
Testing Protein Stress Signals in Peripheral Immunocytes Under the Same Treatment Capable of Decreasing the Incidence of Alzheimer’s Disease in Bladder Cancer Patients


Abstract
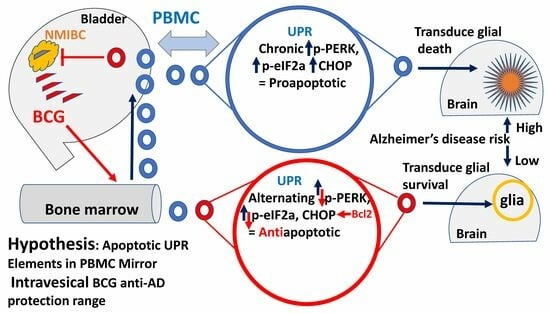
1. Introduction
1.1. Alzheimer’s Disease Prevention by Bacillus Calmette–Guérin
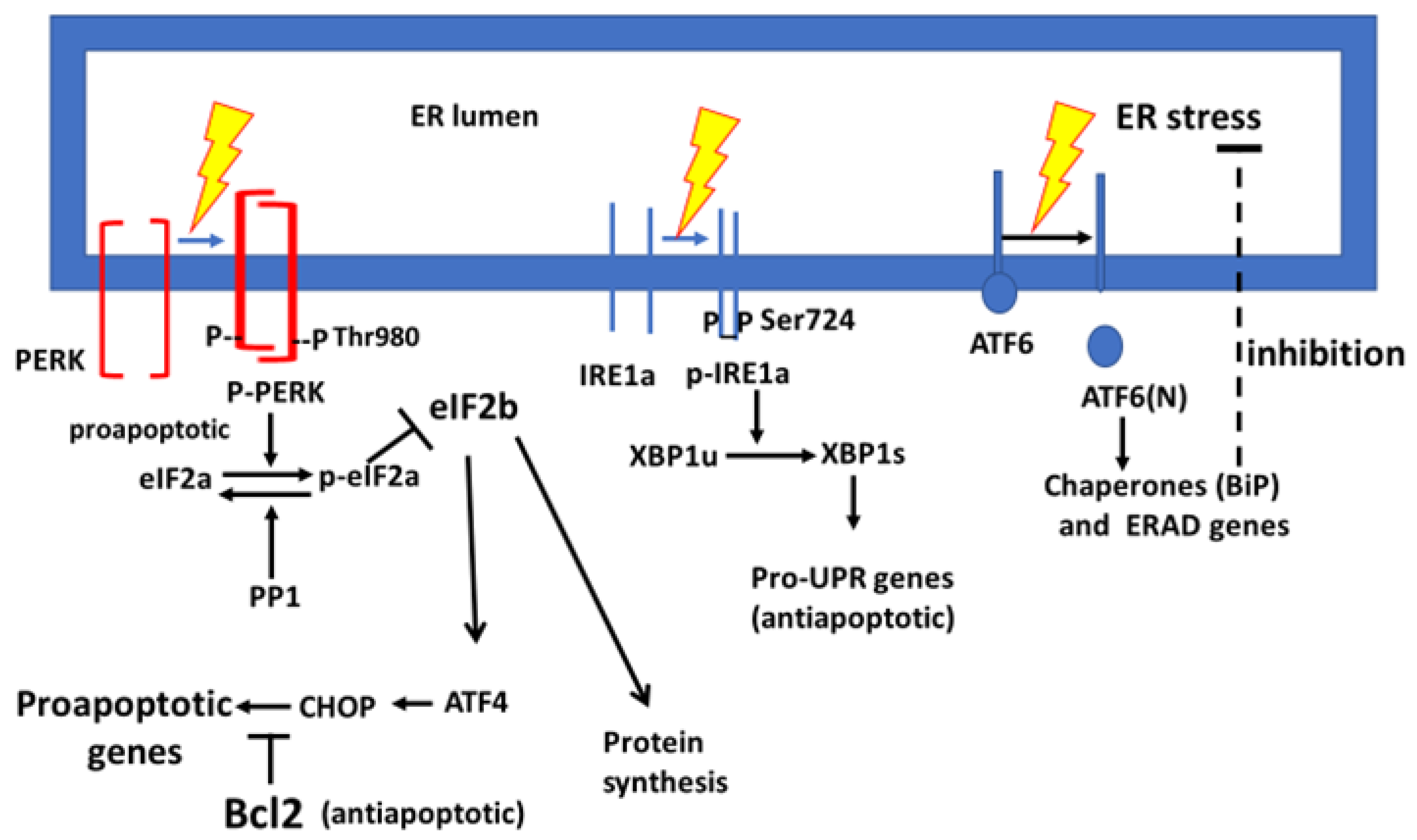
1.2. Involvement of the Unfolded Protein Response in Alzheimer’s Disease
2. Materials and Methods
2.1. Treatment Protocol
2.2. Peripheral Blood Mononuclear Cell Sampling
2.3. Thawing of Peripheral Blood Mononuclear Cells and Protein Extraction
2.4. Preparation of Immune-Electrophoresis Samples
2.5. Antibodies
2.6. Analysis of the Results
2.7. Presentation of the Results
2.8. Calculation of the Score
3. Results
3.1. Presentation of the BCG Impact on the UPR
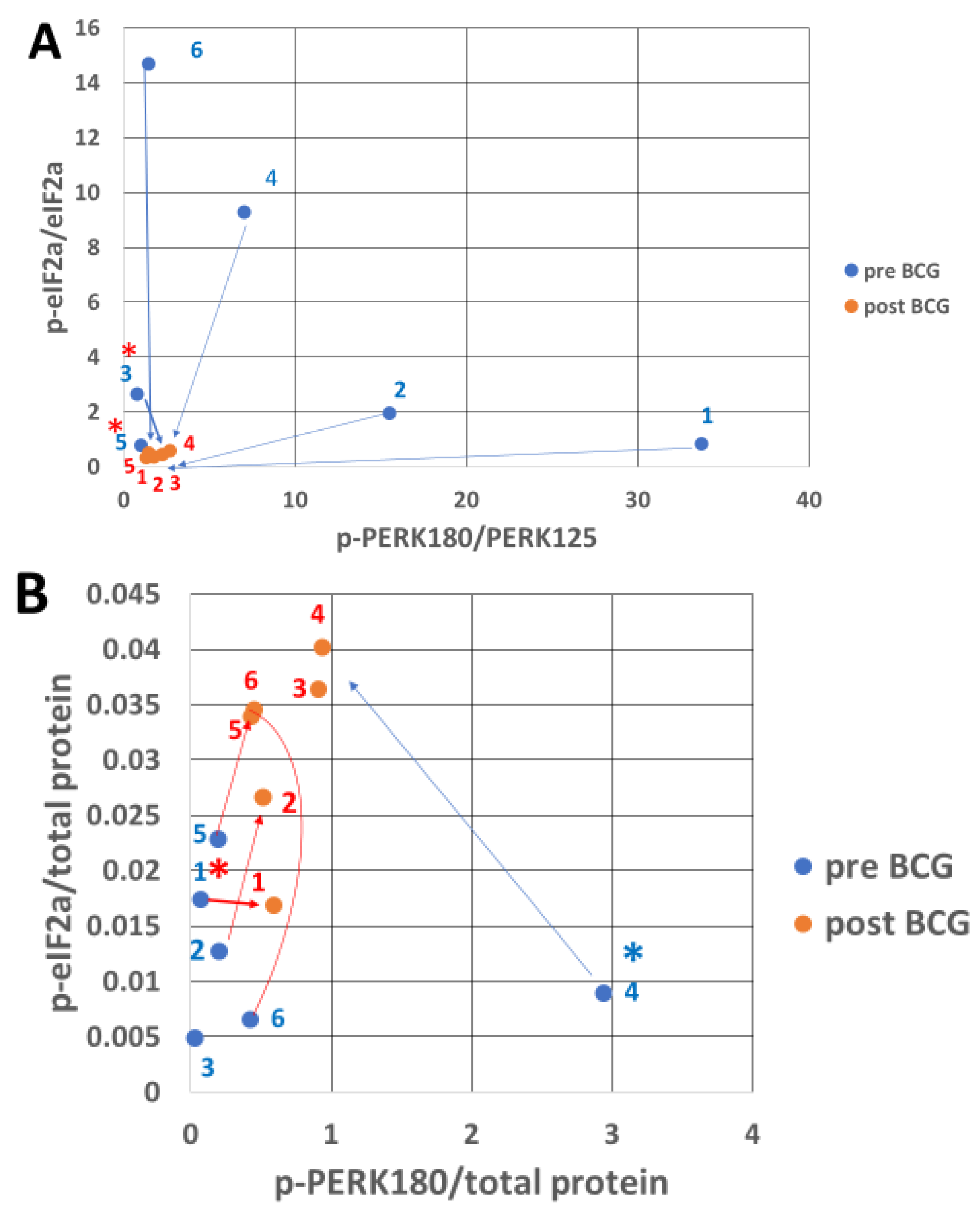
3.2. PERK Response to Endoplasmic Reticulum Stress After Bacillus Calmette–Guérin Treatment
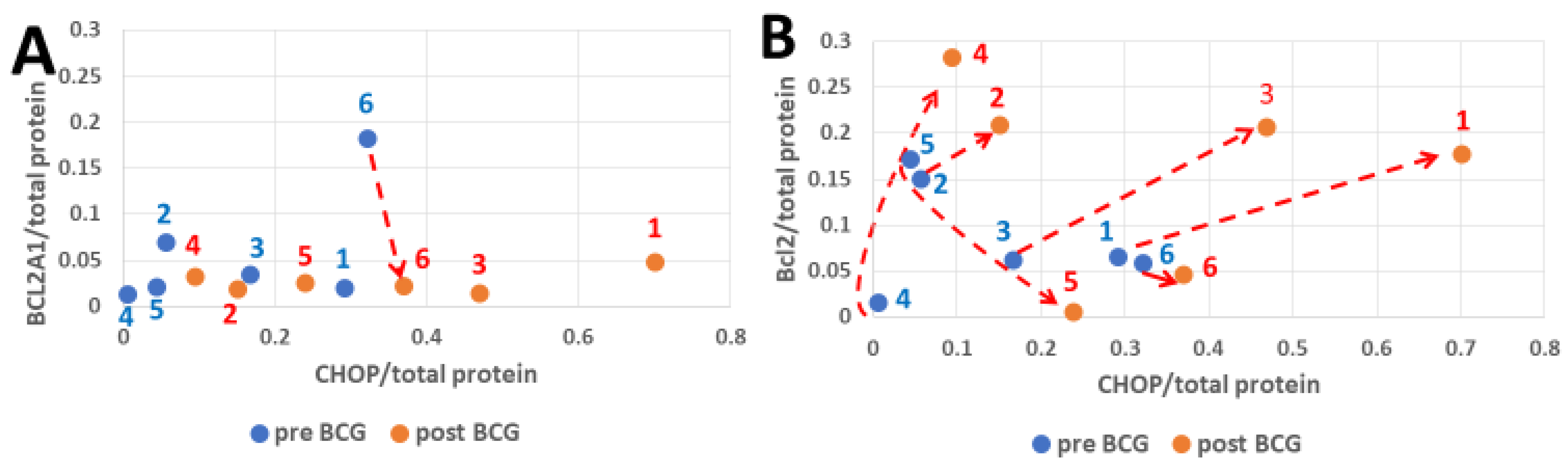
3.3. CHOP and BCL2 Expression After Bacillus Calmette–Guérin Treatment
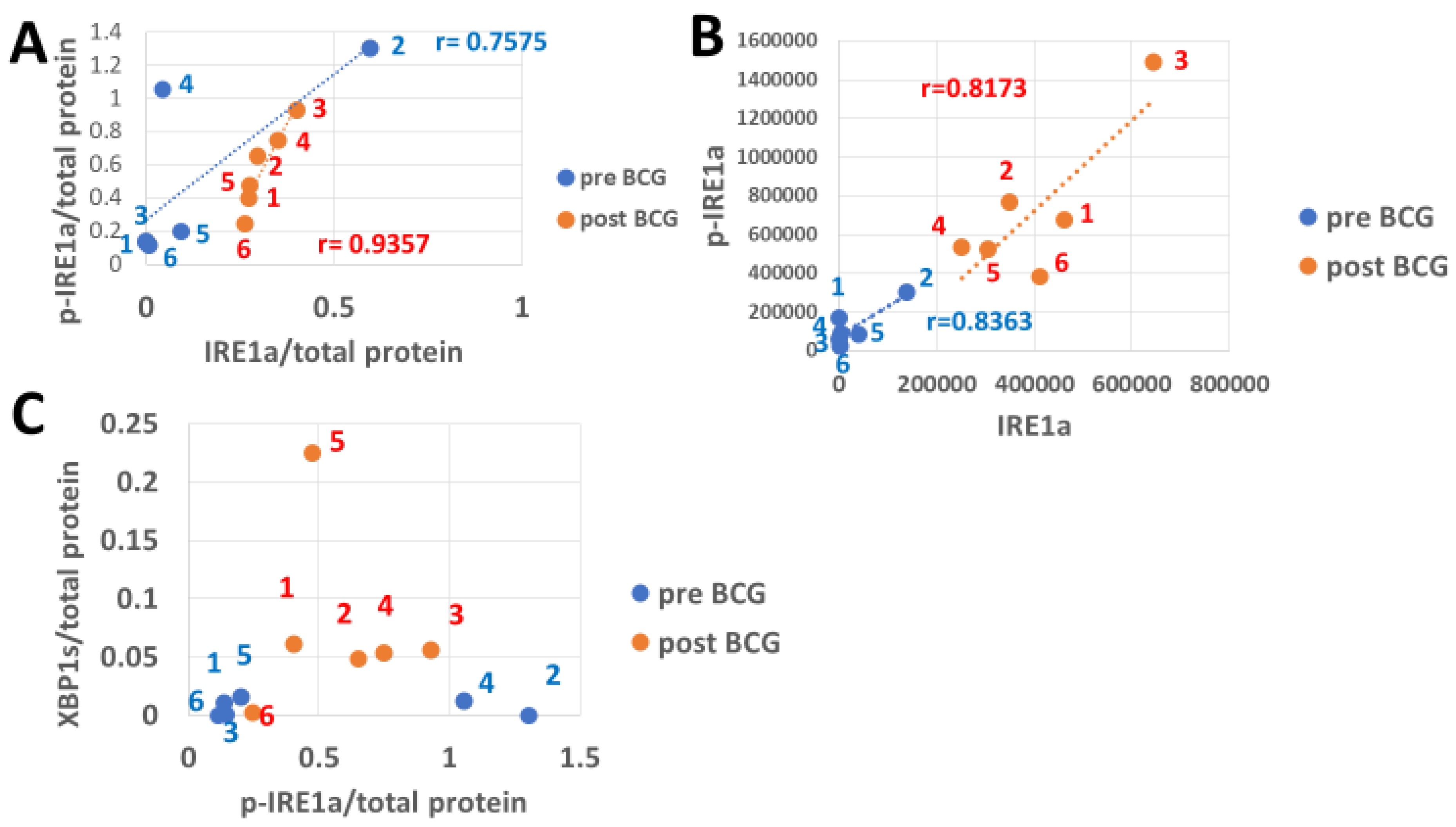
3.4. The Response of IRE1a and Its XBP1 Substrate to Bacillus Calmette–Guérin
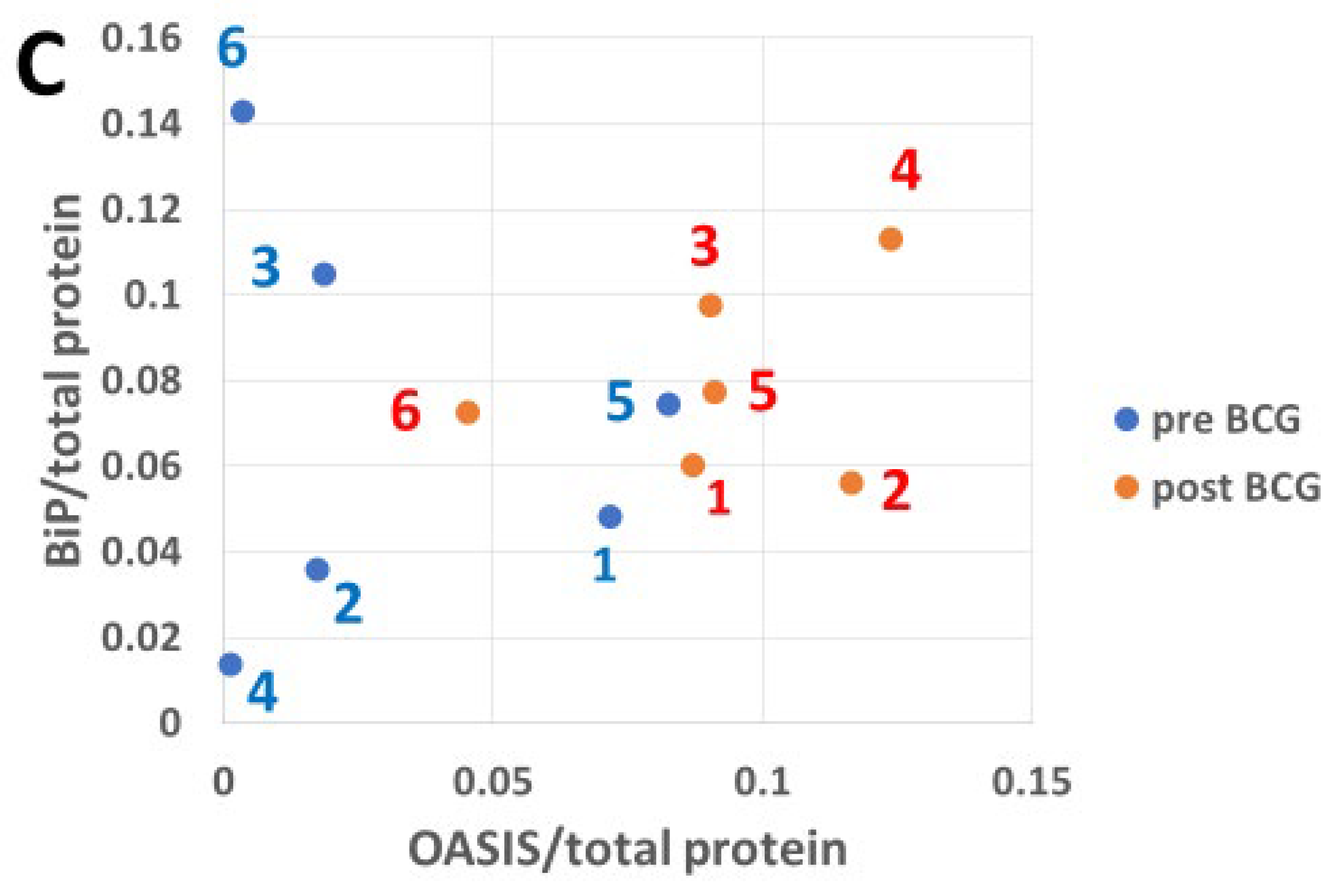
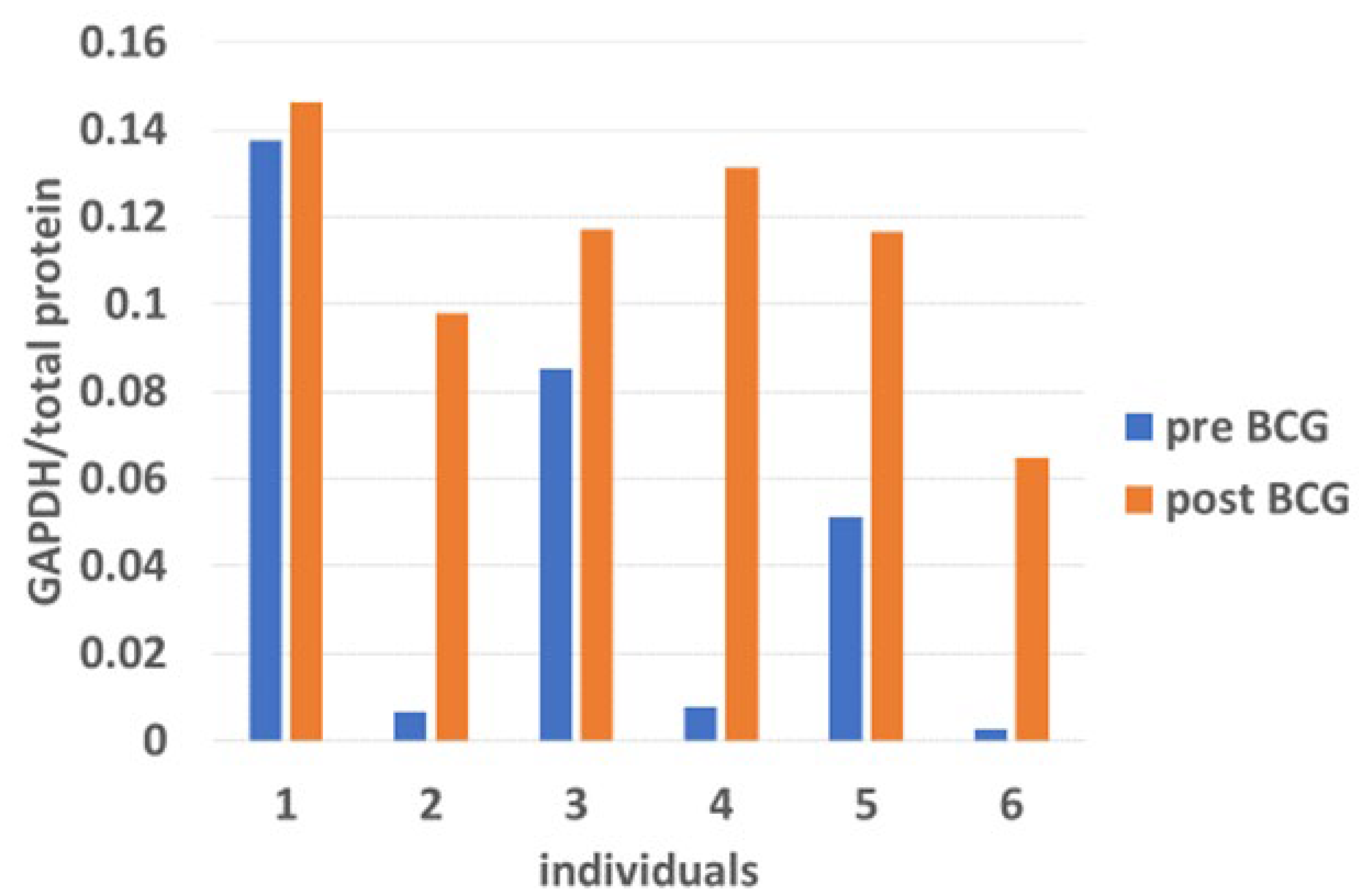
3.5. The Impact of Bacillus Calmette–Guérin on BiP Expression in Response to Endoplasmic Reticulum Stress-Induced Transcription Factors
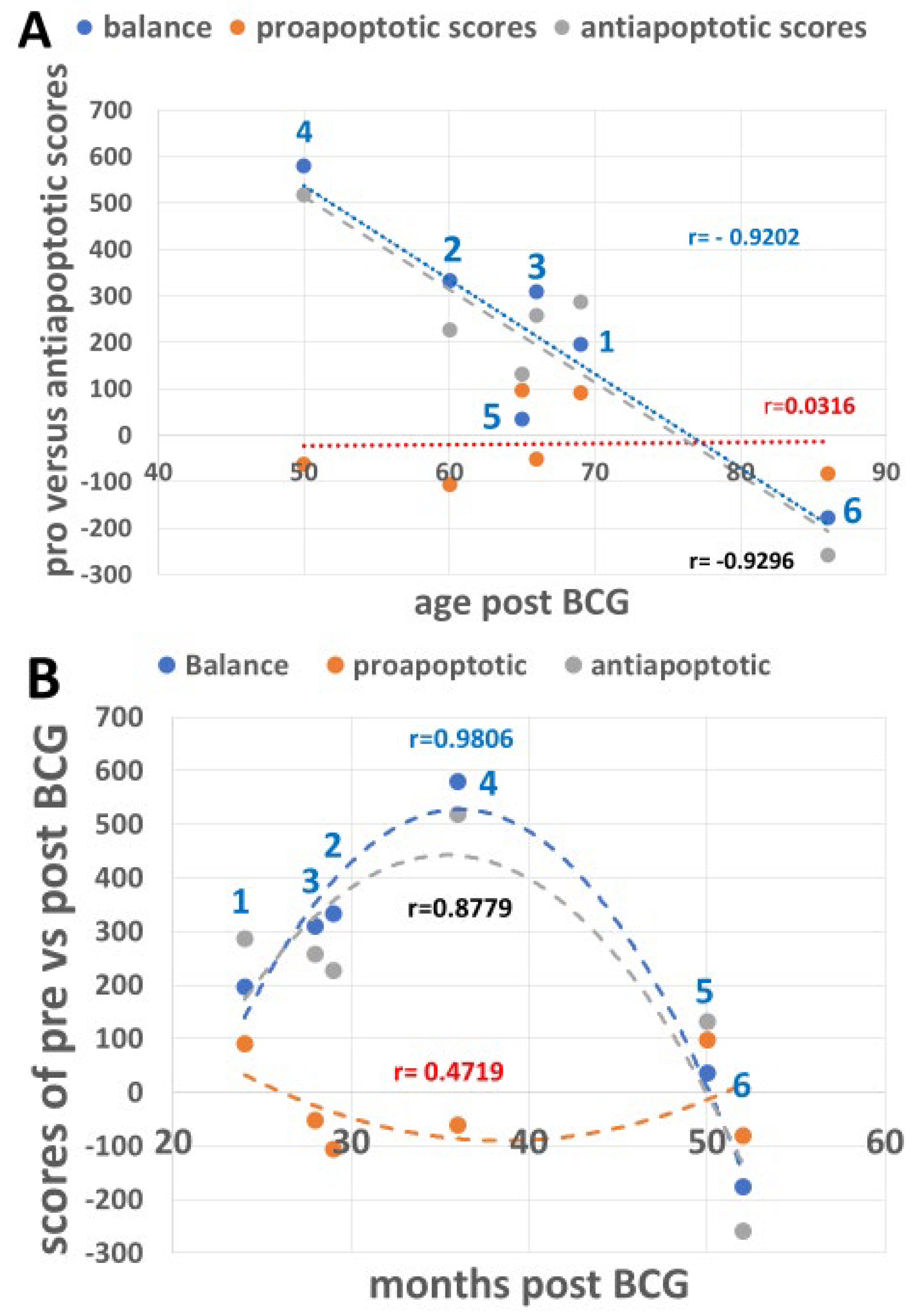
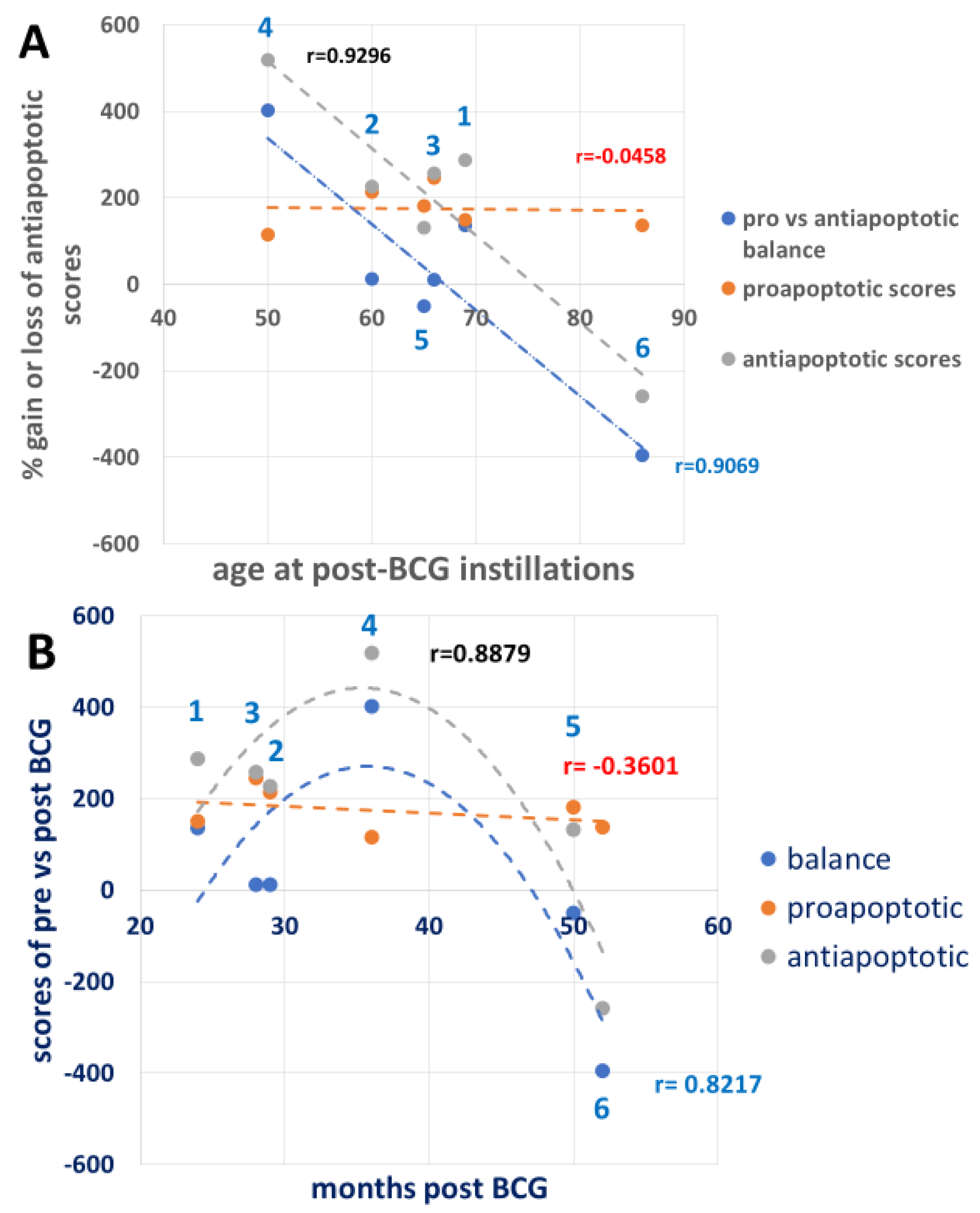
3.6. Combining the Bacillus Calmette–Guérin Apoptotic and Antiapoptotic Effects
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Milosevic, R.; Milovic, N.; Aleksic, P.; Lazic, M.; Cerovic, S.; Bancevic, V.; Kosevic, B.; Maric, P.; Spasic, A.; Simic, D.; et al. Difference in recurrence frequencies of non-muscle-invasive-bladder tumors depending on optimal usage of intravesical immunotherapy of bacillus Calmette-Guerin. Vojnosanit. Pregl. 2015, 72, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Redelman-Sidi, G. BCG in Bladder Cancer Immunotherapy. Cancers 2022, 14, 3073. [Google Scholar] [CrossRef]
- Beatty, J.D.; Islam, S.; North, M.E.; Knight, S.C.; Ogden, C.W. Urine dendritic cells: A noninvasive probe for immune activity in bladder cancer? BJU Int. 2004, 94, 1377–1383. [Google Scholar] [CrossRef]
