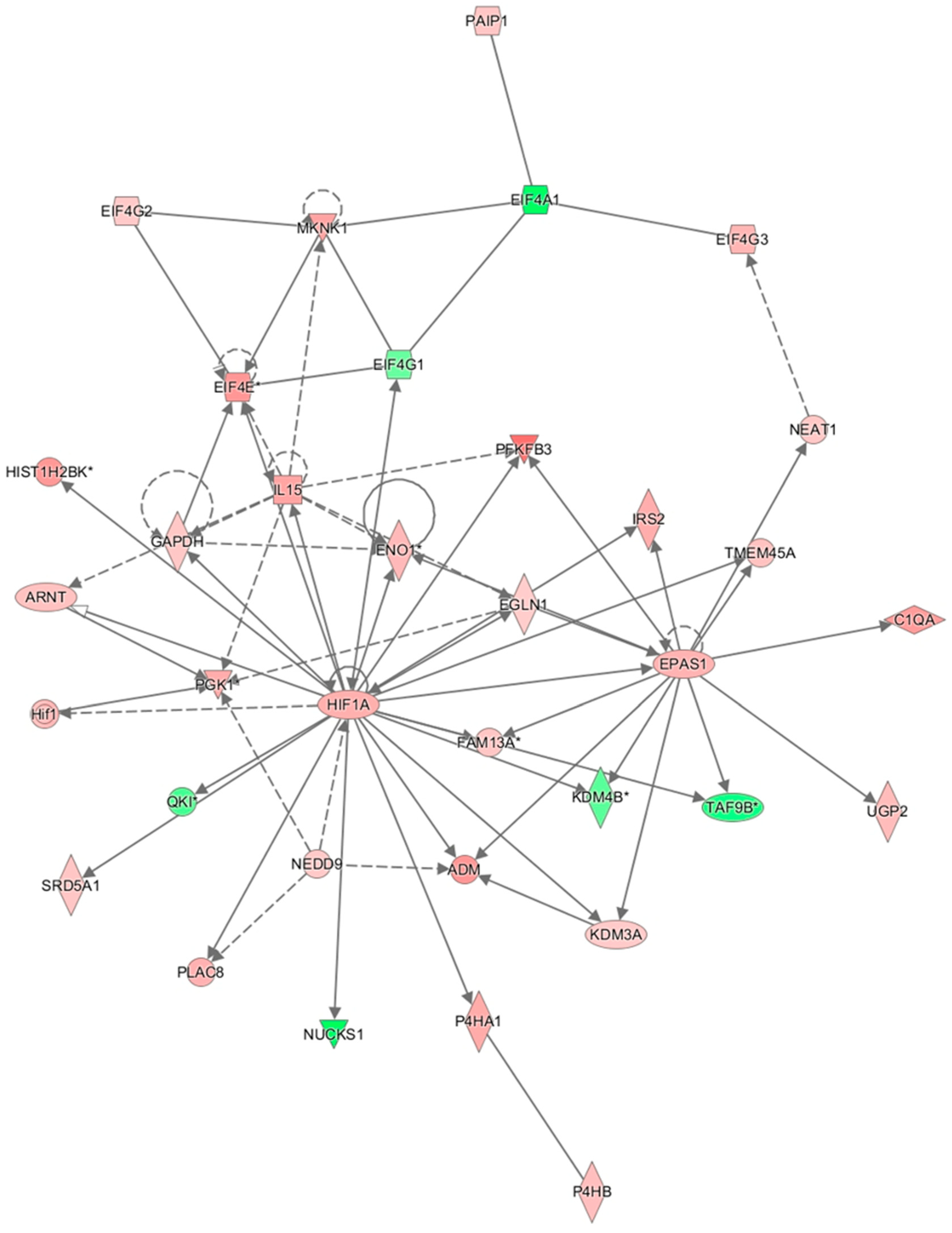
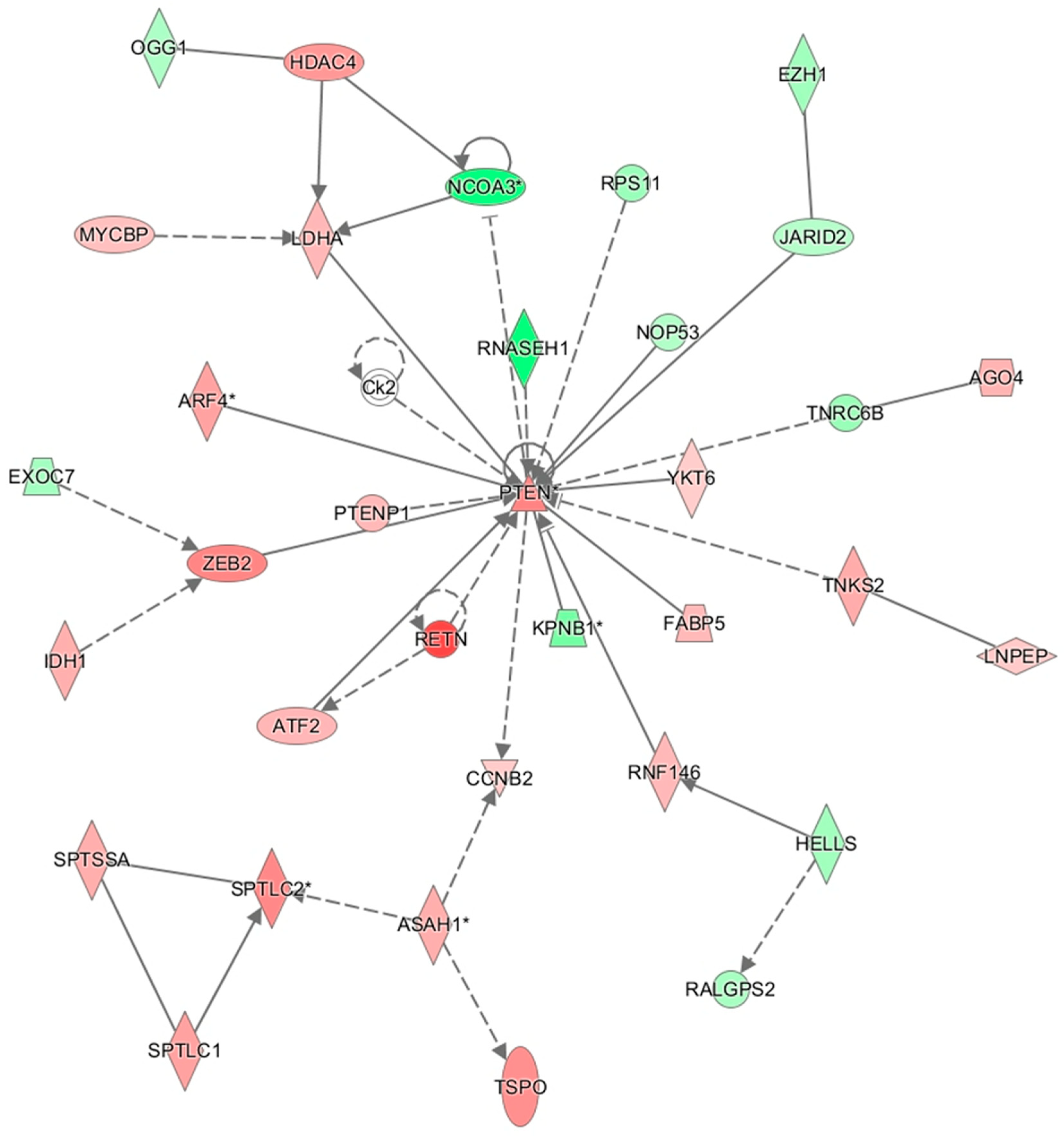
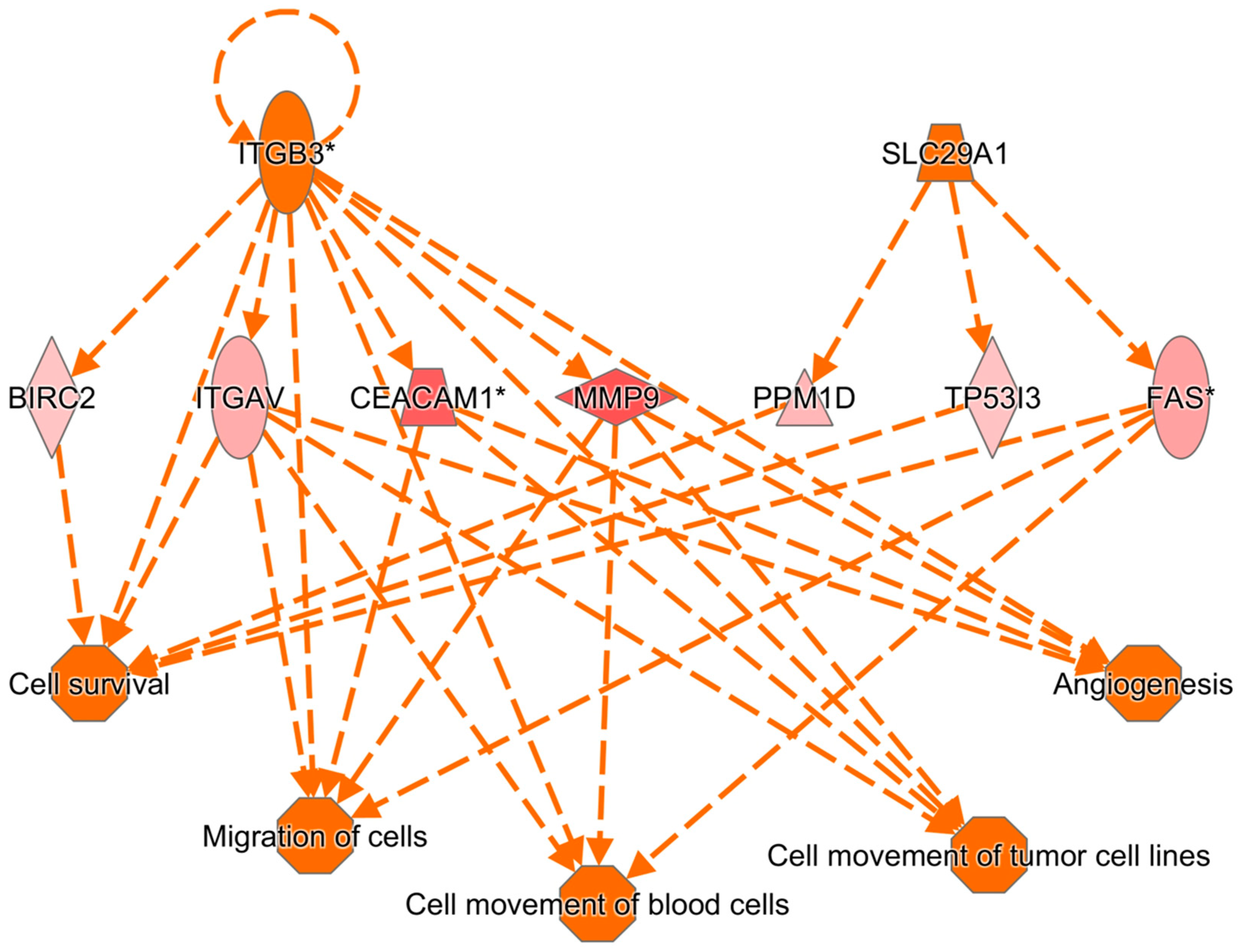
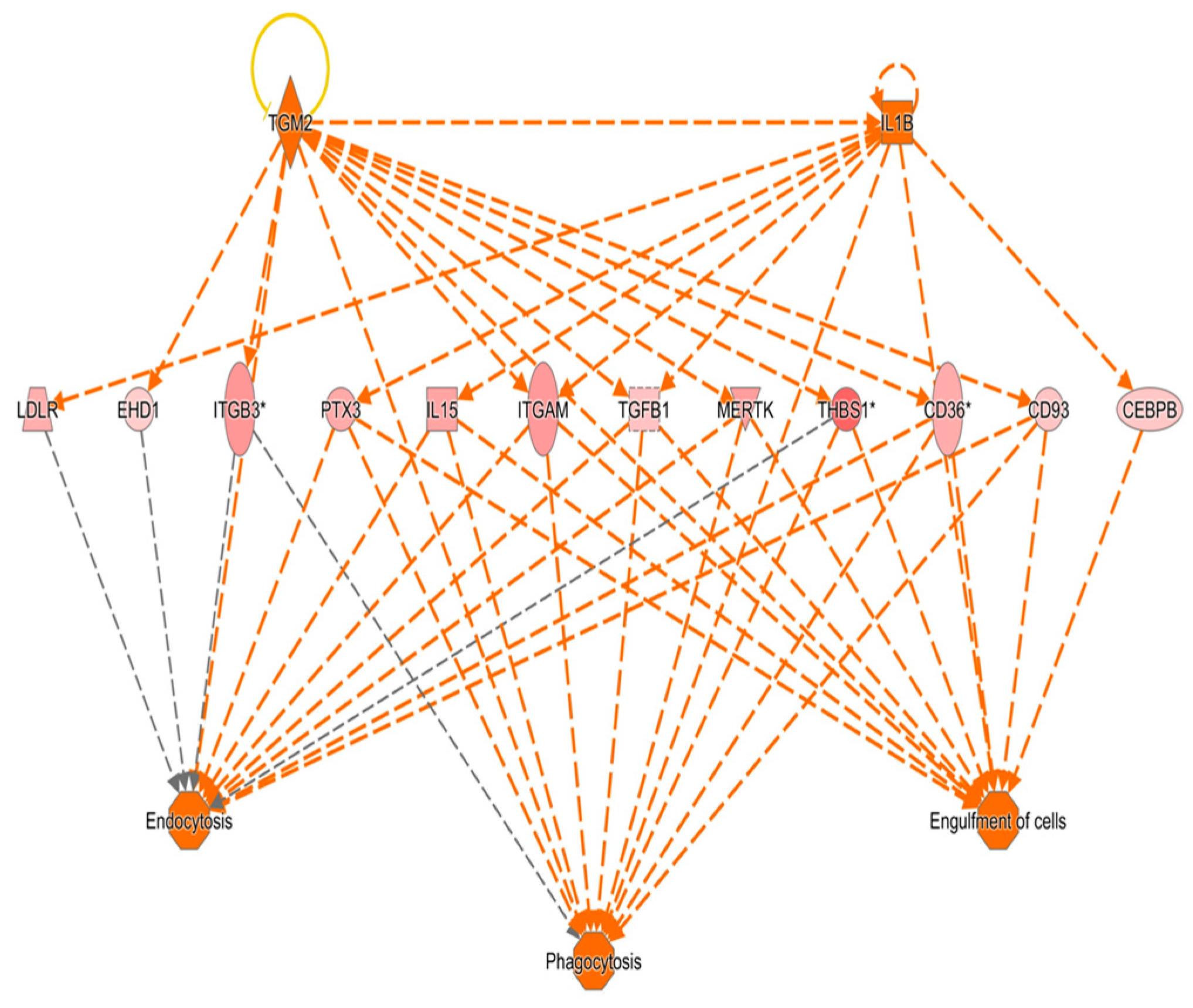
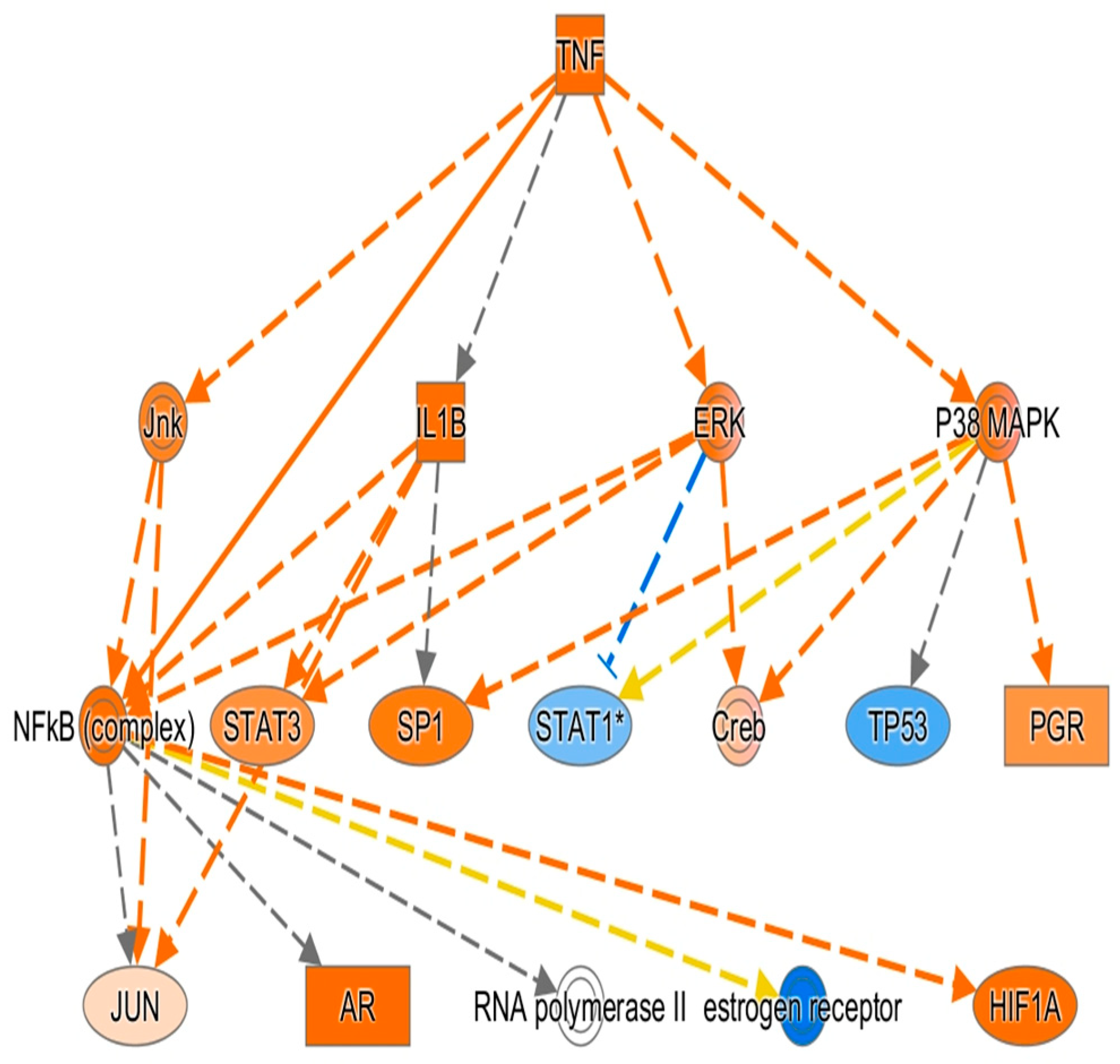
3.9. Results of Ingenuity Pathway Analysis
4. Discussion
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abraham, E.; Anzueto, A.; Gutierrez, G.; Tessler, S.; San Pedro, G.; Wunderink, R.; Dal Nogare, A.; Nasraway, S.; Berman, S.; Cooney, R.; et al. Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. NORASEPT II Study Group. Lancet 1998, 351, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.J., Jr.; Agosti, J.M.; Opal, S.M.; Lowry, S.F.; Balk, R.A.; Sadoff, J.C.; Abraham, E.; Schein, R.M.; Benjamin, E. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The Soluble TNF Receptor Sepsis Study Group. N. Engl. J. Med. 1996, 334, 1697–1702. [Google Scholar] [CrossRef] [PubMed]
- Opal, S.M.; Fisher, C.J., Jr.; Dhainaut, J.F.; Vincent, J.L.; Brase, R.; Lowry, S.F.; Sadoff, J.C.; Slotman, G.J.; Levy, H.; Balk, R.A.; et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: A phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit. Care Med. 1997, 25, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, K.; Karzai, W. Anti-tumor necrosis factor therapy in sepsis: Update on clinical trials and lessons learned. Crit. Care Med. 2001, 29, S121–S125. [Google Scholar] [CrossRef]
- Hu, W.C. A Framework of All Discovered Immunological Pathways and Their Roles for Four Specific Types of Pathogens and Hypersensitivities. Front. Immunol. 2020, 11, 1992. [Google Scholar] [CrossRef]
- Hu, W.C. Human immune responses to Plasmodium falciparum infection: Molecular evidence for a suboptimal THαβ and TH17 bias over ideal and effective traditional TH1 immune response. Malar. J. 2013, 12, 392. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.T.; Liao, M.T.; Tsai, K.W.; Lu, K.C.; Hu, W.C. Interplay of Chemokines Receptors, Toll-like Receptors, and Host Immunological Pathways. Biomedicines 2023, 11, 2384. [Google Scholar] [CrossRef]
- Huang, Y.M.; Shih, L.J.; Hsieh, T.W.; Tsai, K.W.; Lu, K.C.; Liao, M.T.; Hu, W.C. Type 2 hypersensitivity disorders, including systemic lupus erythematosus, Sjögren’s syndrome, Graves’ disease, myasthenia gravis, immune thrombocytopenia, autoimmune hemolytic anemia, dermatomyositis, and graft-versus-host disease, are THαβ-dominant autoimmune diseases. Virulence 2024, 15, 2404225. [Google Scholar] [CrossRef]
- Lee, Y.H.; Tsai, K.W.; Lu, K.C.; Shih, L.J.; Hu, W.C. Cancer as a Dysfunctional Immune Disorder: Pro-Tumor TH1-like Immune Response and Anti-Tumor THαβ Immune Response Based on the Complete Updated Framework of Host Immunological Pathways. Biomedicines 2022, 10, 2497. [Google Scholar] [CrossRef]
- Lu, K.C.; Tsai, K.W.; Wang, Y.K.; Hu, W.C. Types of cell death and their relations to host immunological pathways. Aging 2024, 16, 11755–11768. [Google Scholar] [CrossRef]
