
The Role of Mitochondrial Energy Metabolism in the Mechanism of Exercise Improving Depression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
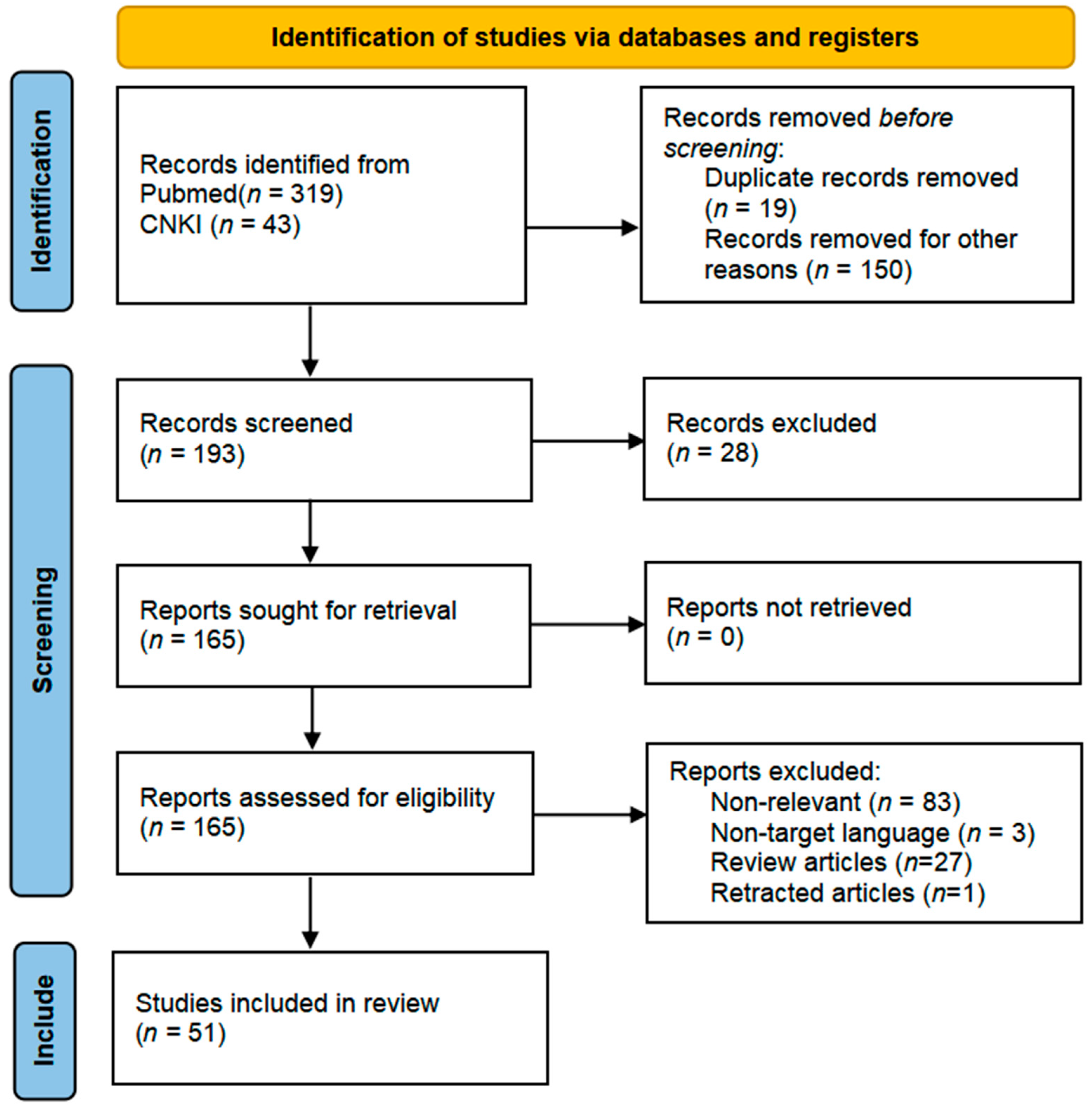
2. Method
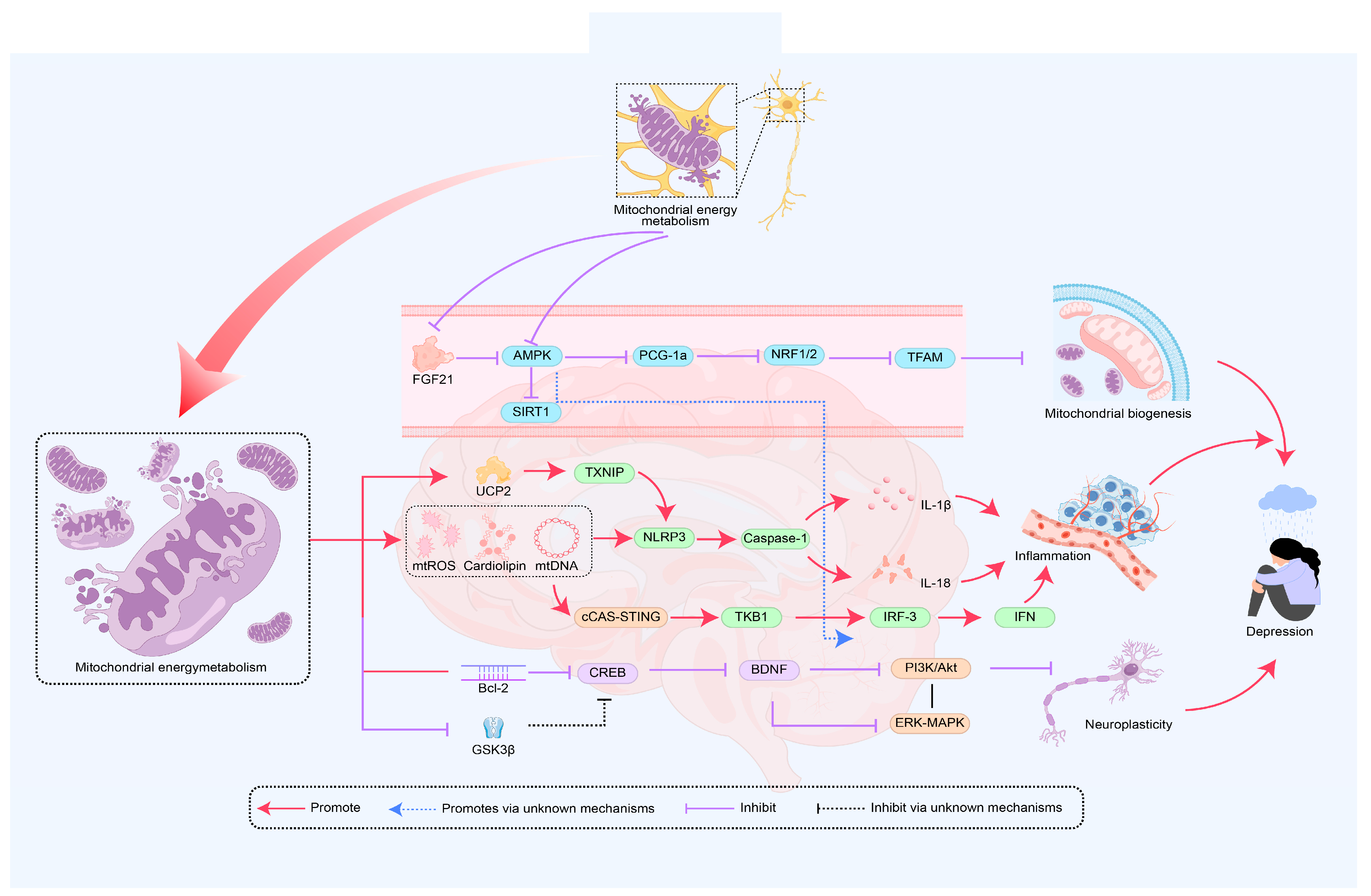
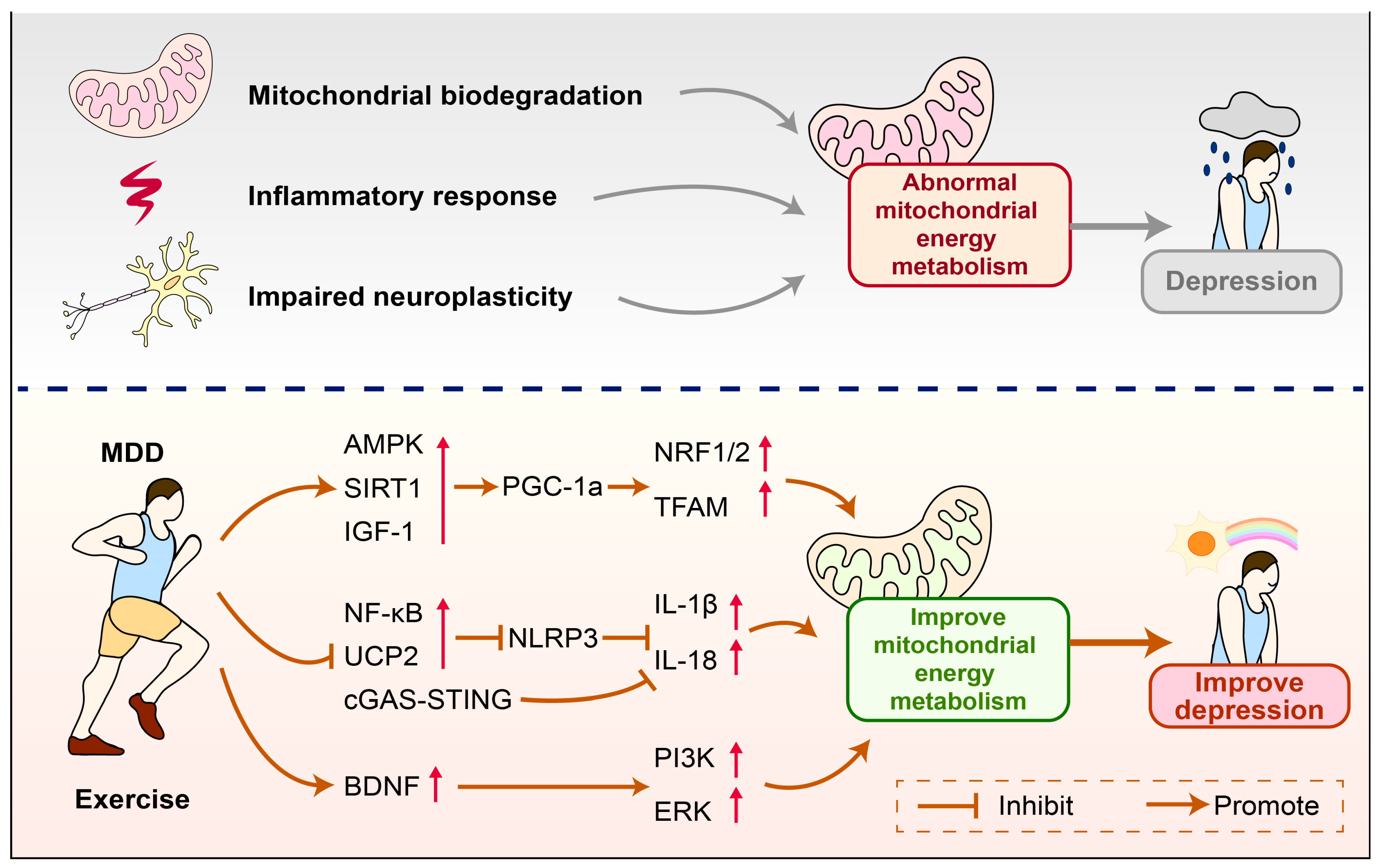
3. The Role of Mitochondrial Energy Metabolism in the Mechanism of Depression
3.1. Mitochondrial Energy Metabolism Improves Depression by Regulating Biogenesis
3.2. Mitochondrial Energy Metabolism Improves Depression by Mediating Immune Inflammation
3.3. Mitochondrial Energy Metabolism Improves Depression by Promoting Neural Plasticity
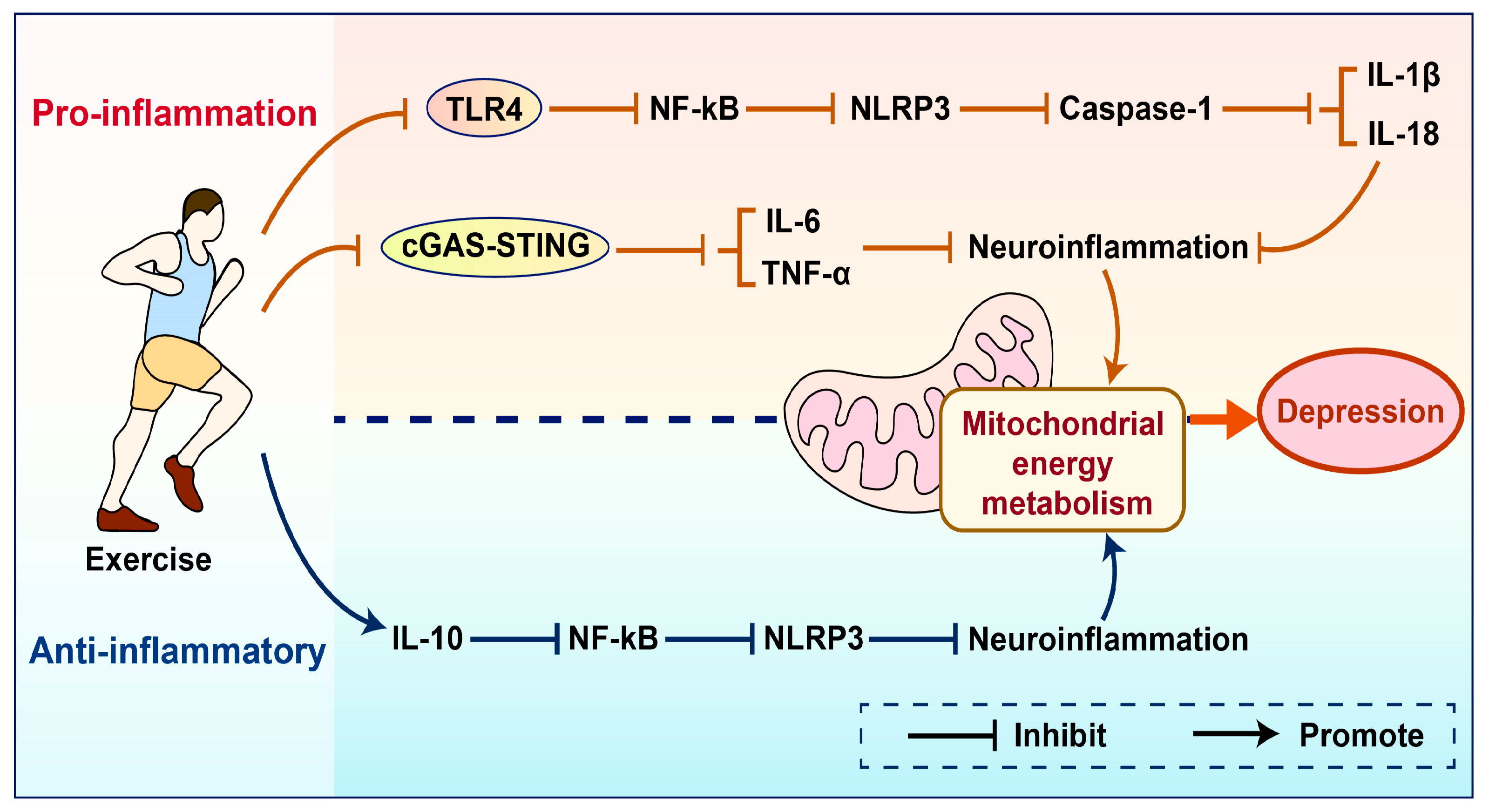
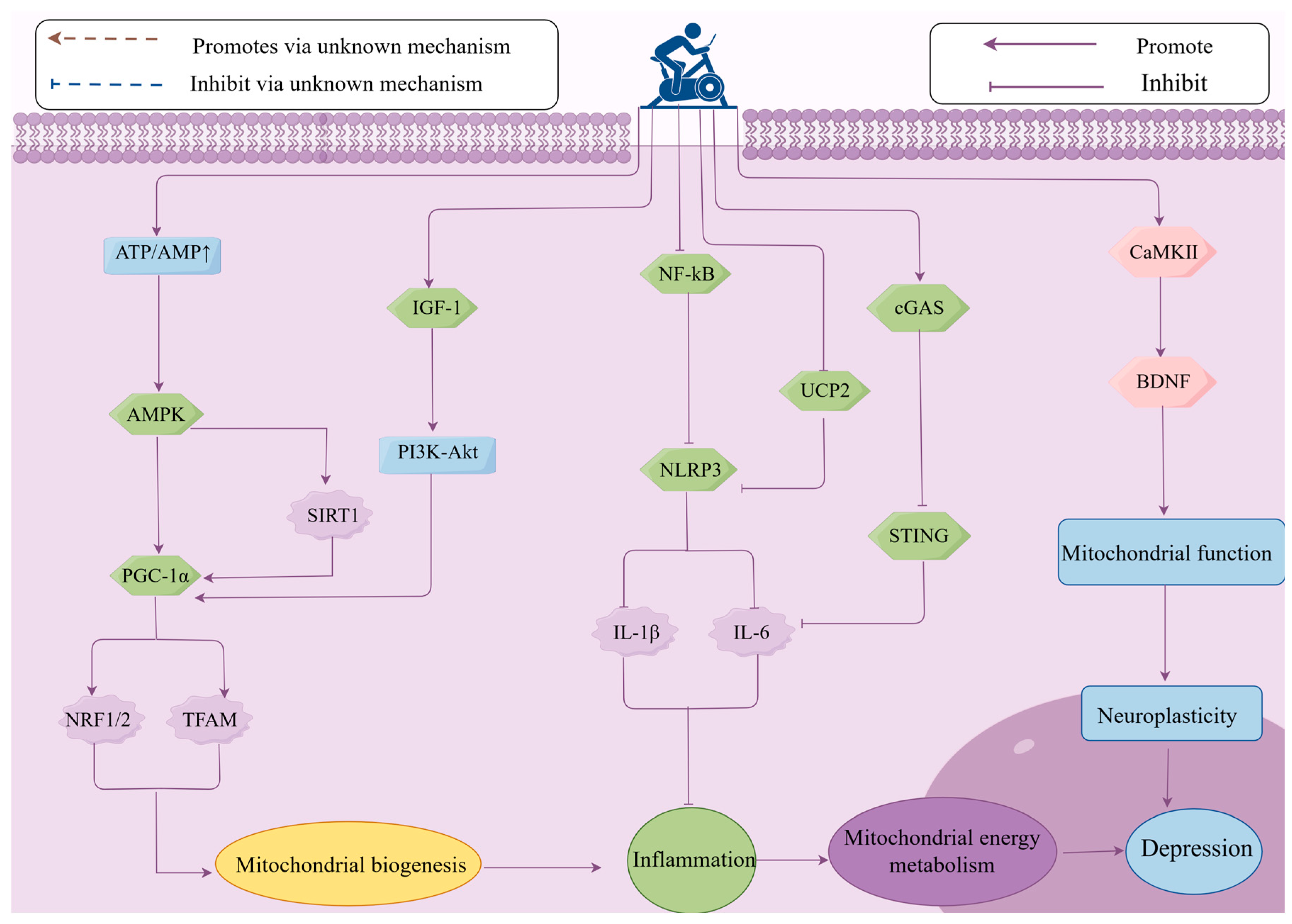
4. The Role of Mitochondrial Energy Metabolism in Exercise-Induced Improvement of Depression
4.1. Mitochondrial Energy Metabolism Mediates the Improvement of Depression Through Biogenesis Induced by Exercise
4.2. Mitochondrial Energy Metabolism Mediates the Improvement of Depression Through Inflammatory Responses Induced by Exercise
4.3. Mitochondrial Energy Metabolism Mediates Exercise-Induced Improvement of Depression Through Neural Plasticity
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Smith, K. Mental health: A world of depression. Nature 2014, 515, 181. [Google Scholar] [CrossRef] [PubMed]
- Klinedinst, N.J.; Regenold, W.T. A mitochondrial bioenergetic basis of depression. J. Bioenerg. Biomembr. 2015, 47, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Cheng, H. Research progress of mitochondrial energy metabolism involved in the pathogenesis of depression. Chin. J. Clin. Pharmacol. Ther. 2021, 26, 1193–1199. [Google Scholar]
- Fattal, O.; Budur, K.; Vaughan, A.J.; Franco, K. Review of the literature on major mental disorders in adult patients with mitochondrial diseases. Psychosomatics 2006, 47, 1–7. [Google Scholar] [CrossRef]
- Martin, J.L.; Magistretti, P.J.; Allaman, I. Regulation of neurotrophic factorsand energy metabolism by antidepressants in astrocytes. Curr. Drug. Targets. 2013, 14, 1308–1321. [Google Scholar] [CrossRef]
- Martins-de-Souza, D.; Guest, P.C.; Harris, L.W.; Vanattou-Saifoudine, N.; Webster, M.J.; Rahmoune, H.; Bahn, S. Identification of proteomic signaturesassociated with depression and psychotic depression in post-mortem brains from major depression patients. Transl. Psychiatry. 2012, 2, e87. [Google Scholar] [CrossRef]
- Caruncho, H.J.; Brymer, K.; Romay-Tallón, R.; Mitchell, M.A.; Rivera-Baltanás, T.; Botterill, J.; Olivares, J.M.; Kalynchuk, L.E. Reelin-Related disturbances in depression: Implications for translational studies. Front. Cell. Neurosci. 2016, 10, 48. [Google Scholar] [CrossRef]
- Zitkovsky, E.K.; Daniels, T.E.; Tyrka, A.R. Mitochondria and early-life adversity. Mitochondrion 2021, 57, 213–221. [Google Scholar] [CrossRef]
