

Genome-Wide Detection of Leukemia Biomarkers from lincRNA–Protein-Coding Gene Interaction Networks in the Three-Dimensional Chromatin Structure

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Protein-Coding Genes and lincRNA Genes
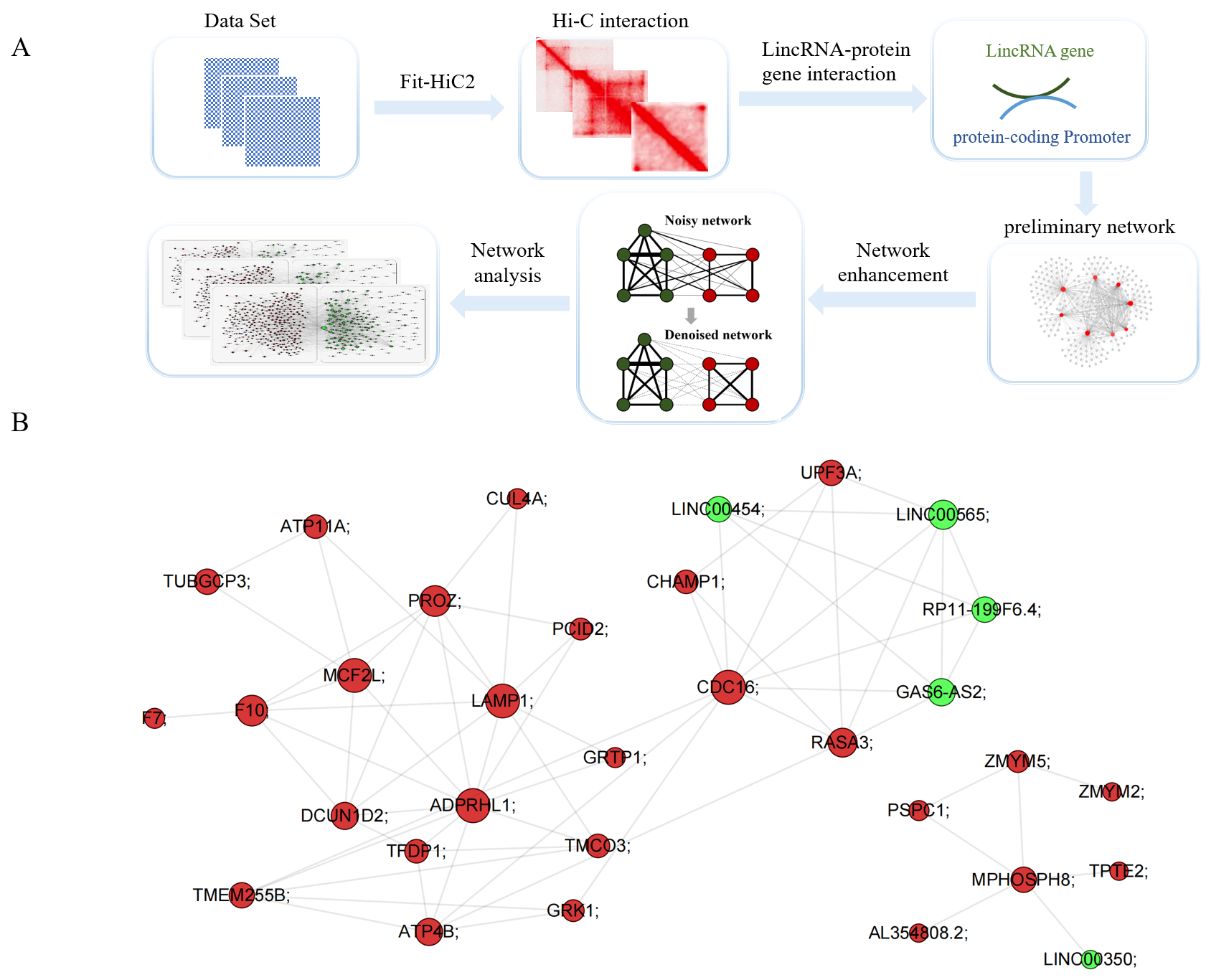
2.2. Construction of Interaction Network of Protein Genes and lincRNA Genes
2.3. Network Enhancement
2.4. POLII ChIP-Seq Analysis
2.5. Biology Experiments
2.6. DRIP-Seq Data Analysis
2.7. Gene Enrichment Analysis
2.8. TCGA Data Download, Processing, and Analysis
2.9. Single-Cell RNA Analysis
2.10. The Interaction Between lincRNA and miRNA
2.11. Potential Small Molecule Drugs Targeting GPCR
3. Results
3.1. LincRNA–Protein-Coding Gene Interaction Network
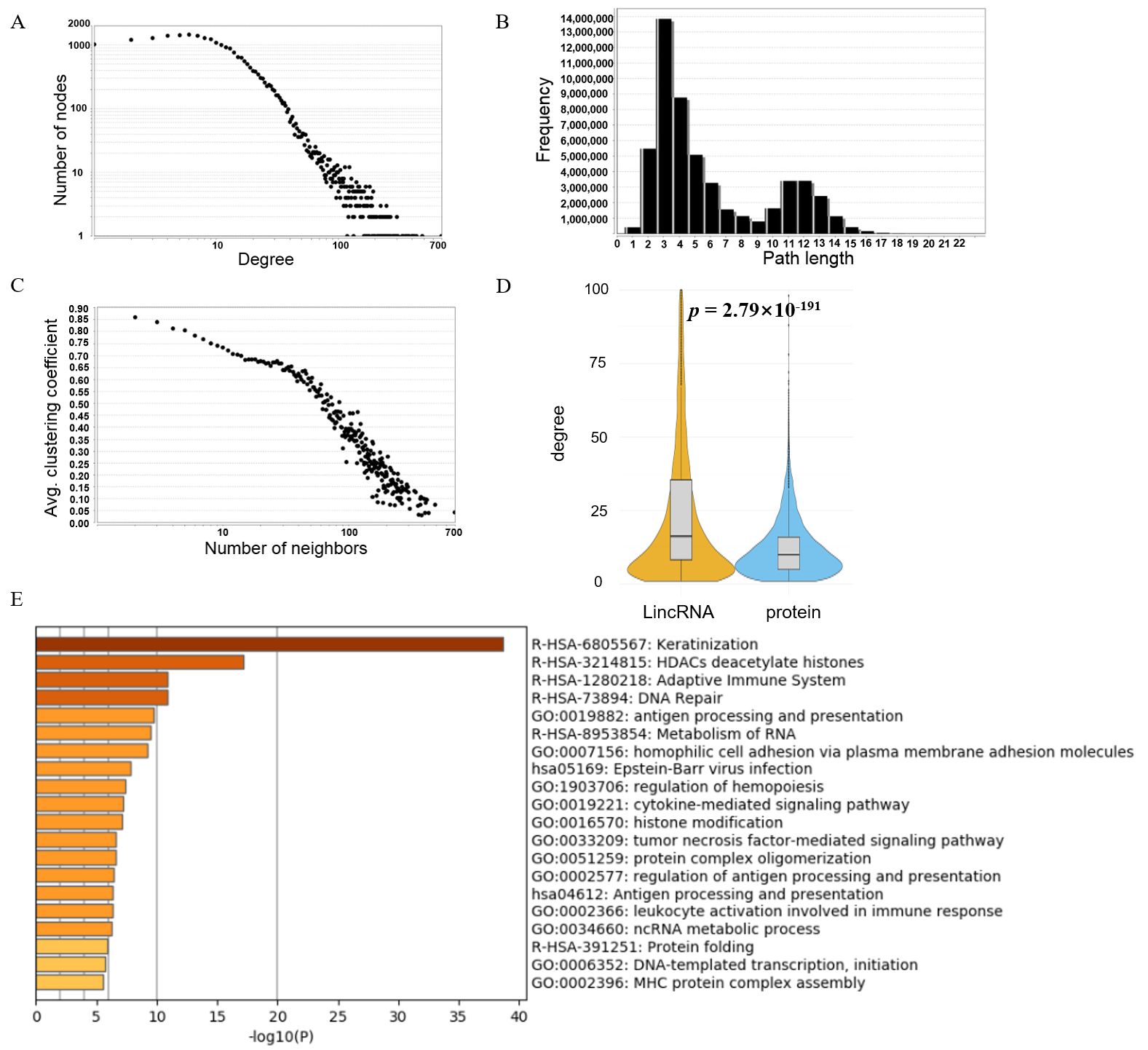
3.2. Topology Analysis of the lincRNA–Protein-Coding Gene Interaction Network
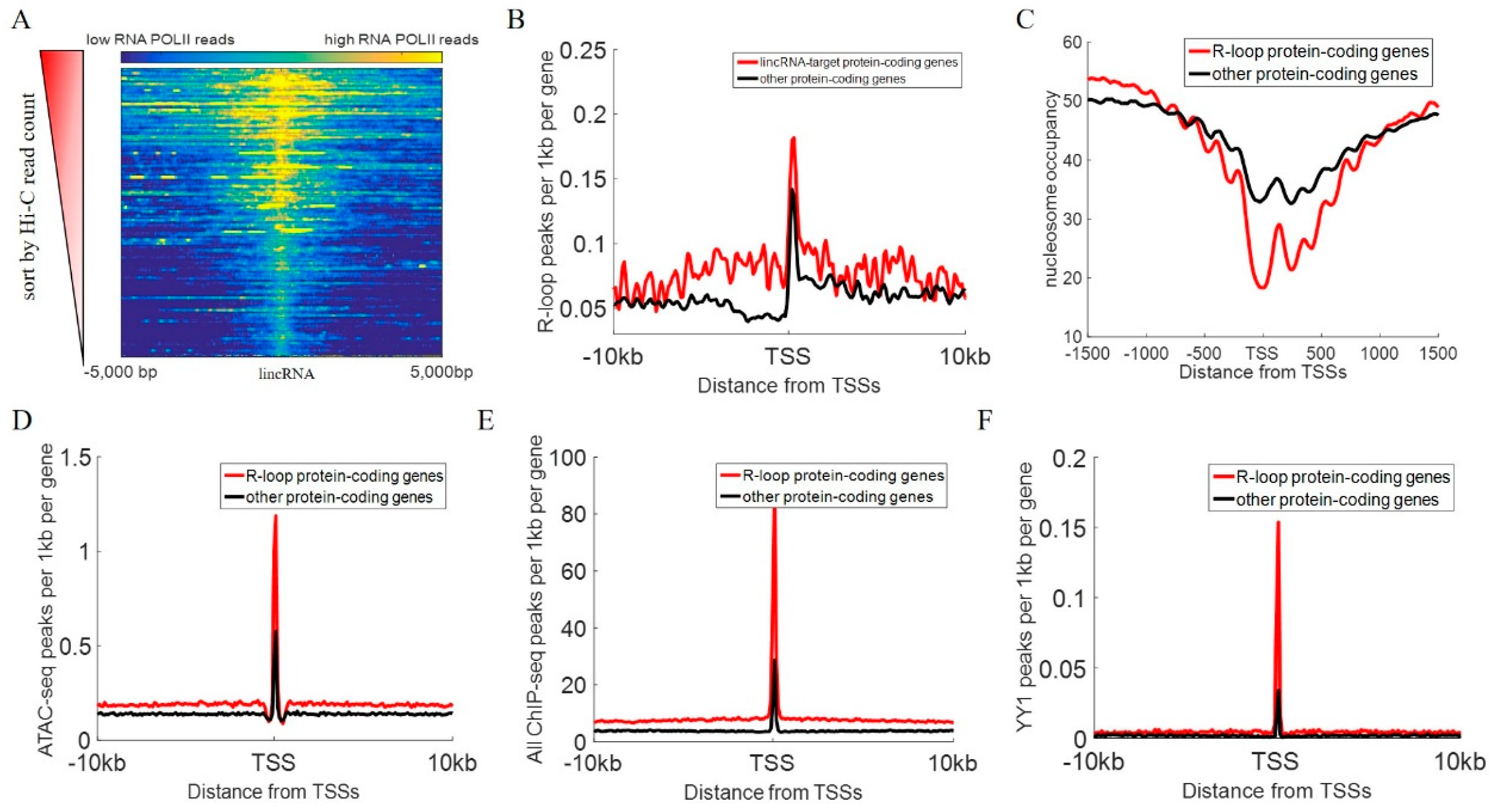
3.3. Effects of lincRNA on Chromatin Status Around Target Genes
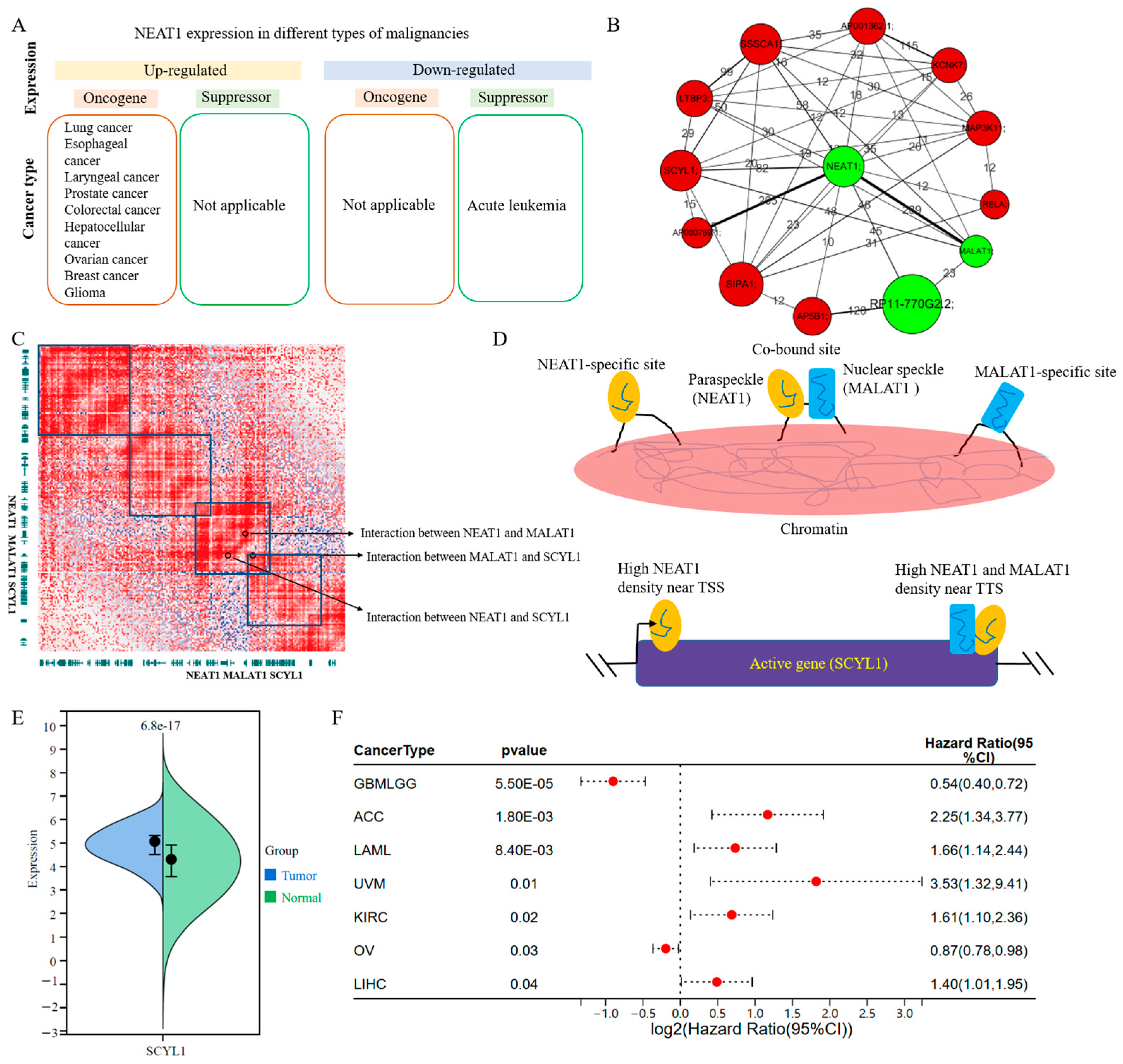
3.4. Biological Significance of the lincRNA–Promoter Network in Leukemia
3.5. Potential Biomarkers of Leukemia from GPCRs Based on the lincRNA–Promoter Network
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long non-coding RNAs: Definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 2023, 24, 430–447. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef]
- Carlevaro-Fita, J.; Lanzos, A.; Feuerbach, L.; Hong, C.; Mas-Ponte, D.; Pedersen, J.S.; Drivers, P.; Functional Interpretation, G.; Johnson, R.; Consortium, P. Cancer LncRNA Census reveals evidence for deep functional conservation of long noncoding RNAs in tumorigenesis. Commun. Biol. 2020, 3, 56. [Google Scholar] [CrossRef]