- Lu, K.C.; Tsai, K.W.; Hu, W.C. Role of TGFβ-producing regulatory T cells in scleroderma and end-stage organ failure. Heliyon 2024, 10, e35590. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.H.; Tsai, K.W.; Wu, Y.J.; Liao, M.T.; Lu, K.C.; Hu, W.C. The Framework for Human Host Immune Responses to Four Types of Parasitic Infections and Relevant Key JAK/STAT Signaling. Int. J. Mol. Sci. 2021, 22, 3310. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, L.; Peng, W.; Deng, H.; Ni, H.; Tong, H.; Hu, H.; Wang, S.; Qian, J.; Liang, A.; et al. Th17/Treg balance: The bloom and wane in the pathophysiology of sepsis. Front. Immunol. 2024, 15, 1356869. [Google Scholar] [CrossRef] [PubMed]
- Rendon, J.L.; Choudhry, M.A. Th17 cells: Critical mediators of host responses to burn injury and sepsis. J. Leukoc. Biol. 2012, 92, 529–538. [Google Scholar] [CrossRef]
- Howrylak, J.A.; Dolinay, T.; Lucht, L.; Wang, Z.; Christiani, D.C.; Sethi, J.M.; Xing, E.P.; Donahoe, M.P.; Choi, A.M. Discovery of the gene signature for acute lung injury in patients with sepsis. Physiol. Genom. 2009, 37, 133–139. [Google Scholar] [CrossRef]
- American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit. Care Med. 1992, 20, 864–874. [CrossRef]
- Rotunno, M.; Hu, N.; Su, H.; Wang, C.; Goldstein, A.M.; Bergen, A.W.; Consonni, D.; Pesatori, A.C.; Bertazzi, P.A.; Wacholder, S.; et al. A gene expression signature from peripheral whole blood for stage I lung adenocarcinoma. Cancer Prev. Res. 2011, 4, 1599–1608. [Google Scholar] [CrossRef]
- Docke, W.D.; Randow, F.; Syrbe, U.; Krausch, D.; Asadullah, K.; Reinke, P.; Volk, H.D.; Kox, W. Monocyte deactivation in septic patients: Restoration by IFN-gamma treatment. Nat. Med. 1997, 3, 678–681. [Google Scholar] [CrossRef]
- Kim, H.; Seed, B. The transcription factor MafB antagonizes antiviral responses by blocking recruitment of coactivators to the transcription factor IRF3. Nat. Immunol. 2010, 11, 743–750. [Google Scholar] [CrossRef]
- Ho, I.C.; Lo, D.; Glimcher, L.H. c-maf promotes T helper cell type 2 (Th2) and attenuates Th1 differentiation by both interleukin 4-dependent and -independent mechanisms. J. Exp. Med. 1998, 188, 1859–1866. [Google Scholar] [CrossRef]
- Ruan, Q.; Kameswaran, V.; Zhang, Y.; Zheng, S.; Sun, J.; Wang, J.; DeVirgiliis, J.; Liou, H.C.; Beg, A.A.; Chen, Y.H. The Th17 immune response is controlled by the Rel-RORgamma-RORgamma T transcriptional axis. J. Exp. Med. 2011, 208, 2321–2333. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, S.; Tanaka, J.; Sugita, J.; Kosugi, M.; Shiratori, S.; Wakasa, K.; Shono, Y.; Shigematsu, A.; Kondo, T.; Kobayashi, T.; et al. Expansion of CD4(+)CD25 (+) regulatory T cells from cord blood CD4(+) cells using the common gamma-chain cytokines (IL-2 and IL-15) and rapamycin. Ann. Hematol. 