- Chung, J.K.; Lee, S.Y.; Park, M.; Joo, E.J.; Kim, S.A. Investigation of mitochondrial DNA copy number in patients with major depressive disorder. Psychiatry. Res. 2019, 282, 112616. [Google Scholar] [CrossRef]
- Petschner, P.; Gonda, X.; Baksa, D.; Eszlari, N.; Trivaks, M.; Juhasz, G.; Bagdy, G. Genes linking mitochondrial function, cognitive impairment and depression are associated with endophenotypes serving precision medicine. Neuroscience 2018, 370, 207–217. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Rappeneau, V.; Wilmes, L.; Touma, C. Molecular correlates of mitochondrial dysfunctions in major depression: Evidence from clinical and rodent studies. Mol. Cell. Neurosci. 2020, 109, 103555. [Google Scholar] [CrossRef]
- Halling, J.F.; Pilegaard, H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl. Physiol. Nutr. Metab. 2020, 45, 927–936. [Google Scholar] [CrossRef]
- Nierenberg, A.A.; Ghaznavi, S.A.; Sande Mathias, I.; Ellard, K.K.; Janos, J.A.; Sylvia, L.G. Peroxisome Proliferator-Activated Receptor Gamma Coactivator-1 Alpha as a Novel Target for Bipolar Disorder and Other Neuropsychiatric Disorders. Biol. Psychiatry 2018, 83, 761–769. [Google Scholar] [CrossRef]
- Markham, A.; Bains, R.; Franklin, P.; Spedding, M. Changes in mitochondrial function are pivotal in neurodegenerative and psychiatric disorders: How important is BDNF? Br. J. Pharm. 2014, 171, 2206–2229. [Google Scholar] [CrossRef]
- Kumar, R.; Harilal, S.; Parambi, D.G.T.; Kanthlal, S.K.; Rahman, M.A.; Alexiou, A.; Batiha, G.E.; Mathew, B. The role of mitochondrial genes in neurodegenerative disorders. Curr. Neuropharmacol. 2022, 20, 824–835. [Google Scholar] [CrossRef]
- Song, Y.; Cao, H.; Zuo, C.; Gu, Z.; Huang, Y.; Miao, J.; Fu, Y.; Guo, Y.; Jiang, Y.; Wang, F. Mitochondrial dysfunction: A fatal blow in depression. Biomed. Pharmacother. 2023, 167, 115652. [Google Scholar] [CrossRef]
- Khan, M.; Baussan, Y.; Hebert-Chatelain, E. Connecting dots between mitochondrial dysfunction and depression. Biomolecules 2023, 13, 695. [Google Scholar] [CrossRef]
- Hu, S.; Tucker, L.; Wu, C.; Yang, L. Beneficial effects of exercise on depression and anxiety during the Covid-19 pandemic: A narrative review. Front. Psychiatry 2020, 11, 587557. [Google Scholar] [CrossRef]
- Noetel, M.; Sanders, T.; Gallardo-Gómez, D.; Taylor, P.; Del Pozo Cruz, B.; van den Hoek, D.; Smith, J.J.; Mahoney, J.; Spathis, J.; Moresi, M.; et al. Effect of exercise for depression: Systematic review and network meta-analysis of randomised controlled trials. BMJ 2024, 385, q1024. [Google Scholar] [CrossRef]
- Luo, J.; Tang, C.; Chen, X.; Ren, Z.; Qu, H.; Chen, R.; Tong, Z. Impacts of aerobic exercise on depression-like behaviors in chronic unpredictable mild stress mice and related factors in the AMPK/PGC-1α Pathway. Int. J. Environ. Res. Public Health 2020, 17, 2042. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of skeletal muscle mitochondria in health, exercise, and aging. Annu. Rev. Physiol. 2019, 81, 19–41. [Google Scholar] [CrossRef] [PubMed]