- Yang, Y.; Zhong, Z.; Zhao, Y.; Ren, K.; Li, N. LincRNA-SLC20A1 (SLC20A1) promotes extracellular matrix degradation in nucleus pulposus cells in human intervertebral disc degeneration by targeting the miR-31-5p/MMP3 axis. Int. J. Clin. Exp. Pathol. 2019, 12, 3632–3643. [Google Scholar]
- Yuan, J.; He, X.; Wang, Y. G-quadruplex DNA contributes to RNA polymerase II-mediated 3D chromatin architecture. Nucleic Acids Res. 2023, 51, 8434–8446. [Google Scholar] [CrossRef]
- Oh, S.; Shao, J.; Mitra, J.; Xiong, F.; D’Antonio, M.; Wang, R.; Garcia-Bassets, I.; Ma, Q.; Zhu, X.; Lee, J.H.; et al. Enhancer release and retargeting activates disease-susceptibility genes. Nature 2021, 595, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Vasquez, K.M. Dynamic alternative DNA structures in biology and disease. Nat. Rev. Genet. 2022, 24, 211–234. [Google Scholar] [CrossRef]
- Heinz, S.; Texari, L.; Hayes, M.G.; Urbanowski, M.; Chang, M.W.; Givarkes, N.; Rialdi, A.; White, K.M.; Albrecht, R.A.; Pache, L.; et al. Transcription Elongation Can Affect Genome 3D Structure. Cell 2018, 174, 1522–1536.e1522. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lu, K.; Hou, Y.; You, Z.; Shu, C.; Wei, X.; Wu, T.; Shi, N.; Zhang, G.; Wu, J.; et al. YY1 complex in M2 macrophage promotes prostate cancer progression by upregulating IL-6. J. Immunother. Cancer 2023, 11, e006020. [Google Scholar] [CrossRef]
- Ke, Y.; Xu, Y.; Chen, X.; Feng, S.; Liu, Z.; Sun, Y.; Yao, X.; Li, F.; Zhu, W.; Gao, L.; et al. 3D Chromatin Structures of Mature Gametes and Structural Reprogramming during Mammalian Embryogenesis. Cell 2017, 170, 367–381.e320. [Google Scholar] [CrossRef]
