


IL-1R2 as a Precision Therapeutic Target in Sepsis: Molecular Insights into Immune Regulation



Abstract
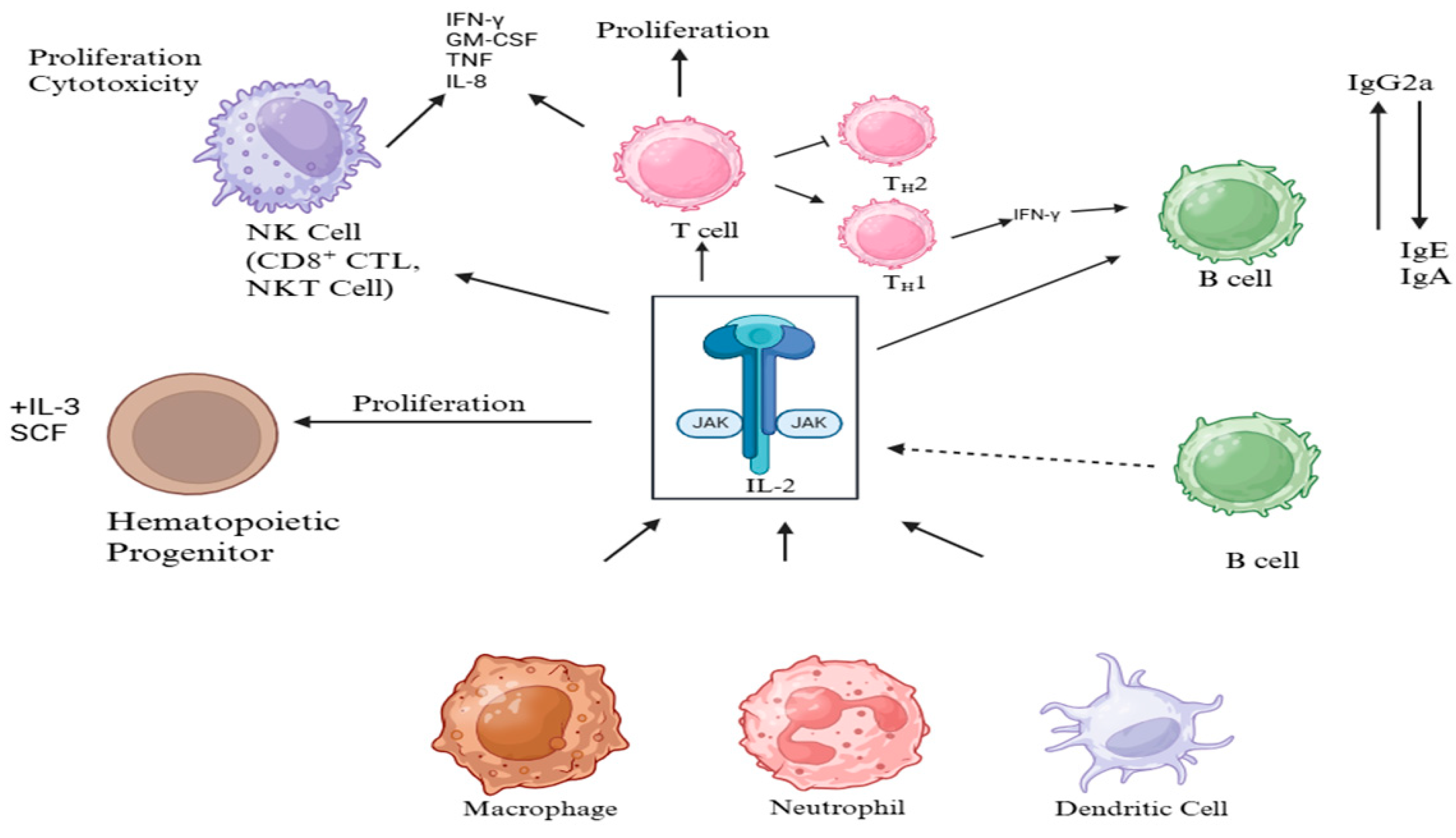
1. Introduction
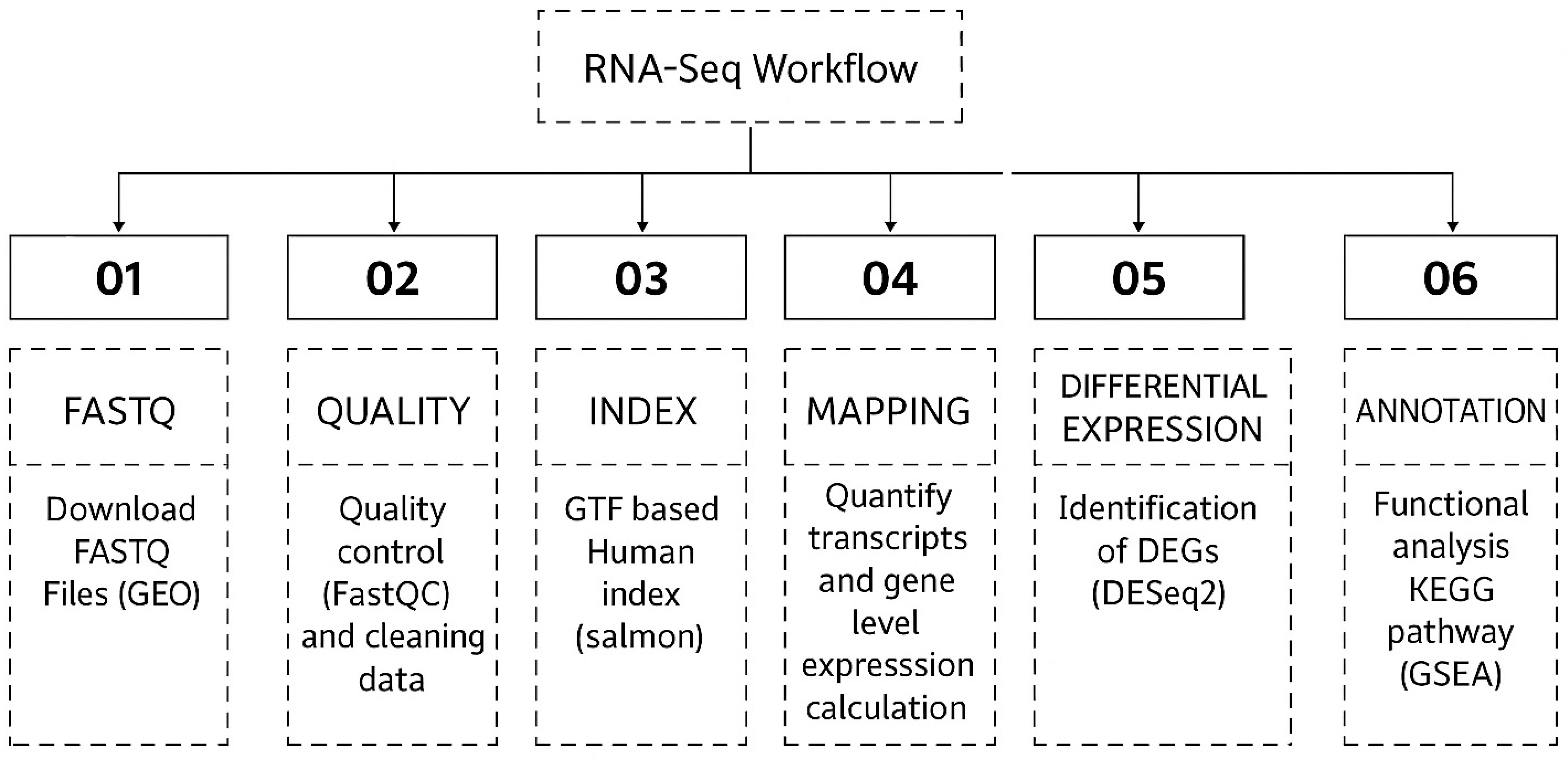
2. Materials and Methods
2.1. Data Collection
2.2. Sequence Alignment and Transcriptome Assembly
3. Results
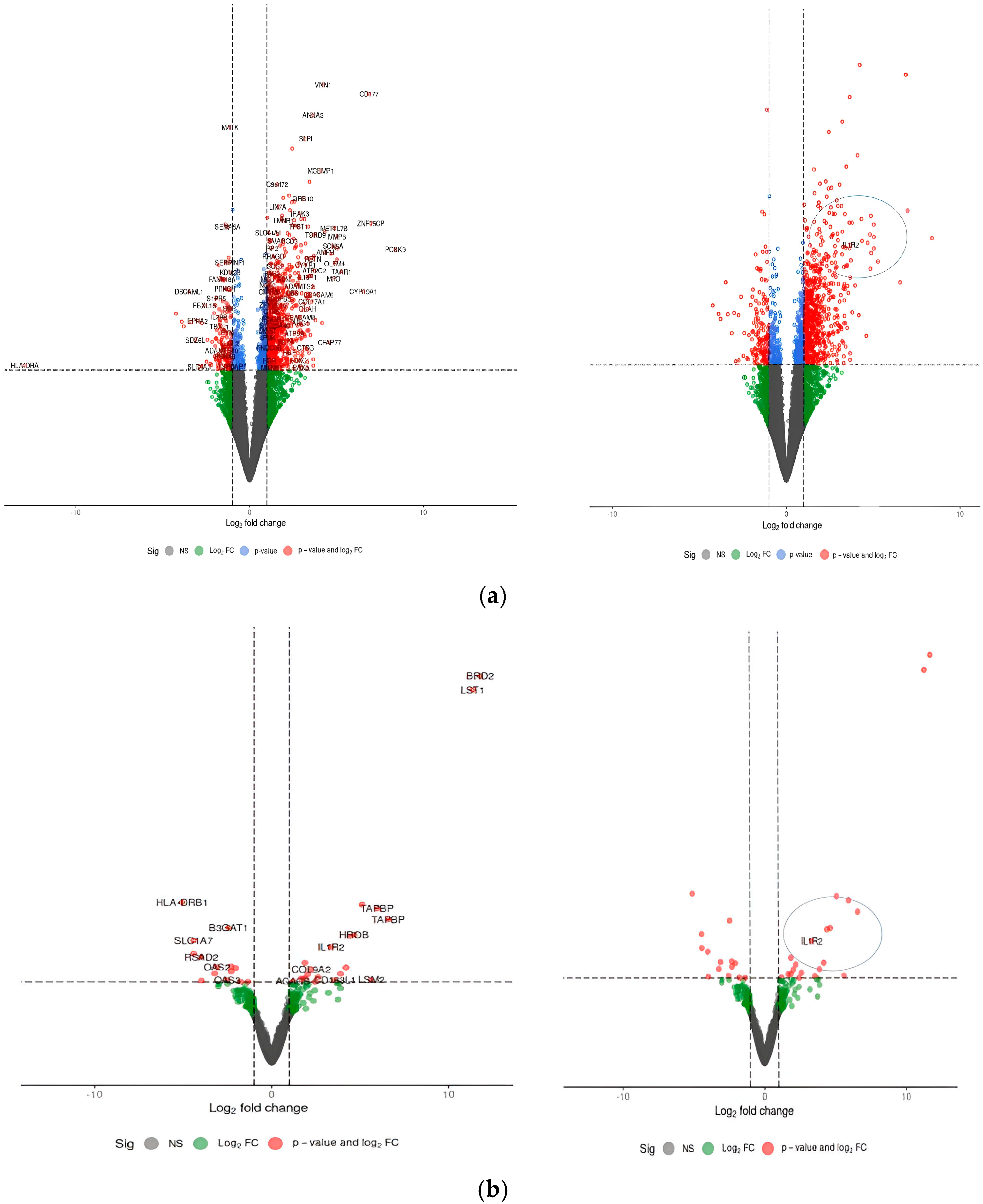
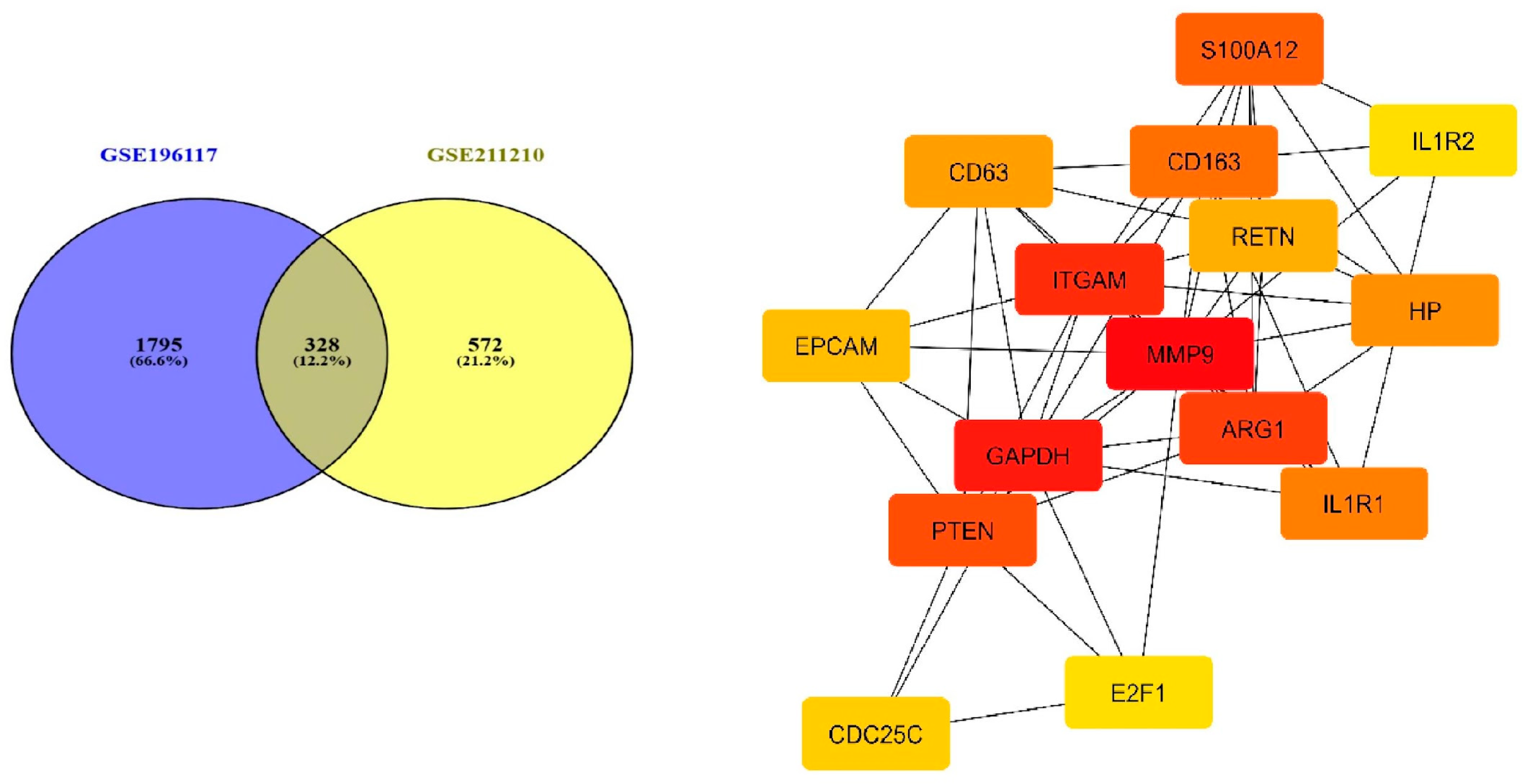
3.1. Determination of Common Differentially Expressed Genes
3.2. Analysis of the Pathways Associated with Significantly Dysregulated Transcriptome Data of Both the Datasets
3.3. Gene Ontologies and Enrichment
3.4. Network Analysis and Key Inflammatory Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NLRP3 | NLR Family Pyrin Domain Containing 3 |
| ROS | Reactive Oxygen Species |