- Suttmann, H.; Riemensberger, J.; Bentien, G.; Schmaltz, D.; Stöckle, M.; Jocham, D.; Böhle, A.; Brandau, S. Neutrophil granulocytes are required for effective Bacillus Calmette-Guerin immunotherapy of bladder cancer and orchestrate local immune responses. Cancer Res. 2006, 66, 8250–8257. [Google Scholar] [CrossRef]
- Moorlag, S.J.; Rodriguez-Rosales, Y.A.; Gillard, J.; Fanucchi, S.; Theunissen, K.; Novakovic, B.; de Bont, C.M.; Negishi, Y.; Fok, E.T.; Kalafati, L.; et al. BCG Vaccination Induces Long-Term Functional Reprogramming of Human Neutrophils. Cell Rep. 2020, 33, 108387. [Google Scholar] [CrossRef] [PubMed]
- Benn, C.S.; Fisker, A.B.; Rieckmann, A.; Sorup, S.; Aaby, P. Vaccinology: Time to change the paradigm? Lancet Infect. Dis. 2020, 20, e274–e283. [Google Scholar] [CrossRef]
- Lange, C.; Aaby, P.; Behr, M.A.; Donald, P.R.; Kaufmann, S.H.E.; Netea, M.G.; Mandalakas, A.M. 100 years of Mycobacterium bovis bacille Calmette-Guerin. Lancet Infect. Dis. 2022, 22, e2–e12. [Google Scholar] [CrossRef]
- Gofrit, O.N.; Bercovier, H.; Klein, B.Y.; Cohen, I.R.; Ben-Hur, T.; Greenblatt, C.L. Can immunization with Bacillus Calmette-Guerin (BCG) protect against Alzheimer’s disease? Med. Hypotheses 2019, 123, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Gofrit, O.N.; Klein, B.Y.; Cohen, I.R.; Ben-Hur, T.; Greenblatt, C.L.; Bercovier, H. Bacillus Calmette-Guerin (BCG) therapy lowers the incidence of Alzheimer’s disease in bladder cancer patients. PLoS ONE 2019, 14, e0224433. [Google Scholar] [CrossRef]
- Gofrit, O.N.; Greenblatt, C.L.; Klein, B.Y.; Ben-Hur, T.; Bercovier, H. Can immunization with BCG delay the onset of Alzheimer’s disease? Geriatr. Gerontol. Int. 2023, 23, 889–890. [Google Scholar] [CrossRef]
- Scherrer, J.F.; Salas, J.; Wiemken, T.L.; Hoft, D.F.; Jacobs, C.; Morley, J.E. Impact of herpes zoster vaccination on incident dementia: A retrospective study in two patient cohorts. PLoS ONE 2021, 16, e0257405. [Google Scholar] [CrossRef] [PubMed]
- Poynton-Smith, E.; Orrell, M.; Morley, J.E.; Scherrer, J.F. Could routine vaccinations prevent dementia? Int. J. Geriatr. Psychiatry 2023, 38, e5978. [Google Scholar] [CrossRef]
- Adams, K.; Weber, K.S.; Johnson, S.M. Exposome and Immunity Training: How Pathogen Exposure Order Influences Innate Immune Cell Lineage Commitment and Function. Int. J. Mol. Sci. 2020, 21, 8462. [Google Scholar] [CrossRef] [PubMed]