- Paolucci, E.M.; Loukov, D.; Bowdish, D.M.E.; Heisz, J.J. Exercise reduces depression and inflammation but intensity matters. Biol. Psychol. 2018, 133, 79–84. [Google Scholar] [CrossRef]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Aguiar, A.S., Jr.; Stragier, E.; da Luz Scheffer, D.; Remor, A.P.; Oliveira, P.A.; Prediger, R.D.; Latini, A.; Raisman-Vozari, R.; Mongeau, R.; Lanfumey, L. Effects of exercise on mitochondrial function, neuroplasticity and anxio-depressive behavior of mice. Neuroscience 2014, 271, 56–63. [Google Scholar] [CrossRef]
- Vaynman, S.; Ying, Z.; Gomez-Pinilla, F. Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur. J. Neurosci. 2004, 20, 2580–2590. [Google Scholar] [CrossRef]
- Park, S.S.; Park, H.S.; Kim, C.J.; Baek, S.S.; Kim, T.W. Exercise attenuates maternal separation-induced mood disorder-like behaviors by enhancing mitochondrial functions and neuroplasticity in the dorsal raphe. Behav. Brain Res. 2019, 372, 112049. [Google Scholar] [CrossRef]
- Zhou, H.; Ren, J.; Toan, S.; Mui, D. Role of mitochondrial quality surveillance in myocardial infarction: From bench to bedside. Ageing Res. Rev. 2021, 66, 101250. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1alpha. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders-A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Panes, J.D.; Wendt, A.; Ramirez-Molina, O.; Castro, P.A.; Fuentealba, J. Deciphering the role of PGC-1α in neurological disorders: From mitochondrial dysfunction to synaptic failure. Neural. Regen. Res. 2022, 17, 237–245. [Google Scholar] [CrossRef]
- Alcocer-Gómez, E.; Núñez-Vasco, J.; Casas-Barquero, N.; Williams, M.R.; Navarro-Pando, J.M.; Bullón, P.; Cordero, M.D. Gene expression profile in major depressive disorder shows reduced mitochondrial biogenesis. CNS Neurosci. Ther. 2016, 22, 636–638. [Google Scholar] [CrossRef]
- Jin, X.; Zhu, L.; Lu, S.; Li, C.; Bai, M.; Xu, E.; Shen, J.; Li, Y. Baicalin ameliorates CUMS-induced depression-like behaviors through activating AMPK/PGC-1α pathway and enhancing NIX-mediated mitophagy in mice. Eur. J. Pharmacol. 2023, 938, 175435. [Google Scholar] [CrossRef]
- Głombik, K.; Stachowicz, A.; Ślusarczyk, J.; Trojan, E.; Budziszewska, B.; Suski, M.; Kubera, M.; Lasoń, W.; Wędzony, K.; Olszanecki, R.; et al. Maternal stress predicts altered biogenesis and the profile of mitochondrial proteins in the frontal cortex and hippocampus of adult offspring rats. Psychoneuroendocrinology 2015, 60, 151–162. [Google Scholar] [CrossRef]
- Chen, M.; Yan, R.; Luo, J.; Ning, J.; Zhou, R.; Ding, L. The role of PGC-1α-mediated mitochondrial biogenesis in neurons. Neurochem. Res. 2023, 48, 2595–2606. [Google Scholar] [CrossRef]