| OxS | Oxidative Stress |
| Th17 | T-helper 17 Cells |
| PBMCs | Peripheral Blood Mononuclear Cells |
References
- Caraballo, C.; Jaimes, F. Organ dysfunction in sepsis: An ominous trajectory from infection to death. Yale J. Biol. Med. 2019, 92, 629. [Google Scholar]
- Remick, D.G. Pathophysiology of sepsis. Am. J. Pathol. 2007, 170, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Remick, D.G.; Bolgos, G.; Copeland, S.; Siddiqui, J. Role of interleukin-6 in mortality from and physiologic response to sepsis. Infect. Immun. 2005, 73, 2751–2757. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Cai, J.-P.; Luo, Y.-L.; Chen, C.; Zhang, S. The specific roles of JAK/STAT signaling pathway in sepsis. Inflammation 2015, 38, 1599–1608. [Google Scholar] [CrossRef]
- Chaudhry, H.; Zhou, J.; Zhong, Y.; Ali, M.M.; McGuire, F.; Nagarkatti, P.S.; Nagarkatti, M. Role of cytokines as a double-edged sword in sepsis. In Vivo 2013, 27, 669–684. [Google Scholar]
- Ge, Y.; Huang, M.; Yao, Y.-M. Recent advances in the biology of IL-1 family cytokines and their potential roles in development of sepsis. Cytokine Growth Factor. Rev. 2019, 45, 24–34. [Google Scholar] [CrossRef]
- Liu, D.; Huang, S.-Y.; Sun, J.-H.; Zhang, H.-C.; Cai, Q.-L.; Gao, C.; Li, L.; Cao, J.; Xu, F.; Zhou, Y. Sepsis-induced immunosuppression: Mechanisms, diagnosis and current treatment options. Mil. Med. Res. 2022, 9, 56. [Google Scholar] [CrossRef]
- Ye, C.; Huang, X.; Tong, Y.; Chen, Y.; Zhao, X.; Xie, W.; Wang, Y.; Wang, J.; Zhang, A.; Mo, Y. Overexpression of ALKBH5 Alleviates LPS-Induced Neuroinflammation via Increasing NFKBIA. Int. Immunopharmacol. 2025, 144, 113598. [Google Scholar] [CrossRef]