2011, 90, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.C.; Nadeau, K.C.; Tu, W.; Hwa, V.; Dionis, K.; Bezrodnik, L.; Teper, A.; Gaillard, M.; Heinrich, J.; Krensky, A.M.; et al. Cutting edge: Decreased accumulation and regulatory function of CD4+ CD25(high) T cells in human STAT5b deficiency. J. Immunol. 2006, 177, 2770–2774. [Google Scholar] [CrossRef]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Gonda, H.; Sugai, M.; Nambu, Y.; Katakai, T.; Agata, Y.; Mori, K.J.; Yokota, Y.; Shimizu, A. The balance between Pax5 and Id2 activities is the key to AID gene expression. J. Exp. Med. 2003, 198, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, K.; Nishizumi, H.; Umemori, H.; Aizawa, S.; Takatsu, K.; Yamamoto, T. Distinctive roles of Fyn and Lyn in IgD- and IgM-mediated signaling. Int. Immunol. 1999, 11, 1441–1449. [Google Scholar] [CrossRef]
- Kirstetter, P.; Thomas, M.; Dierich, A.; Kastner, P.; Chan, S. Ikaros is critical for B cell differentiation and function. Eur. J. Immunol. 2002, 32, 720–730. [Google Scholar] [CrossRef]
- Netea, M.G.; Azam, T.; Lewis, E.C.; Joosten, L.A.; Wang, M.; Langenberg, D.; Meng, X.; Chan, E.D.; Yoon, D.Y.; Ottenhoff, T.; et al. Mycobacterium tuberculosis induces interleukin-32 production through a caspase- 1/IL-18/interferon-gamma-dependent mechanism. PLoS Med. 2006, 3, e277. [Google Scholar] [CrossRef]
- Yang, C.Y.; Best, J.A.; Knell, J.; Yang, E.; Sheridan, A.D.; Jesionek, A.K.; Li, H.S.; Rivera, R.R.; Lind, K.C.; D’Cruz, L.M.; et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat. Immunol. 2011, 12, 1221–1229. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Karl, I.E. The pathophysiology and treatment of sepsis. N. Engl. J. Med. 2003, 348, 138–150. [Google Scholar] [CrossRef]
- Oberholzer, A.; Oberholzer, C.; Moldawer, L.L. Sepsis syndromes: Understanding the role of innate and acquired immunity. Shock 2001, 16, 83–96. [Google Scholar] [CrossRef]
- Girardin, E.; Grau, G.E.; Dayer, J.M.; Roux-Lombard, P.; Lambert, P.H. Tumor necrosis factor and interleukin-1 in the serum of children with severe infectious purpura. N. Engl. J. Med. 1988, 319, 397–400. [Google Scholar] [CrossRef]
- Thomas, L. Germs. N. Engl. J. Med. 1972, 287, 553–555. [Google Scholar] [CrossRef]
- Warren, H.S. Strategies for the treatment of sepsis. N. Engl. J. Med. 1997, 336, 952–953. [Google Scholar] [CrossRef]
- Zeni, F.; Freeman, B.; Natanson, C. Anti-inflammatory therapies to treat sepsis and septic shock: A reassessment. Crit. Care Med. 1997, 25, 1095–1100. [Google Scholar] [CrossRef]
- Ward, P.A. Immunosuppression in sepsis. JAMA 2011, 306, 2618–2619. [Google Scholar] [CrossRef]
- Kasten, K.R.; Tschop, J.; Adediran, S.G.; Hildeman, D.A.; Caldwell, C.C. T cells are potent early mediators of the host response to sepsis. Shock 2010, 34, 327–336. [Google Scholar] [CrossRef]
- Hein, F.; Massin, F.; Cravoisy-Popovic, A.; Barraud, D.; Levy, B.; Bollaert, P.E.; Gibot, S. The relationship between CD4+CD25+CD127-regulatory T cells and inflammatory response and outcome during shock states. Crit. Care 2010, 14, R19. [Google Scholar] [CrossRef]