- Gabriele, M.; Brandao, H.B.; Grosse-Holz, S.; Jha, A.; Dailey, G.M.; Cattoglio, C.; Hsieh, T.S.; Mirny, L.; Zechner, C.; Hansen, A.S. Dynamics of CTCF- and cohesin-mediated chromatin looping revealed by live-cell imaging. Science 2022, 376, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Chen, C.; Teng, L.; Tan, K. Global view of enhancer-promoter interactome in human cells. Proc. Natl. Acad. Sci. USA 2014, 111, E2191–E2199. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Duan, Z.; Andronescu, M.; Schutz, K.; McIlwain, S.; Kim, Y.J.; Lee, C.; Shendure, J.; Fields, S.; Blau, C.A.; Noble, W.S. A three-dimensional model of the yeast genome. Nature 2010, 465, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Pourshafeie, A.; Zitnik, M.; Zhu, J.; Bustamante, C.D.; Batzoglou, S.; Leskovec, J. Network enhancement as a general method to denoise weighted biological networks. Nat. Commun. 2018, 9, 3108. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Smyth, G.K.; Michaud, J.; Scott, H.S. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 2005, 21, 2067–2075. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Shu, C.; Wang, C.; Chen, S.; Huang, X.; Cui, J.; Li, W.; Xu, B. ERR-activated GPR35 promotes immune infiltration level of macrophages in gastric cancer tissues. Cell Death Discov. 2022, 8, 444. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Li, G.; Ruan, X.; Auerbach, R.K.; Sandhu, K.S.; Zheng, M.; Wang, P.; Poh, H.M.; Goh, Y.; Lim, J.; Zhang, J.; et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 2012, 148, 84–98. [Google Scholar] [CrossRef]