- Zhao, P.; Yao, R.; Yang, J.; Wen, W.; Yao, Y.; Du, X. Efficacy and safety of clarithromycin for patients with sepsis or septic shock: A systematic review and meta-analysis. Emerg. Crit. Care Med. 2024, 4, 90–96. [Google Scholar] [CrossRef]
- Karakike, E.; Scicluna, B.P.; Roumpoutsou, M.; Mitrou, I.; Karampela, N.; Karageorgos, A.; Psaroulis, K.; Massa, E.; Pitsoulis, A.; Chaloulis, P.; et al. Effect of intravenous clarithromycin in patients with sepsis, respiratory and multiple organ dysfunction syndrome: A randomized clinical trial. Crit. Care 2022, 26, 183. [Google Scholar] [CrossRef]
- Yang, N.; Zhang, L.; Lv, J.; Niu, Z.; Liu, J.; Li, P.; Zhang, Z. ALKBH8 Suppresses Oxidative Stress and Ameliorates Inflammation via Blocking NFκB/NLRP3 Axis in Sepsis. SSRN Electron. J. 2021. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Mbandi, S.K.; Hesse, U.; Rees, D.J.G.; Christoffels, A. A glance at quality score: Implication for de novo transcriptome reconstruction of Illumina reads. Front. Genet. 2014, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Kryuchkova-Mostacci, N.; Robinson-Rechavi, M. A benchmark of gene expression tissue-specificity metrics. Brief. Bioinform. 2017, 18, 205–214. [Google Scholar] [CrossRef]
- Yu, G. enrichplot: Visualization of Functional Enrichment Result. 4. In Biomedical Knowledge Mining Book; R Package Version 1.24; 2024. Available online: https://yulab-smu.top/biomedical-knowledge-mining-book/ (accessed on 30 May 2025).