- Kalafati, L.; Kourtzelis, I.; Schulte-Schrepping, J.; Li, X.; Hatzioannou, A.; Grinenko, T.; Hagag, E.; Sinha, A.; Has, C.; Dietz, S.; et al. Innate Immune Training of Granulopoiesis Promotes Anti-tumor Activity. Cell 2020, 183, 771–785.e12. [Google Scholar] [CrossRef]
- Ansari, N.; Khodagholi, F. Molecular mechanism aspect of ER stress in Alzheimer’s disease: Current approaches and future strategies. Curr. Drug Targets. 2013, 14, 114–122. [Google Scholar] [CrossRef]
- Cornejo, V.H.; Hetz, C. The unfolded protein response in Alzheimer’s disease. Semin. Immunopathol. 2013, 35, 277–292. [Google Scholar] [CrossRef]
- Di Domenico, F.; Coccia, R.; Cocciolo, A.; Murphy, M.P.; Cenini, G.; Head, E.; Butterfield, D.A.; Giorgi, A.; Schinina, M.E.; Mancuso, C.; et al. Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: Redox proteomics analysis of human brain. Biochim. Biophys. Acta. 2013, 1832, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef]
- Karran, E.; De Strooper, B. The amyloid cascade hypothesis: Are we poised for success or failure? J. Neurochem. 2016, 139 (Suppl. 2), 237–252. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Endres, K.; Reinhardt, S. ER-stress in Alzheimer’s disease: Turning the scale? Am. J. Neurodegener. Dis. 2013, 2, 247–265. [Google Scholar] [PubMed]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.I.; Kaufman, R.J.; Back, S.H.; Moon, J.Y. Development of a Reporter System Monitoring Regulated Intramembrane Proteolysis of the Transmembrane bZIP Transcription Factor ATF6alpha. Mol. Cells 2019, 42, 783–793. [Google Scholar]
- Urra, H.; Hetz, C. Fine-tuning PERK signaling to control cell fate under stress. Nat. Struct. Mol. Biol. 2017, 24, 789–790. [Google Scholar] [CrossRef]
- Hetz, C.; Martinon, F.; Rodriguez, D.; Glimcher, L.H. The unfolded protein response: Integrating stress signals through the stress sensor IRE1alpha. Physiol. Rev. 2011, 91, 1219–1243. [Google Scholar] [CrossRef] [PubMed]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef]
- Hong, F.; Liu, B.; Wu, B.X.; Morreall, J.; Roth, B.; Davies, C.; Sun, S.; Diehl, J.A.; Li, Z. CNPY2 is a key initiator of the PERK-CHOP pathway of the unfolded protein response. Nat. Struct. Mol. Biol. 2017, 24, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef]
- Vellanki, R.N.; Zhang, L.; Volchuk, A. OASIS/CREB3L1 is induced by endoplasmic reticulum stress in human glioma cell lines and contributes to the unfolded protein response, extracellular matrix production, and cell migration. PLoS ONE 2013, 8, e54060. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Mendez, A.; Almeida, A.; Fernandez, E.; Maestre, C.; Moncada, S.; Bolanos, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef]