- Chen, D.; Fu, L.Y.; Zhang, Z.; Li, G.; Zhang, H.; Jiang, L.; Harrison, A.P.; Shanahan, H.P.; Klukas, C.; Zhang, H.Y.; et al. Dissecting the chromatin interactome of microRNA genes. Nucleic Acids Res. 2014, 42, 3028–3043. [Google Scholar] [CrossRef] [PubMed]
- Paralkar, V.R.; Taborda, C.C.; Huang, P.; Yao, Y.; Kossenkov, A.V.; Prasad, R.; Luan, J.; Davies, J.O.; Hughes, J.R.; Hardison, R.C.; et al. Unlinking an lncRNA from Its Associated cis Element. Mol. Cell 2016, 62, 104–110. [Google Scholar] [CrossRef]
- Tang, Z.; Luo, O.J.; Li, X.; Zheng, M.; Zhu, J.J.; Szalaj, P.; Trzaskoma, P.; Magalska, A.; Wlodarczyk, J.; Ruszczycki, B.; et al. CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 2015, 163, 1611–1627. [Google Scholar] [CrossRef]
- Sanz, L.A.; Hartono, S.R.; Lim, Y.W.; Steyaert, S.; Rajpurkar, A.; Ginno, P.A.; Xu, X.; Chedin, F. Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol. Cell 2016, 63, 167–178. [Google Scholar] [CrossRef]
- Chedin, F. Nascent Connections: R-Loops and Chromatin Patterning. Trends Genet. TIG 2016, 32, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, S.C.; Wang, S.; Ma, W.K.; Al Husini, N.; Dhoondia, Z.; Ansari, A.; Pascuzzi, P.E.; Tran, E.J. Regulated Formation of lncRNA-DNA Hybrids Enables Faster Transcriptional Induction and Environmental Adaptation. Mol. Cell 2016, 62, 148. [Google Scholar] [CrossRef]
- Mavrich, T.N.; Ioshikhes, I.P.; Venters, B.J.; Jiang, C.; Tomsho, L.P.; Qi, J.; Schuster, S.C.; Albert, I.; Pugh, B.F. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008, 18, 1073–1083. [Google Scholar] [CrossRef]
- Shivaswamy, S.; Bhinge, A.; Zhao, Y.; Jones, S.; Hirst, M.; Iyer, V.R. Dynamic remodeling of individual nucleosomes across a eukaryotic genome in response to transcriptional perturbation. PLoS Biol. 2008, 6, e65. [Google Scholar] [CrossRef] [PubMed]