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Xu, S.; Hu, E.; Cai, Y.; Xie, Z.; Luo, X.; Zhan, L.; Tang, W.; Wang, Q.; Liu, B.; Wang, R.; et al. Using clusterProfiler to characterize multiomics data. Nat. Protoc. 2024, 19, 3292–3320. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.I.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Khoshbakht, T.; Hussen, B.M.; Taheri, M.; Arefian, N. Regulatory Role of Non-Coding RNAs on Immune Responses During Sepsis. Front. Immunol. 2021, 12, 798713. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING Database in 2023: Protein–Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res. 2023, 51, D638–D664. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef]
- Lang, Y.; Jiang, Y.; Gao, M.; Wang, W.; Wang, N.; Wang, K.; Zhang, H.; Chen, G.; Liu, K.; Liu, M. Interleukin-1 receptor 2: A new biomarker for sepsis diagnosis and gram-negative/gram-positive bacterial differentiation. Shock 2017, 47, 119–124. [Google Scholar] [CrossRef]
- Giri, J.G.; Wells, J.; Dower, S.K.; McCall, C.E.; Guzman, R.N.; Slack, J.; Bird, T.A.; Shanebeck, K.; Grabstein, K.H.; Sims, J.E. Elevated levels of shed type II IL-1 receptor in sepsis. Potential role for type II receptor in regulation of IL-1 responses. J. Immunol. 1994, 153, 5802–5809. [Google Scholar] [CrossRef]
- Aksu, M.D.; van der Ent, T.; Zhang, Z.; Riza, A.L.; de Nooijer, A.H.; Ricaño-Ponce, I.; Janssen, N.; Engel, J.J.; Streata, I.; Dijkstra, H.; et al. Regulation of plasma soluble receptors of TNF and IL-1 in patients with COVID-19 differs from that observed in sepsis. J. Infect. 2024, 89, 106300. [Google Scholar] [CrossRef]
- Xue, M.; Xie, J.; Liu, L.; Huang, Y.; Guo, F.; Xu, J.; Yang, Y.; Qiu, H. Early and dynamic alterations of Th2/Th1 in previously immunocompetent patients with community-acquired severe sepsis: A prospective observational study. J. Transl. Med. 2019, 17, 57. [Google Scholar] [CrossRef]
- Mithal, L.B.; Arshad, M.; Swigart, L.R.; Khanolkar, A.; Ahmed, A.; Coates, B.M. Mechanisms and modulation of sepsis-induced immune dysfunction in children. Pediatr. Res. 2022, 91, 447–453. [Google Scholar] [CrossRef]
- Delano, M.J.; Ward, P.A. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol. Rev. 2016, 274, 330–353. [Google Scholar] [CrossRef] [PubMed]
- Zanders, L.; Kny, M.; Hahn, A.; Schmidt, S.; Wundersitz, S.; Todiras, M.; Lahmann, I.; Bandyopadhyay, A.; Wollersheim, T.; Kaderali, L.; et al. Sepsis induces interleukin 6, gp130/JAK2/STAT3, and muscle wasting. J. Cachexia Sarcopenia Muscle 2022, 13, 713–727. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Dong, H.; Wu, C.; Zhong, Y.; Li, J. The role of NLRP3 inflammasome in sepsis: A potential therapeutic target. Int. Immunopharmacol. 2023, 115, 109697. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Shiono, S.; Joo, A.; Yoshimura, A. Control mechanism of JAK/STAT signal transduction pathway. FEBS Lett. 2003, 534, 190–196. [Google Scholar] [CrossRef]