- Ayala, A.; Chung, C.S.; Xu, Y.X.; Evans, T.A.; Redmond, K.M.; Chaudry, I.H. Increased inducible apoptosis in CD4+ T lymphocytes during polymicrobial sepsis is mediated by Fas ligand and not endotoxin. Immunology 1999, 97, 45–55. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Tinsley, K.W.; Swanson, P.E.; Grayson, M.H.; Osborne, D.F.; Wagner, T.H.; Cobb, J.P.; Coopersmith, C.; Karl, I.E. Depletion of dendritic cells, but not macrophages, in patients with sepsis. J. Immunol. 2002, 168, 2493–2500. [Google Scholar] [CrossRef]
- Unsinger, J.; Herndon, J.M.; Davis, C.G.; Muenzer, J.T.; Hotchkiss, R.S.; Ferguson, T.A. The role of TCR engagement and activation-induced cell death in sepsis-induced T cell apoptosis. J. Immunol. 2006, 177, 7968–7973. [Google Scholar] [CrossRef] [PubMed]
- Manjuck, J.; Saha, D.C.; Astiz, M.; Eales, L.J.; Rackow, E.C. Decreased response to recall antigens is associated with depressed costimulatory receptor expression in septic critically ill patients. J. Lab. Clin. Med. 2000, 135, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Kelly-Scumpia, K.M.; Scumpia, P.O.; Weinstein, J.S.; Delano, M.J.; Cuenca, A.G.; Nacionales, D.C.; Wynn, J.L.; Lee, P.Y.; Kumagai, Y.; Efron, P.A.; et al. B cells enhance early innate immune responses during bacterial sepsis. J. Exp. Med. 2011, 208, 1673–1682. [Google Scholar] [CrossRef]
- Hammer, A.M.; Morris, N.L.; Cannon, A.R.; Khan, O.M.; Gagnon, R.C.; Movtchan, N.V.; van Langeveld, I.; Li, X.; Gao, B.; Choudhry, M.A. Interleukin-22 Prevents Microbial Dysbiosis and Promotes Intestinal Barrier Regeneration Following Acute Injury. Shock 2017, 48, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.H.; Zheng, Z.N.; Xiao, C.X.; Liu, X.; Lin, X.Q. IL-22 relieves sepsis-induced liver injury via activating JAK/STAT3 signaling pathway. J. Biol. Regul. Homeost. Agents 2020, 34, 1719–1727. [Google Scholar] [CrossRef]
- Yu, C.; Ling, Q.; Jiao, J.; Liu, J.; Huang, Z.; Wang, F.; Sun, X.; Kong, X. Interleukin-22 protects from endotoxemia by inducing suppressive F4/80(+)Ly6G(hi)Ly6C(hi) cells population. BMC Immunol. 2022, 23, 45. [Google Scholar] [CrossRef]
- Wu, Y.; Wu, G.; Li, M.; Chang, Y.; Yu, M.; Meng, Y.; Wan, X. Prediction of Th17/Treg cell balance on length of stay in intensive care units of patients with sepsis. J. Intensive Med. 2024, 4, 240–246. [Google Scholar] [CrossRef]
- Naylor, J.M.; Kronfeld, D.S. In vivo studies of hypoglycemia and lactic acidosis in endotoxic shock. Am. J. Physiol. 1985, 248, E309–E316. [Google Scholar] [CrossRef]
- Hornef, M.W.; Wick, M.J.; Rhen, M.; Normark, S. Bacterial strategies for overcoming host innate and adaptive immune responses. Nat. Immunol. 2002, 3, 1033–1040. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probe ID | p-Value | Arrow | Fold | Gene |
|---|---|---|---|---|
| 201743_at | 1.37 × 10−4 | up | 2.18 | CD14 |
| 204924_at | 1.45 × 10−10 | up | 3.38 | TLR2 |