- Reja, R.; Vinayachandran, V.; Ghosh, S.; Pugh, B.F. Molecular mechanisms of ribosomal protein gene coregulation. Genes. Dev. 2015, 29, 1942–1954. [Google Scholar] [CrossRef]
- Cheng, X.; Hou, Y.; Nie, Y.; Zhang, Y.; Huang, H.; Liu, H.; Sun, X. Nucleosome Positioning of Intronless Genes in the Human Genome. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 15, 1111–1121. [Google Scholar] [CrossRef]
- Cheneby, J.; Gheorghe, M.; Artufel, M.; Mathelier, A.; Ballester, B. ReMap 2018: An updated atlas of regulatory regions from an integrative analysis of DNA-binding ChIP-seq experiments. Nucleic Acids Res. 2018, 46, D267–D275. [Google Scholar] [CrossRef] [PubMed]
- Sigova, A.A.; Abraham, B.J.; Ji, X.; Molinie, B.; Hannett, N.M.; Guo, Y.E.; Jangi, M.; Giallourakis, C.C.; Sharp, P.A.; Young, R.A. Transcription factor trapping by RNA in gene regulatory elements. Science 2015, 350, 978–981. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, Z.; Zheng, H.; Chan, M.T.; Wu, W.K. NEAT1: A novel cancer-related long non-coding RNA. Cell Prolif. 2017, 50, e12329. [Google Scholar] [CrossRef]
- Tian, X.; Tian, J.; Tang, X.; Ma, J.; Wang, S. Long non-coding RNAs in the regulation of myeloid cells. J. Hematol. Oncol. 2016, 9, 99. [Google Scholar] [CrossRef]
- Wen, F.; Cao, Y.X.; Luo, Z.Y.; Liao, P.; Lu, Z.W. LncRNA MALAT1 promotes cell proliferation and imatinib resistance by sponging miR-328 in chronic myelogenous leukemia. Biochem. Biophys. Res. Commun. 2018, 507, 1–8. [Google Scholar] [CrossRef]