- Valero, T. Mitochondrial biogenesis: Pharmacological approaches. Curr. Pharm. Des. 2014, 20, 5507–5509. [Google Scholar] [CrossRef]
- Shi, H.J.; Xu, C.; Liu, M.Y.; Wang, B.K.; Liu, W.B.; Chen, D.H.; Zhang, L.; Xu, C.Y.; Li, X.F. Resveratrol Improves the Energy Sensing and Glycolipid Metabolism of Blunt Snout Bream Megalobrama amblycephala Fed High-Carbohydrate Diets by Activating the AMPK-SIRT1-PGC-1α Network. Front. Physiol. 2018, 9, 1258. [Google Scholar] [CrossRef]
- Ryan, K.M.; Patterson, I.; McLoughlin, D.M. Peroxisome proliferator-activated receptor gamma co-activator-1 alpha in depression and the response to electroconvulsive therapy. Psychol. Med. 2019, 49, 1859–1868. [Google Scholar] [CrossRef]
- Xu, W.; Yan, J.; Ocak, U.; Lenahan, C.; Shao, A.; Tang, J.; Zhang, J.; Zhang, J.H. Melanocortin 1 receptor attenuates early brain injury following subarachnoid hemorrhage by controlling mitochondrial metabolism via AMPK/SIRT1/PGC-1α pathway in rats. Theranostics 2021, 11, 522–539. [Google Scholar] [CrossRef]
- Wenz, T. Regulation of mitochondrial biogenesis and PGC-1α under cellular stress. Mitochondrion 2013, 13, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Arcaro, A.; Guerreiro, A.S. The phosphoinositide 3-kinase pathway in human cancer: Genetic alterations and therapeutic implications. Curr. Genom. 2007, 8, 271–306. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Lonze, B.E.; Ginty, D.D. Function and regulation of CREB family transcription factors in the nervous system. Neuron 2002, 35, 605–623. [Google Scholar] [CrossRef]
- Cardoso, S.; López, I.P.; Piñeiro-Hermida, S.; Pichel, J.G.; Moreira, P.I. IGF1R deficiency modulates brain signaling pathways and disturbs mitochondria and redox homeostasis. Biomedicines 2021, 9, 158. [Google Scholar] [CrossRef]
- Yang, C.; Sui, G.; Li, D.; Wang, L.; Zhang, S.; Lei, P.; Chen, Z.; Wang, F. Exogenous IGF-1 alleviates depression-like behavior and hippocampal mitochondrial dysfunction in high-fat diet mice. Physiol. Behav. 2021, 229, 113236. [Google Scholar] [CrossRef]
- Guan, X.; Yan, Q.; Wang, D.; Du, G.; Zhou, J. IGF-1 Signaling regulates mitochondrial remodeling during myogenic differentiation. Nutrients 2022, 14, 1249. [Google Scholar] [CrossRef]
- Weng, G.; Zhou, B.; Liu, T.; Huang, Z.; Yang, H. Sitagliptin promotes mitochondrial biogenesis in human SH-SY5Y cells by increasing the expression of PGC-1α/NRF1/TFAM. IUBMB Life 2019, 71, 1515–1521. [Google Scholar] [CrossRef]
- Peng, K.; Yang, L.; Wang, J.; Ye, F.; Dan, G.; Zhao, Y.; Cai, Y.; Cui, Z.; Ao, L.; Liu, J.; et al. The Interaction of mitochondrial biogenesis and fission/fusion mediated by PGC-1α regulates rotenone-Induced dopaminergic neurotoxicity. Mol. Neurobiol. 2017, 54, 3783–3797. [Google Scholar] [CrossRef]
- Nisoli, E.; Tonello, C.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Falcone, S.; Valerio, A.; Cantoni, O.; Clementi, E.; et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 2005, 310, 314–317. [Google Scholar] [CrossRef]