| 210166_at | 9.16 × 10−8 | up | 2.40 | TLR5 |
| 210176_at | 0.001131 | up | 2.07 | TLR1 |
| 213817_at | 3.14 × 10−13 | up | 21.04 | IRAK3 |
| 219618_at | 1.89 × 10−9 | up | 2.69 | IRAK4 |
| 220832_at | 4.76 × 10−9 | up | 5.16 | TLR8 |
| 221060_s_at | 6.62 × 10−7 | up | 3.33 | TLR4 |
| 212184_s_at | 2.03 × 10−5 | up | 2.61 | TAB2 |
| 221705_s_at | 8.46 × 10−10 | down | 2.08 | SIKE1 |
| 205118_at | 1.05 × 10−10 | down | 7.61 | FPR1 |
| 210772_at | 2.06 × 10−8 | up | 4.78 | FPR2 |
| Probe ID | p-Value | Arrow | Fold | Gene |
|---|---|---|---|---|
| 201137_s_at | 5.80 × 10−4 | down | 2.09 | HLA-DPB1 |
| 203290_at | 2.56 × 10−8 | down | 5.19 | HLA-DQA1 |
| 204670_x_at | 6.77 × 10−8 | down | 2.85 | HLA-DRB1/B4 |
| 205671_s_at | 1.27 × 10−4 | down | 2.02 | HLA-DOB |
| 208306_x_at | 1.53 × 10−6 | down | 2.44 | HLA-DRB1 |
| 208894_at | 8.06 × 10−7 | down | 2.76 | HLA-DRA |
| 209312_x_at | 1.24 × 10−6 | down | 2.67 | HLA-DRB1/B4/B5 |
| 209823_x_at | 8.65 × 10−4 | down | 2.08 | HLA-DQB1 |
| 210294_at | 7.08 × 10−10 | down | 2.25 | TAPBP |
| 210528_at | 1.28 × 10−5 | down | 2.57 | MR1 |
| 211948_x_at | 3.66 × 10−28 | down | 11.75 | BAT2L2 |
| 211990_at | 5.10 × 10−6 | down | 3.19 | HLA-DPA1 |
| 212384_at | 8.83 × 10−15 | down | 2.98 | HLABAT1 |
| 212671_s_at | 0.002545 | down | 2.27 | HLA-DQA1/A2 |
| 214055_x_at | 1.16 × 10−24 | down | 9.42 | BAT2L2 |
| 215193_x_at | 2.90 × 10−6 | down | 2.52 | HLA-DRB1/B3/B4 |
| 221491_x_at | 1.50 × 10−6 | down | 2.25 | HLA-DRB1/B3/B4/B5 |
| Probe ID | p-Value | Arrow | Fold | Gene |
|---|---|---|---|---|
| 201473_at | 3.65 × 10−9 | up | 2.46 | JUNB |
| 201502_s_at | 9.16 × 10−7 | down | 2.36 | NFKBIA |
| 202527_s_at | 5.77 × 10−9 | up | 3.24 | SMAD4 |
| 203077_s_at | 4.90 × 10−7 | up | 2.37 | SMAD2 |
| 203574_at | 4.37 × 10−10 | up | 5.18 | NFIL3 |
| 204039_at | 4.62 × 10−8 | up | 2.06 | CEBPA |
| 204203_at | 9.92 × 10−7 | up | 2.17 | CEBPG |
| 205841_at | 1.02 × 10−13 | up | 4.66 | JAK2 |
| 206036_s_at | 8.46 × 10−12 | down | 4.75 | REL |
| 206359_at | 3.22 × 10−7 | up | 2.09 | SOCS3 |
| 206363_at | 9.68 × 10−6 | down | 2.26 | MAF |
| 208991_at | 1.49 × 10−13 | down | 3.22 | STAT3 |
| 209604_s_at | 2.74 × 10−19 | down | 6.55 | GATA3 |
| 209969_s_at | 2.12 × 10−8 | down | 4.75 | STAT1 |
| 210479_s_at | 5.21 × 10−15 | down | 7.85 | RORA |
| 212501_at | 1.73 × 10−7 | up | 2.17 | CEBPB |
| 212550_at | 7.19 × 10−10 | up | 2.52 | STAT5B |
| 213006_at | 6.03 × 10−10 | up | 4.21 | CEBPD |
| 218221_at | 1.49 × 10−11 | up | 2.35 | ARNT |
| 218559_s_at | 9.49 × 10−7 | up | 3.35 | MAFB |
| 218880_at | 5.34 × 10−11 | up | 3.75 | FOSL2 |
| 208808_s_at | 1.07 × 10−11 | up | 9.12 | HMGB |
| 200989_at | 1.17 × 10−6 | up | 3.00 | HIF1A |
| 221969_at | 9.96 × 10−13 | down | 4.20 | PAX5 |
| 203140_at | 3.09 × 10−10 | up | 3.69 | BCL6 |
| 210105_s_at | 9.37 × 10−10 | down | 3.32 | FYN |
| 210754_s_at | 2.98 × 10−10 | down | 3.55 | LYN |
| 217620_s_at | 4.31 × 10−12 | down | 2.79 | PIK3CB |