- Allaman, I.; Belanger, M.; Magistretti, P.J. Astrocyte-neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87. [Google Scholar] [CrossRef]
- Belanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Berry, K.; Farias-Itao, D.S.; Grinberg, L.T.; Plowey, E.D.; Schneider, J.A.; Rodriguez, R.D.; Suemoto, C.K.; Buckwalter, M.S. B and T Lymphocyte Densities Remain Stable with Age in Human Cortex. ASN Neuro 2021, 13, 17590914211018117. [Google Scholar] [CrossRef]
- Medina-Rodriguez, E.M.; Han, D.; Lowell, J.; Beurel, E. Stress promotes the infiltration of peripheral immune cells to the brain. Brain Behav. Immun. 2023, 111, 412–423. [Google Scholar] [CrossRef]
- Nguyen, J.N.; Chauhan, A. Bystanders or not? Microglia and lymphocytes in aging and stroke. Neural Regen. Res. 2023, 18, 1397–1403. [Google Scholar]
- van der Meer, J.W.; Joosten, L.A.; Riksen, N.; Netea, M.G. Trained immunity: A smart way to enhance innate immune defence. Mol. Immunol. 2015, 68, 40–44. [Google Scholar] [CrossRef]
- Kuhtreiber, W.M.; Faustman, D.L. BCG Therapy for Type 1 Diabetes: Restoration of Balanced Immunity and Metabolism. Trends Endocrinol. Metab. 2019, 30, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Kühtreiber, W.M.; Takahashi, H.; Keefe, R.C.; Song, Y.; Tran, L.; Luck, T.G.; Shpilsky, G.; Moore, L.; Sinton, S.M.; Graham, J.C.; et al. BCG Vaccinations Upregulate Myc, a Central Switch for Improved Glucose Metabolism in Diabetes. iScience 2020, 23, 101085. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M. Can immunotherapy treat neurodegeneration? Cancer Immunotherapy could guide treatment for Alzheimer’s disease. Science 2017, 357, 254–255. [Google Scholar] [CrossRef] [PubMed]
- Abellanas, M.A.; Purnapatre, M.; Burgaletto, C.; Schwartz, M. Monocyte-derived macrophages act as reinforcements when microglia fall short in Alzheimer’s disease. Nat. Neurosci. 2025, 28, 436–445. [Google Scholar] [CrossRef]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Scheper, W. Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neurodegener. Dis. 2012, 10, 212–215. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; Veerhuis, R.; Van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar] [CrossRef]