- Chinen, T.; Kannan, A.K.; Levine, A.G.; Fan, X.; Klein, U.; Zheng, Y.; Gasteiger, G.; Feng, Y.; Fontenot, J.D.; Rudensky, A.Y. An essential role for the IL-2 receptor in Treg cell function. Nat. Immunol. 2016, 17, 1322–1333. [Google Scholar] [CrossRef]
- Ying, L.; Yao, Y.; Lv, H.; Lu, G.; Zhang, Q.; Yang, Y.; Zhou, J. IL-17A contributes to skeletal muscle atrophy in lung cancer-induced cachexia via JAK2/STAT3 pathway. Am. J. Physiol. Cell Physiol. 2022, 322, C814–C824. [Google Scholar] [CrossRef]
- Mizgerd, J.P.; Skerrett, S.J.; Allen, E.; Shapiro, S.D.; Doerschuk, C.M. Modulation of endotoxin-induced NF-κB activation in lung and liver through TNF type 1 and IL-1 receptors. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L1095–L1100. [Google Scholar]
- Supino, D.; Minute, L.; Mariancini, A.; Riva, F.; Magrini, E.; Garlanda, C. Negative regulation of the IL-1 system by IL-1R2 and IL-1R8: Relevance in pathophysiology and disease. Front. Immunol. 2022, 13, 804641. [Google Scholar] [CrossRef]
- Hofmaenner, D.A.; Kleyman, A.; Press, A.; Bauer, M.; Singer, M. The many roles of cholesterol in sepsis: A review. Am. J. Respir. Crit. Care Med. 2022, 205, 388–396. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. The Metabolic Basis of Immune Dysfunction Following Sepsis and Trauma. Front. Immunol. 2020, 11, 1043. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J.; Chen, J.; Li, J.; Zhang, Y.; Wang, Y.; Wang, Y. Monocyte-Macrophage Membrane Expression of IL-1R2 Is a Severity Marker in Sepsis. Front. Immunol. 2024, 15, 11982311. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO Accession | GSE196117 | GSE211210 |
|---|---|---|
| Sample No | n = 40 | n = 10 |
| Title of study | Transcriptional profiling of whole blood leukocytes obtained from critically ill patients with sepsis treated with clarithromycin, or not, and healthy participants | ALKBH8 suppresses oxidative stress and ameliorates inflammation via blocking NFκB/NLRP3 axis in Sepsis |
| Differential Genes with adjusted p-value < 0.05 | 2315 genes Upregulated | 932 genes Upregulated |
| 1728 genes down regulated | 707 genes down regulated |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dave, K.; Munteanu, C.R. IL-1R2 as a Precision Therapeutic Target in Sepsis: Molecular Insights into Immune Regulation. Curr. Issues Mol. Biol. 2025, 47, 429. https://doi.org/10.3390/cimb47060429
Dave K, Munteanu CR. IL-1R2 as a Precision Therapeutic Target in Sepsis: Molecular Insights into Immune Regulation. Current Issues in Molecular Biology. 2025; 47(6):429. https://doi.org/10.3390/cimb47060429
Chicago/Turabian StyleDave, Kirtan, and Cristian R. Munteanu. 2025. "IL-1R2 as a Precision Therapeutic Target in Sepsis: Molecular Insights into Immune Regulation" Current Issues in Molecular Biology 47, no. 6: 429. https://doi.org/10.3390/cimb47060429
APA StyleDave, K., & Munteanu, C. R. (2025). IL-1R2 as a Precision Therapeutic Target in Sepsis: Molecular Insights into Immune Regulation. Current Issues in Molecular Biology, 47(6), 429. https://doi.org/10.3390/cimb47060429