- West, J.A.; Davis, C.P.; Sunwoo, H.; Simon, M.D.; Sadreyev, R.I.; Wang, P.I.; Tolstorukov, M.Y.; Kingston, R.E. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell 2014, 55, 791–802. [Google Scholar] [CrossRef]
- Kim, J.; Piao, H.L.; Kim, B.J.; Yao, F.; Han, Z.; Wang, Y.; Xiao, Z.; Siverly, A.N.; Lawhon, S.E.; Ton, B.N.; et al. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat. Genet. 2018, 50, 1705–1715. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, Z.; Zhang, J.; Ma, X.; Bi, X.; Xu, L.; Feng, K.; Wu, Z.; Ma, X.; Zhuang, L. RNA-binding protein QKI promotes the progression of HCC by interacting with long non-coding RNA EGOT. Int. Immunopharmacol. 2024, 136, 112297. [Google Scholar] [CrossRef]
- Wang, Y.; Li, X.; Huang, Y.; Gang, Q.; Liu, M.; Zhang, H.; Shen, S.; Qi, Y.; Zhang, J. The Prognostic Value and Potential Immune Mechanisms of lncRNAs Related to Immunogenic Cell Death in Papillary Thyroid Carcinoma. J. Inflamm. Res. 2024, 17, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Gomez, H.R.; DeVries, A.; Castillo, P.; von Roemeling, C.; Qdaisat, S.; Stover, B.D.; Xie, C.; Weidert, F.; Zhao, C.; Moor, R.; et al. RNA aggregates harness the danger response for potent cancer immunotherapy. Cell 2024, 187, 2521–2535.e2521. [Google Scholar] [CrossRef] [PubMed]
- Maksoud, R.; Magawa, C.; Eaton-Fitch, N.; Thapaliya, K.; Marshall-Gradisnik, S. Biomarkers for myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): A systematic review. BMC Med. 2023, 21, 189. [Google Scholar] [CrossRef]
- Henry, N.L.; Hayes, D.F. Cancer biomarkers. Mol. Oncol. 2012, 6, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, C.; Xu, X.; Shu, C.; Cao, C.; Wang, Z.; Fu, Y.; Xu, L.; Xu, K.; Xu, J.; et al. APAF1-Binding Long Noncoding RNA Promotes Tumor Growth and Multidrug Resistance in Gastric Cancer by Blocking Apoptosome Assembly. Adv. Sci. 2022, 9, e2201889. [Google Scholar] [CrossRef]