- Nijholt, D.A.; van Haastert, E.S.; Rozemuller, A.J.; Scheper, W.; Hoozemans, J.J. The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J. Pathol. 2012, 226, 693–702. [Google Scholar] [CrossRef]
- Doody, R.S.; Farlow, M.; Aisen, P.S. Alzheimer’s Disease Cooperative Study Data Analysis and Publication Committee Phase 3 Trials of Solanezumab and Bapineuzumab for Alzheimer’s Disease. N. Engl. J. Med. 2014, 370, 1460. [Google Scholar]
- Abisambra, J.F.; Jinwal, U.K.; Blair, L.J.; O’Leary, J.C., 3rd; Li, Q.; Brady, S.; Wang, L.; Guidi, C.E.; Zhang, B.; Nordhues, B.A.; et al. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 2013, 33, 9498–9507. [Google Scholar] [CrossRef]
- Kates, M.; Matoso, A.; Choi, W.; Baras, A.S.; Daniels, M.J.; Lombardo, K.; Brant, A.; Mikkilineni, N.; McConkey, D.J.; Kamat, A.M.; et al. Adaptive Immune Resistance to Intravesical BCG in Non-Muscle Invasive Bladder Cancer: Implications for Prospective BCG-Unresponsive Trials. Clin. Cancer Res. 2020, 26, 882–891. [Google Scholar] [CrossRef]
- Bedke, J.; Black, P.C.; Szabados, B.; Guerrero-Ramos, F.; Shariat, S.F.; Xylinas, E.; Brinkmann, J.; Blake-Haskins, J.A.; Cesari, R.; Redorta, J.P. Optimizing outcomes for high-risk, non-muscle-invasive bladder cancer: The evolving role of PD-(L)1 inhibition. Urol. Oncol. 2023, 41, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Reineke, L.C.; Komar, A.A.; Caprara, M.G.; Merrick, W.C. A small stem loop element directs internal initiation of the URE2 internal ribosome entry site in Saccharomyces cerevisiae. J. Biol. Chem. 2008, 283, 19011–19025. [Google Scholar] [CrossRef]
- Reineke, L.C.; Cao, Y.; Baus, D.; Hossain, N.M.; Merrick, W.C. Insights into the role of yeast eIF2A in IRES-mediated translation. PLoS ONE 2011, 6, e24492. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Xiao, B.; Shi, Y.; Han, F. PERK signalling pathway mediates single prolonged stress-induced dysfunction of medial prefrontal cortex neurons. Apoptosis 2017, 22, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Gow, A.; Wrabetz, L. CHOP and the endoplasmic reticulum stress response in myelinating glia. Curr. Opin. Neurobiol. 2009, 19, 505–510. [Google Scholar] [CrossRef]
- Hetz, C.A. ER stress signaling and the BCL-2 family of proteins: From adaptation to irreversible cellular damage. Antioxid. Redox Signal. 2007, 9, 2345–2355. [Google Scholar] [CrossRef]