| 221756_at | 5.52 × 10−10 | down | 2.62 | PIK3IP1 |
| 204054_at | 2.56 × 10−10 | up | 5.51 | PTEN |
| 206370_at | 2.95 × 10−9 | down | 2.44 | PIK3CG |
| 212240_s_at | 4.29 × 10−13 | down | 4.51 | PIK3R1 |
| Probe ID | p-Value | Arrow | Fold | Gene |
|---|---|---|---|---|
| 201110_s_at | 2.02 × 10−9 | up | 8.27 | THBS1 |
| 203085_s_at | 1.57 × 10−8 | up | 2.33 | TGFB1 |
| 203828_s_at | 7.88 × 10−5 | down | 2.13 | IL32 |
| 205016_at | 8.33 × 10−10 | up | 4.86 | TGFA |
| 205992_s_at | 4.40 × 10−6 | up | 3.58 | IL15 |
| 208114_s_at | 7.75 × 10−20 | down | 5.85 | ISG20L2 |
| 208200_at | 3.06 × 10−11 | down | 4.80 | IL1A |
| 212195_at | 3.90 × 10−6 | up | 2.67 | IL6ST |
| 209555_s_at | 2.87 × 10−5 | up | 3.18 | CD36 |
| 212657_s_at | 2.96 × 10−7 | up | 2.31 | IL1RN |
| Probe ID | p-Value | Arrow | Fold | Gene |
|---|---|---|---|---|
| 201642_at | 1.42 × 10−9 | up | 2.32 | IFNGR2 |
| 202948_at | 5.77 × 10−10 | up | 6.46 | IL1R1 |
| 203233_at | 2.36 × 10−10 | up | 3.27 | IL4R |
| 204191_at | 2.98 × 10−7 | up | 2.06 | IFNAR1 |
| 204731_at | 7.48 × 10−21 | down | 11.93 | TGFBR3 |
| 204786_s_at | 5.23 × 10−19 | down | 6.86 | IFNAR2 |
| 205227_at | 2.89 × 10−5 | up | 2.68 | IL1RAP |
| 205291_at | 2.89 × 10−8 | down | 2.44 | IL2RB |
| 205707_at | 1.73 × 10−9 | down | 2.41 | IL17RA |
| 205798_at | 2.48 × 10−24 | down | 31.79 | IL7R |
| 205926_at | 1.06 × 10−9 | down | 2.19 | IL27RA |
| 205945_at | 1.49 × 10−22 | down | 16.69 | IL6R |
| 206618_at | 4.70 × 10−9 | up | 12.92 | IL18R1 |
| 207072_at | 5.22 × 10−8 | up | 4.93 | IL18RAP |
| 211372_s_at | 1.76 × 10−8 | up | 10.68 | IL1R2 |
| 211676_s_at | 6.66 × 10−9 | up | 4.61 | IFNGR1 |
| 205159_at | 1.17 × 10−6 | up | 2.51 | CSF2RB |
| 210340_s_at | 4.36 × 10−10 | up | 2.30 | CSF2RA |
| Probe ID | p-Value | Arrow | Fold | Gene |
|---|---|---|---|---|
| 200602_at | 3.75 × 10−12 | up | 4.38 | APP |
| 206157_at | 8.31 × 10−8 | up | 3.27 | PTX3 |
| 208691_at | 0.001264 | up | 2.49 | TFRC |
| 208703_s_at | 1.26 × 10−7 | up | 3.05 | APLP2 |
| 219890_at | 1.43 × 10−12 | up | 7.83 | CLEC5A |
| 220496_at | 2.59 × 10−7 | up | 3.33 | CLEC1B |
| 205033_s_at | 1.17 × 10−5 | up | 4.79 | DEFA1/A1B/A3 |
| 207269_at | 2.87 × 10−5 | up | 6.67 | DEFA4 |
| 201943_s_at | 7.91 × 10−12 | up | 6.94 | CPD |
| 204961_s_at | 7.26 × 10−8 | up | 2.02 | NCF1/1B/1C |
| 207677_s_at | 5.88 × 10−10 | up | 2.66 | NCF4 |
| 214084_x_at | 1.31 × 10−8 | up | 2.25 | NCF1C |
| Probe ID | p-Value | Arrow | Fold | Gene |
|---|---|---|---|---|
| 200985_s_at | 4.85 × 10−11 | up | 6.59 | CD59 |
| 201925_s_at | 2.14 × 10−7 | up | 5.61 | CD55 |
| 202953_at | 7.01 × 10−6 | up | 2.53 | C1QB |
| 205786_s_at | 5.02 × 10−13 | up | 4.05 | ITGAM |
| 206244_at | 6.06 × 10−12 | up | 6.76 | CR1 |
| 208783_s_at | 0.004769 | up | 2.21 | CD46 |
| 209906_at | 7.48 × 10−9 | up | 4.34 | C3AR1 |
| 210184_at | 1.17 × 10−6 | up | 2.07 | ITGAX |
| 218232_at | 1.52 × 10−8 | up | 3.97 | C1QA |