- Maxwell, M.B.; Hom-Tedla, M.S.; Yi, J.W.; Li, S.T.; Rivera, S.A.; Yu, J.T.; Burns, M.J.; Mcrae, H.M.; Stevenson, B.T.; Coakley, K.E.; et al. ARID1A suppresses R-loop-mediated STING-type I interferon pathway activation of anti-tumor immunity. Cell 2024, 187, 3390–3408. [Google Scholar] [CrossRef]
- Zhang, S.C.; Liu, Y.; Sun, Y.C.; Liu, Q.; Gu, Y.; Huang, Y.; Zeng, Z.; Tang, F.Z.; Ouyang, Y. Aberrant R-loop-mediated immune evasion, cellular communication, and metabolic reprogramming affect cancer progression: A single-cell analysis. Mol. Cancer 2024, 23, 11. [Google Scholar] [CrossRef]
- Kannan, A.; Gangadharan Leela, S.; Branzei, D.; Gangwani, L. Role of senataxin in R-loop-mediated neurodegeneration. Brain Commun. 2024, 6, fcae239. [Google Scholar] [CrossRef]
- Wei, C.; Gao, Y.; Chen, X.; Zhao, C.; Li, P. ZNF668: A new diagnostic predictor of kidney renal clear cell carcinoma. Anti-Cancer Drugs 2022, 33, e491–e499. [Google Scholar] [CrossRef]
- Li, P.; Wang, C.; Chen, G.; Han, Y.; Lu, H.; Li, N.; Lv, Y.; Chu, C.; Peng, X. Molecular mechanisms of Tetrastigma hemsleyanum Diels&Gilg against lung squamous cell carcinoma: From computational biology and experimental validation. J. Ethnopharmacol. 2024, 331, 118326. [Google Scholar] [PubMed]
- Sun, Y.; Wu, Y.S.; Pang, G.Z.; Huang, J.R.; Sheng, M.Y.; Xie, J.Y.; Chen, P.Y.; Wang, Y.; Yin, D.R.; Zhao, G.J.; et al. STING is crucial for the survival of RUNX1::RUNX1T1 leukemia cells. Leukemia 2024, 38, 2102–2114. [Google Scholar] [CrossRef] [PubMed]