- Wang, X.; Olberding, K.E.; White, C.; Li, C. Bcl-2 proteins regulate ER membrane permeability to luminal proteins during ER stress-induced apoptosis. Cell Death Differ. 2011, 18, 38–47. [Google Scholar] [CrossRef]
- Klein, B.Y.; Ben-David, I.; Gofrit, O.N.; Greenblatt, C.L. Repurposing peripheral immunocytes of Bacillus Calmette-Guérin-vaccinated melanoma patients to reveal Alzheimer’s disease mechanisms, possibly via the unfolded protein response. J. Alzheimer’s Dis. Rep. 2025, 9, 25424823241309664. [Google Scholar] [CrossRef]
- Martin, R.; Nora, M.; Anna, L.; Olivia, P.; Leif, B.; Gunilla, W.-T.; Ellen, T.; Anna-Karin, L.-C. Altered hypoxia-induced cellular responses and inflammatory profile in lung fibroblasts from COPD patients compared to control subjects. Respir. Res. 2024, 25, 282. [Google Scholar] [CrossRef]
- Angelidou, A.; Diray-Arce, J.; Conti, M.-G.; Netea, M.G.; Blok, B.A.; Liu, M.; Sanchez-Schmitz, G.; Ozonoff, A.; van Haren, S.D.; Levy, O. Human Newborn Monocytes Demonstrate Distinct BCG-Induced Primary and Trained Innate Cytokine Production and Metabolic Activation In Vitro. Front. Immunol. 2021, 12, 674334. [Google Scholar] [CrossRef]
- Dias, C.; Fernandes, E.; Barbosa, R.M.; Laranjinha, J.; Ledo, A. Astrocytic aerobic glycolysis provides lactate to support neuronal oxidative metabolism in the hippocampus. Biofactors 2023, 49, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, C.L.; Bercovier, H.; Klein, B.Y.; Gofrit, O.N. Intravesical Bacille Calmette-Guerin (BCG) Vaccine Affects Cognition. J. Alzheimers Dis. 2024, 100, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Kühtreiber, W.M.; Tran, L.; Kim, T.; Dybala, M.; Nguyen, B.; Plager, S.; Huang, D.; Janes, S.; Defusco, A.; Baum, D.; et al. Long-term reduction in hyperglycemia in advanced type 1 diabetes: The value of induced aerobic glycolysis with BCG vaccinations. Npj Vaccines 2018, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Dias, H.F.; Kuhtreiber, W.M.; Nelson, K.J.; Ng, N.C.; Zheng, H.; Faustman, D.L. Epigenetic changes related to glucose metabolism in type 1 diabetes after BCG vaccinations: A vital role for KDM2B. Vaccine 2022, 40, 1540–1554. [Google Scholar] [CrossRef]
- Klinger, D.; Hill, B.L.; Barda, N.; Halperin, E.; Gofrit, O.N.; Greenblatt, C.L.; Rappoport, N.; Linial, M.; Bercovier, H. Bladder Cancer Immunotherapy by BCG Is Associated with a Significantly Reduced Risk of Alzheimer’s Disease and Parkinson’s Disease. Vaccines 2021, 9, 491. [Google Scholar] [CrossRef]