- Mäkelä, J.; Tselykh, T.V.; Maiorana, F.; Eriksson, O.; Do, H.T.; Mudò, G.; Korhonen, L.T.; Belluardo, N.; Lindholm, D. Fibroblast growth factor-21 enhances mitochondrial functions and increases the activity of PGC-1α in human dopaminergic neurons via Sirtuin-1. SpringerPlus 2014, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Ma, J.; Mu, D.; Li, B.; Lian, B.; Sun, C. FGF21 protects dopaminergic neurons in parkinson’s disease models via repression of neuroinflammation. Neurotox. Res. 2020, 37, 616–627. [Google Scholar] [CrossRef]
- Heo, J.; Noble, E.E.; Call, J.A. The role of exerkines on brain mitochondria: A mini-review. J. Appl. Physiol. 2023, 134, 28–35. [Google Scholar] [CrossRef]
- Hu, S.; Wu, X.; Xing, J.; Shen, F.; Fang, D. Ameliorating effects and mechanisms of exerkines on mitochondrial dysfunction of neurons. China Sport Sci. Technol. 2024, 60, 61–72. [Google Scholar]
- Bansa, Y.; Kuhad, A. Mitochondrial dysfunction in depression. Curr. Neuropharmacol. 2016, 14, 610–618. [Google Scholar] [CrossRef]
- Alcocer-Gómez, E.; De Miguel, M.; Casas-Barquero, N.; Núñez-Vasco, J.; Sánchez-Alcazar, J.A.; Fernández-Rodríguez, A.; Cordero, M.D. NLRP3 inflammasome is activated in mononuclear blood cells from patients with major depressive disorder. Brain Behav. Immun. 2014, 36, 111–117. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Peng, Y.L.; Liu, Y.Z.; Wu, T.Y.; Shen, X.L.; Zhou, J.R.; Sun, D.Y.; Huang, A.J.; Wang, X.; et al. Involvement of inflammasome activation in lipopolysaccharide-induced mice depressive-like behaviors. CNS. Neurosci. Ther. 2014, 20, 119–124. [Google Scholar] [CrossRef]
- Franklin, T.C.; Xu, C.; Duman, R.S. Depression and sterile inflammation: Essential role of danger associated molecular patterns. Brain Behav. Immun. 2018, 72, 2–13. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Liu, Y.Z.; Shen, X.L.; Wu, T.Y.; Zhang, T.; Wang, W.; Wang, Y.X.; Jiang, C.L. NLRP3 Inflammasome mediates chronic mild stress-induced depression in mice via neuroinflammation. Int. J. Neuropsychopharmacol. 2015, 18, pyv006. [Google Scholar] [CrossRef]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.K. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell. Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef]
- Bao, P.; Gong, Y.; Wang, Y.; Xu, M.; Qian, Z.; Ni, X.; Lu, J. Hydrogen sulfide prevents LPS-Induced depression-like behavior through the Suppression of NLRP3 Inflammasome and pyroptosis and the improvement of mitochondrial function in the hippocampus of mice. Biology 2023, 12, 1092. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Oxenkrug, G.F. Genetic and hormonal regulation of tryptophan kynurenine metabolism: Implications for vascular cognitive impairment, major depressive disorder, and aging. Ann. N. Y. Acad. Sci. 2007, 1122, 35–49. [Google Scholar] [CrossRef]