| 218983_at | 7.83 × 10−8 | up | 2.64 | C1RL |
| 220088_at | 9.13 × 10−8 | up | 2.49 | C5AR1 |
| 202910_s_at | 3.42 × 10−7 | up | 2.26 | CD97 |
| Probe ID | p-Value | Arrow | Fold | Gene |
|---|---|---|---|---|
| 203167_at | 1.02 × 10−13 | up | 3.14 | TIMP2 |
| 203936_s_at | 2.89 × 10−16 | up | 10.59 | MMP9 |
| 206871_at | 1.04 × 10−6 | up | 5.39 | ELANE |
| 207329_at | 3.41 × 10−11 | up | 32.06 | MMP8 |
| 207890_s_at | 1.30 × 10−11 | up | 3.11 | MMP25 |
| 202833_s_at | 2.83 × 10−9 | up | 2.78 | SERPINA1 |
| 204614_at | 5.64 × 10−8 | up | 3.07 | SERPINB2 |
| 212268_at | 8.64 × 10−11 | up | 5.64 | SERPINB1 |
| Probe ID | p-Value | Arrow | Fold | Gene |
|---|---|---|---|---|
| 203305_at | 2.16 × 10−4 | up | 2.18 | F13A1 |
| 204714_s_at | 1.87 × 10−8 | up | 3.93 | F5 |
| 205756_s_at | 2.79 × 10−5 | up | 2.08 | F8 |
| 205871_at | 7.54 × 10−7 | down | 3.12 | PLGLA/B1/B2 |
| 206655_s_at | 2.25 × 10−8 | up | 5.37 | GP1BB/SEPT5 |
| 207808_s_at | 6.30 × 10−8 | up | 2.88 | PROS1 |
| 211924_s_at | 5.53 × 10−7 | up | 2.33 | PLAUR |
| 212245_at | 6.18 × 10−7 | up | 2.30 | MCFD2 |
| 213258_at | 1.07 × 10−6 | up | 2.35 | TFPI |
| 213506_at | 0.002877 | up | 2.35 | F2RL1 |
| 214415_at | 1.30 × 10−9 | down | 5.54 | PLGLB1/B2 |
| 216956_s_at | 4.64 × 10−5 | up | 2.39 | ITGA2B |
| 218718_at | 2.79 × 10−10 | up | 9.39 | PDGFC |
| 204627_s_at | 1.30 × 10−6 | up | 4.18 | ITGB3 |
| 203887_s_at | 8.66 × 10−9 | up | 4.53 | THBD |
| Probe ID | p-Value | Arrow | Fold | Gene |
|---|---|---|---|---|
| 200650_s_at | 2.02 × 10−9 | up | 2.71 | LDHA |
| 200737_at | 2.94 × 10−11 | up | 3.17 | PGK1 |
| 201030_x_at | 9.45 × 10−5 | down | 2.02 | LDHB |
| 201251_at | 2.51 × 10−10 | up | 2.67 | PKM2 |
| 202464_s_at | 6.45 × 10−9 | up | 7.30 | PFKFB3 |
| 202934_at | 9.80 × 10−14 | up | 4.77 | HK2 |
| 202990_at | 2.15 × 10−12 | up | 4.20 | PYGL |
| 203502_at | 1.24 × 10−4 | up | 3.67 | BPGM |
| 205936_s_at | 5.17 × 10−12 | up | 4.99 | HK3 |
| 206348_s_at | 9.53 × 10−11 | up | 2.60 | PDK3 |
| 208308_s_at | 3.92 × 10−9 | up | 2.22 | GPI |
| 209992_at | 3.99 × 10−9 | up | 11.77 | PFKFB2 |
| 213453_x_at | 2.13 × 10−12 | up | 2.18 | GAPDH |
| 217294_s_at | 3.28 × 10−6 | up | 2.62 | ENO1 |
| 218273_s_at | 1.01 × 10−7 | down | 2.25 | PDP1 |
| Probe ID | p-Value | Arrow | Fold | Gene |
|---|---|---|---|---|
| 200078_s_at | 6.15 × 10−13 | up | 2.53 | ATP6V0B |
| 201171_at | 4.49 × 10−10 | up | 2.48 | ATP6V0E1 |
| 201443_s_at | 5.84 × 10−6 | up | 2.33 | ATP6AP2 |
| 201971_s_at | 4.45 × 10−13 | down | 5.21 | ATP6V1A |
| 202872_at | 1.95 × 10−10 | up | 6.18 | ATP6V1C1 |
| 202874_s_at | 6.99 × 10−10 | up | 5.72 | ATP6V1C1 |
| 204158_s_at | 5.14 × 10−8 | up | 2.07 | TCIRG1 |
| 208898_at | 2.66 × 10−9 | up | 2.41 | ATP6V1D |
| 213587_s_at | 1.13 × 10−8 | down | 2.07 | ATP6V0E2 |
| 206208_at | 1.00 × 10−11 | up | 3.51 | CA4 |
| 206209_s_at | 4.18 × 10−15 | up | 7.98 | CA4 |
| 209301_at | 2.78 × 10−6 | up | 3.42 | CA2 |
| 212536_at | 4.38 × 10−9 | up | 4.21 | ATP11B |