- Weinberg, M.S.; Zafar, A.; Magdamo, C.; Chung, S.Y.; Chou, W.H.; Nayan, M.; Deodhar, M.; Frendl, D.M.; Feldman, A.S.; Faustman, D.L.; et al. Association of BCG Vaccine Treatment with Death and Dementia in Patients with Non-Muscle-Invasive Bladder Cancer. JAMA Netw. Open. 2023, 6, e2314336. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Number | ||||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| Gender | Male | Male | Male | Male | Male | Female |
| Age of pre-BCG sample donation | 58 | 57 | 64 | 47 | 61 | 82 |
| Age of bladder exposed to BCG | 58 | 57 | 64 | 47 | 61 | 82 |
| Age of post-BCG PBMC sample donation | 60 | 60 | 66 | 50 | 65 | 86 |
| Months from the start of BCG exposure to the PBMC sample donation | 24 | 29 | 28 | 36 | 50 | 52 |
| Diagnostic information | Heavy smoker | Heavy smoker | Heavy smoker | Seasonal allergy | Arthritis reactive to BCG | Crohn’s disease |
| Line No. | UPR Signals | Patients Number | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |||
| 1 | p-eIF2a | −61.1 | −78.9 | −83.3 | −93.8 | −8.66 | −3.1 | Pro-apoptotic |
| 2 | p-PERK | 93.5 | −89.86 | −32.97 | −61.9 | 23.59 | −91.3 | Pro-apoptotic |
| 3 | CHOP | 58.3 | 62.3 | 64.4 | 93.96 | 81.6 | 12.86 | Pro-apoptotic |
| 4 | BCL2 | 63 | 27.9 | 69.77 | 94.6 | −96.87 | −21.2 | Anti-apoptotic |
| 5 | BCL2A1 | 58.3 | -74 | −58.7 | 59.4 | 18.8 | −88.2 | Anti-apoptotic |
| 6 | p-IRE1a | 65.9 | −50 | 84.6 | 99.99 | 58 | −81.7 | Anti-apoptotic |
| 7 | XBP1s | 82 | 99.99 | 98.3 | 77.5 | 93 | 99.9 | Anti-apoptotic |
| 8 | ATF6 | −96.6 | −98.4 | −97.6 | −80.6 | 51.3 | −98.16 | Anti-apoptotic |
| 9 | ATF6(N) | 70.5 | 99.99 | 70.5 | −13.5 | −61 | −65.5 | Anti-apoptotic |
| 10 | OASIS | 17.7 | 91.6 | 70.2 | 99 | 9.4 | −50.3 | Anti-apoptotic |
| 11 | BiP | 20 | 36 | −6.9 | 88 | 3.3 | −49 | Anti-apoptotic |
| 12 | GAPDH | 5.9 | 93 | 27.3 | 94 | 56 | 95.8 | Anti-apoptotic |
| 13 | Sum of lines 1–3 | 90.7 | −106.46 | −51.87 | −61.74 | 96.53 | −81.54 | Pro-apoptotic |
| 14 | Sum of lines 4–12 | 286.7 | 226.48 | 257.47 | 518.89 | 131.93 | −258.06 | Anti-apoptotic |
| 15 | Balance between pro and anti-apoptotic effects. | 196 | 332.94 | 309.34 | 580.13 | 35.4 | −176.82 | Balance |
| Line No. | UPR Signals | Patients Number | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |||
| 1 | p-eIF2a | −3 | 52 | 86.5 | 76.6 | 32 | 80.6 | Pro-apoptotic |