- Erhardt, S.; Lim, C.K.; Linderholm, K.R.; Janelidze, S.; Lindqvist, D.; Samuelsson, M.; Lundberg, K.; Postolache, T.T.; Träskman-Bendz, L.; Guillemin, G.J.; et al. Connecting inflammation with glutamate agonism in suicidality. Neuropsychopharmacology 2013, 38, 743–752. [Google Scholar] [CrossRef]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef]
- Czarny, P.; Wigner, P.; Galecki, P.; Sliwinski, T. The interplay between inflammation, oxidative stress, DNA damage, DNA repair and mitochondrial dysfunction in depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 80, 309–321. [Google Scholar] [CrossRef]
- Fries, G.R.; Saldana, V.A.; Finnstein, J.; Rein, T. Molecular pathways of major depressive disorder converge on the synapse. Mol. Psychiatry 2023, 28, 284–297. [Google Scholar] [CrossRef]
- Bhatt, S.; Nagappa, A.N.; Patil, C.R. Role of oxidative stress in depression. Drug Discov. Today 2020, 25, 1270–1276. [Google Scholar] [CrossRef]
- Beurel, E.; Toups, M.; Nemeroff, C.B. The bidirectional relationship of depression and inflammation: Double trouble. Neuron 2020, 107, 234–256. [Google Scholar] [CrossRef]
- Du, R.H.; Wu, F.F.; Lu, M.; Shu, X.D.; Ding, J.H.; Wu, G.; Hu, G. Uncoupling protein 2 modulation of the NLRP3 inflammasome in astrocytes and its implications in depression. Redox. Biol. 2016, 9, 178–187. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling. Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, J.; Luan, Y.; Li, X.; Meng, X.; Liao, W.; Tang, J.; Wang, Z. cGAS-STING, inflammasomes and pyroptosis: An overview of crosstalk mechanism of activation and regulation. Cell Commun. Signal. 2024, 22, 22. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhong, X.; Yi, Y.; Xie, L.; Zhou, W.; Cao, W.; Chen, L. Prophylactic effects of betaine on depression and anxiety behaviors in mice with dextran sulfate sodium-induced colitis. J. Agric. Food. Chem. 2024, 72, 21041–21051. [Google Scholar] [CrossRef]
- Wu, J.; Li, X.; Zhang, X.; Wang, W.; You, X. What role of the cGAS-STING pathway plays in chronic pain? Front. Mol. Neurosci. 2022, 15, 963206. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. STING-dependent cytosolic DNA sensing pathways. Trends. Immunol. 2014, 35, 88–93. [Google Scholar] [CrossRef]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO. Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Marchi, S.; Guilbaud, E.; Tait, S.W.G.; Yamazaki, T.; Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 2023, 23, 159–173. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef]
- Li, Z.R.; Liu, D.G.; Xie, S.; Wang, Y.H.; Han, Y.S.; Li, C.Y.; Zou, M.S.; Jiang, H.X. Sleep deprivation leads to further impairment of hippocampal synaptic plasticity by suppressing melatonin secretion in the pineal gland of chronically unpredictable stress rats. Eur. J. Pharmacol. 2022, 930, 175149. [Google Scholar] [CrossRef] [PubMed]