| 213582_at | 1.89 × 10−8 | up | 2.24 | ATP11A |
| Probe ID | p-Value | Arrow | Fold | Gene |
|---|---|---|---|---|
| 205255_x_at | 3.09 × 10−8 | down | 2.96 | TCF7 |
| 205456_at | 5.31 × 10−8 | down | 2.88 | CD3E |
| 205488_at | 1.01 × 10−5 | down | 2.87 | GZMA |
| 205495_s_at | 5.33 × 10−10 | down | 4.38 | GNLY |
| 205758_at | 1.20 × 10−7 | down | 3.26 | CD8A |
| 206666_at | 1.84 × 10−7 | down | 3.45 | GZMK |
| 206804_at | 1.10 × 10−15 | down | 5.12 | CD3G |
| 207460_at | 3.78 × 10−9 | down | 2.50 | GZMM |
| 208003_s_at | 5.52 × 10−18 | down | 12.04 | NFAT5 |
| 209671_x_at | 3.58 × 10−8 | down | 2.77 | TRAC |
| 209813_x_at | 1.49 × 10−9 | down | 4.42 | TARP |
| 210164_at | 8.91 × 10−9 | down | 3.76 | GZMB |
| 210321_at | 8.94 × 10−10 | down | 5.80 | GZMH |
| 210370_s_at | 1.34 × 10−7 | down | 2.48 | LY9 |
| 210556_at | 4.68 × 10−8 | down | 2.85 | NFATC3 |
| 210972_x_at | 1.78 × 10−7 | down | 2.88 | TRAC/J17/V20 |
| 211796_s_at | 6.35 × 10−6 | down | 2.93 | TRBC1/C2 |
| 212759_s_at | 3.98 × 10−16 | down | 3.93 | TCF7L2 |
| 213193_x_at | 2.53 × 10−6 | down | 2.92 | TRBC1 |
| 213539_at | 1.00 × 10−8 | down | 3.19 | CD3D |
| 214617_at | 2.22 × 10−6 | down | 2.65 | PRF1 |
| 216191_s_at | 4.71 × 10−7 | down | 4.76 | TRDV3 |
| 216920_s_at | 2.28 × 10−10 | down | 5.34 | TARP/TRGC2 |
| 217143_s_at | 1.26 × 10−8 | down | 6.06 | TRD@ |
| 217527_s_at | 2.12 × 10−13 | down | 5.80 | NFATC2IP |
| 220684_at | 7.39 × 10−9 | down | 2.08 | TBX21 |
| 220704_at | 2.15 × 10−10 | down | 5.69 | IKZF1 |
| 214032_at | 6.60 × 10−8 | down | 2.52 | ZAP70 |
| 204891_s_at | 4.58 × 10−8 | down | 3.31 | LCK |
| 205831_at | 4.40 × 10−10 | down | 3.93 | CD2 |
| 201565_s_at | 8.13 × 10−13 | down | 4.17 | ID2 |
| 213931_at | 7.33 × 10−8 | down | 3.55 | ID2/2B |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-J.; Lu, J.-J.; Lin, C.-P.; Hu, W.-C. Microarray Analysis Reveals Sepsis Is a Syndrome with Hyperactivity of TH17 Immunity, with Over-Presentation of the Treg Cell Cytokine TGF-β. Curr. Issues Mol. Biol. 2025, 47, 435. https://doi.org/10.3390/cimb47060435
Chen Y-J, Lu J-J, Lin C-P, Hu W-C. Microarray Analysis Reveals Sepsis Is a Syndrome with Hyperactivity of TH17 Immunity, with Over-Presentation of the Treg Cell Cytokine TGF-β. Current Issues in Molecular Biology. 2025; 47(6):435. https://doi.org/10.3390/cimb47060435
Chicago/Turabian StyleChen, Yu-Ju, Jang-Jih Lu, Chih-Pei Lin, and Wan-Chung Hu. 2025. "Microarray Analysis Reveals Sepsis Is a Syndrome with Hyperactivity of TH17 Immunity, with Over-Presentation of the Treg Cell Cytokine TGF-β" Current Issues in Molecular Biology 47, no. 6: 435. https://doi.org/10.3390/cimb47060435
APA StyleChen, Y.-J., Lu, J.-J., Lin, C.-P., & Hu, W.-C. (2025). Microarray Analysis Reveals Sepsis Is a Syndrome with Hyperactivity of TH17 Immunity, with Over-Presentation of the Treg Cell Cytokine TGF-β. Current Issues in Molecular Biology, 47(6), 435. https://doi.org/10.3390/cimb47060435