| 2 | p-PERK | 94.7 | 99.99 | 94.98 | −54.7 | 99.99 | 43.4 | Pro-apoptotic |
| 3 | CHOP | 58.3 | 62.3 | 64.4 | 93.96 | 81.6 | 12.86 | Pro-apoptotic |
| 4 | BCL2 | 63 | 27.9 | 69.77 | 94.6 | −96.87 | −21.2 | Anti-apoptotic |
| 5 | BCL2A1 | 58.3 | −74 | −58.7 | 59.4 | 18.8 | −88.2 | Anti-apoptotic |
| 6 | p-IRE1a | 65.9 | −50 | 84.6 | 99.99 | 58 | −81.7 | Anti-apoptotic |
| 7 | XBP1s | 82 | 99.99 | 98.3 | 77.5 | 93 | 99.9 | Anti-apoptotic |
| 8 | ATF6 | −96.6 | −98.4 | −97.6 | −80.6 | 51.3 | −98.16 | Anti-apoptotic |
| 9 | ATF6(N) | 70.5 | 99.99 | 70.5 | −13.5 | −61 | −65.5 | Anti-apoptotic |
| 10 | OASIS | 17.7 | 91.6 | 70.2 | 99 | 9.4 | −50.3 | Anti-apoptotic |
| 11 | BiP | 20 | 36 | −6.9 | 88 | 3.3 | −49 | Anti-apoptotic |
| 12 | GAPDH | 5.9 | 93 | 27.3 | 94 | 56 | 95.8 | Anti-apoptotic |
| 13 | Sum of lines 1–3 | 150 | 214.29 | 245.88 | 115.59 | 181.59 | 136.86 | Pro-apoptotic |
| 14 | Sum of lines 4–12 | 286.7 | 226.48 | 257.47 | 518.89 | 131.93 | −258.06 | Anti-apoptotic |
| 15 | Balance between pro- and anti-apoptotic effects. | 136.7 | 12.19 | 11.59 | 403.03 | −49.66 | −394.92 | Balance |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, B.Y.; Gofrit, O.N.; Greenblatt, C.L. Testing Protein Stress Signals in Peripheral Immunocytes Under the Same Treatment Capable of Decreasing the Incidence of Alzheimer’s Disease in Bladder Cancer Patients. Curr. Issues Mol. Biol. 2025, 47, 392. https://doi.org/10.3390/cimb47060392
Klein BY, Gofrit ON, Greenblatt CL. Testing Protein Stress Signals in Peripheral Immunocytes Under the Same Treatment Capable of Decreasing the Incidence of Alzheimer’s Disease in Bladder Cancer Patients. Current Issues in Molecular Biology. 2025; 47(6):392. https://doi.org/10.3390/cimb47060392
Chicago/Turabian StyleKlein, Benjamin Y., Ofer N. Gofrit, and Charles L. Greenblatt. 2025. "Testing Protein Stress Signals in Peripheral Immunocytes Under the Same Treatment Capable of Decreasing the Incidence of Alzheimer’s Disease in Bladder Cancer Patients" Current Issues in Molecular Biology 47, no. 6: 392. https://doi.org/10.3390/cimb47060392
APA StyleKlein, B. Y., Gofrit, O. N., & Greenblatt, C. L. (2025). Testing Protein Stress Signals in Peripheral Immunocytes Under the Same Treatment Capable of Decreasing the Incidence of Alzheimer’s Disease in Bladder Cancer Patients. Current Issues in Molecular Biology, 47(6), 392. https://doi.org/10.3390/cimb47060392