- Burkhalter, J.; Fiumelli, H.; Allaman, I.; Chatton, J.Y.; Martin, J.L. Brain-derived neurotrophic factor stimulates energy metabolism in developing cortical neurons. J. Neurosci. 2003, 23, 8212–8220. [Google Scholar] [CrossRef]
- Martinowich, K.; Lu, B. Interaction between BDNF and serotonin: Role in mood disorders. Neuropsychopharmacology 2008, 33, 73–83. [Google Scholar] [CrossRef]
- Kim, D.K.; Jeong, H.; Bae, J.; Cha, M.Y.; Kang, M.; Shin, D.; Ha, S.; Hyeon, S.J.; Kim, H.; Suh, K.; et al. Aβ-induced mitochondrial dysfunction in neural progenitors controls KDM5A to influence neuronal differentiation. Exp. Mol. Med. 2022, 54, 1461–1471. [Google Scholar] [CrossRef]
- Markham, A.; Cameron, I.; Bains, R.; Franklin, P.; Kiss, J.P.; Schwendimann, L.; Gressens, P.; Spedding, M. Brain-derived neurotrophic factor-mediated effects on mitochondrial respiratory coupling and neuroprotection share the same molecular signalling pathways. Eur. J. Neurosci. 2012, 35, 366–374. [Google Scholar] [CrossRef]
- Blendy, J.A. The role of CREB in depression and antidepressant treatment. Biol. Psychiatry 2006, 59, 1144–1150. [Google Scholar] [CrossRef]
- Yuan, P.; Zhou, R.; Wang, Y.; Li, X.; Li, J.; Chen, G.; Guitart, X.; Manji, H.K. Altered levels of extracellular signal-regulated kinase signaling proteins in postmortem frontal cortex of individuals with mood disorders and schizophrenia. J. Affect. Disord. 2010, 124, 164–169. [Google Scholar] [CrossRef]
- Li, Y.; Luikart, B.W.; Birnbaum, S.; Chen, J.; Kwon, C.H.; Kernie, S.G.; Bassel-Duby, R.; Parada, L.F. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 2008, 59, 399–412. [Google Scholar] [CrossRef]
- Sun, D.; Li, S.; Huang, H.; Xu, L. Neurotoxicity of melittin: Role of mitochondrial oxidative phosphorylation system in synaptic plasticity dysfunction. Toxicology 2023, 497–498, 153628. [Google Scholar] [CrossRef]
- Malhi, G.S.; Mann, J.J. Depression. Lancet 2018, 392, 2299–2312. [Google Scholar] [CrossRef]
- Takano, K.; Yamasaki, H.; Kawabe, K.; Moriyama, M.; Nakamura, Y. Imipramine induces brain-derived neurotrophic factor mRNA expression in cultured astrocytes. J. Pharmacol. Sci. 2012, 120, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Shi, X.; Liu, N.; Jiang, Z.; Ma, C.; Luo, G.; Liu, S.; Wei, X.; Liu, Y.; Ming, D. Photobiomodulation therapy mitigates depressive-like behaviors by remodeling synaptic links and mitochondrial function. J. Photochem. Photobiol. B 2024, 258, 112998. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Wang, L.; Sheng, H. Mitochondria in depression: The dysfunction of mitochondrial energy metabolism and quality control systems. CNS. Neurosci. Ther. 2024, 30, e14576. [Google Scholar] [CrossRef] [PubMed]
- Carling, D. AMPK signalling in health and disease. Curr. Opin. Cell. Biol. 2017, 45, 31–37. [Google Scholar] [CrossRef]