- Crossley, M.P.; Song, C.L.; Bocek, M.J.; Choi, J.H.; Kousorous, J.; Sathirachinda, A.; Lin, C.Y.; Brickner, J.R.; Bai, G.S.; Lans, H.; et al. R-loop-derived cytoplasmic RNA-DNA hybrids activate an immune response. Nature 2023, 613, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.R.; Aliyari, S.R.; Parvatiyar, K.; Wang, L.L.; Zhen, A.J.; Sun, W.; Han, X.B.; Zhang, A.; Kato, E.; Shi, H.L.; et al. STING directly interacts with PAR to promote apoptosis upon acute ionizing radiation-mediated DNA damage. Cell Death Differ. 2025. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, Y.; Ning, W.; Huhe, M.; Shu, C. Genome-Wide Detection of Leukemia Biomarkers from lincRNA–Protein-Coding Gene Interaction Networks in the Three-Dimensional Chromatin Structure. Curr. Issues Mol. Biol. 2025, 47, 384. https://doi.org/10.3390/cimb47060384
Hou Y, Ning W, Huhe M, Shu C. Genome-Wide Detection of Leukemia Biomarkers from lincRNA–Protein-Coding Gene Interaction Networks in the Three-Dimensional Chromatin Structure. Current Issues in Molecular Biology. 2025; 47(6):384. https://doi.org/10.3390/cimb47060384
Chicago/Turabian StyleHou, Yue, Wei Ning, Muren Huhe, and Chuanjun Shu. 2025. "Genome-Wide Detection of Leukemia Biomarkers from lincRNA–Protein-Coding Gene Interaction Networks in the Three-Dimensional Chromatin Structure" Current Issues in Molecular Biology 47, no. 6: 384. https://doi.org/10.3390/cimb47060384
APA StyleHou, Y., Ning, W., Huhe, M., & Shu, C. (2025). Genome-Wide Detection of Leukemia Biomarkers from lincRNA–Protein-Coding Gene Interaction Networks in the Three-Dimensional Chromatin Structure. Current Issues in Molecular Biology, 47(6), 384. https://doi.org/10.3390/cimb47060384