- Morales-Alamo, D.; Calbet, J.A.L. AMPK signaling in skeletal muscle during exercise: Role of reactive oxygen and nitrogen species. Free. Radic. Biol. Med. 2016, 98, 68–77. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Grahame; Hardie, D. AMP-activated protein kinase: A key regulator of energy balance with many roles in human disease. J. Intern. Med. 2014, 276, 543–559. [Google Scholar] [CrossRef]
- Alizadeh Pahlavani, H.; Laher, I.; Knechtle, B.; Zouhal, H. Exercise and mitochondrial mechanisms in patients with sarcopenia. Front. Physiol. 2022, 13, 1040381. [Google Scholar] [CrossRef]
- Lei, Y.; Wang, J.; Wang, D.; Li, C.; Liu, B.; Fang, X.; You, J.; Guo, M.; Lu, X.Y. SIRT1 in forebrain excitatory neurons produces sexually dimorphic effects on depression-related behaviors and modulates neuronal excitability and synaptic transmission in the medial prefrontal cortex. Mol. Psychiatry 2020, 25, 1094–1111. [Google Scholar] [CrossRef]
- Gurd, B.J. Deacetylation of PGC-1α by SIRT1: Importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl. Physiol. Nutr. Metab. 2011, 36, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Salomone, F.; Barbagallo, I.; Godos, J.; Lembo, V.; Currenti, W.; Cinà, D.; Avola, R.; D’Orazio, N.; Morisco, F.; Galvano, F.; et al. Silibinin restores NAD+ levels and induces the SIRT1/AMPK pathway in non-alcoholic fatty Liver. Nutrients 2017, 9, 1086. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef]
- Gibala, M.J.; McGee, S.L.; Garnham, A.P.; Howlett, K.; Snow, R.J.; Hargreaves, M. Brief intense interval exercise activates AMPK and p38 MAPK signaling and increases the expression of PGC-1alpha in human skeletal muscle. J. Appl. Physiol. 2009, 106, 929–934. [Google Scholar] [CrossRef]
- Bayod, S.; Del Valle, J.; Canudas, A.M.; Lalanza, J.F.; Sanchez-Roige, S.; Camins, A.; Escorihuela, R.M.; Pallàs, M. Long-term treadmill exercise induces neuroprotective molecular changes in rat brain. J. Appl. Physiol. 2011, 111, 1380–1390. [Google Scholar] [CrossRef]
- Qiao, X.; Yan, J.; Zang, Z.; Xi, L.; Zhu, W.; Zhang, E.; Wu, L. Association between IGF-1 levels and MDD: A case-control and meta-analysis. Front. Psychiatry 2024, 15, 1396938. [Google Scholar] [CrossRef]
- Kim, S.; Choi, J.Y.; Moon, S.; Park, D.H.; Kwak, H.B.; Kang, J.H. Roles of myokines in exercise-induced improvement of neuropsychiatric function. Pflugers Arch. 2019, 471, 491–505. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef]
- Al-Jarrah, M.D.; Erekat, N.S. Treadmill exercise training could attenuate the upregulation of Interleukin-1 beta and tumor necrosis factor alpha in the skeletal muscle of mouse model of chronic/progressive Parkinson disease. NeuroRehabilitation 2018, 43, 501–507. [Google Scholar] [CrossRef]
- Zhang, X.; He, Q.; Huang, T.; Zhao, N.; Liang, F.; Xu, B.; Chen, X.; Li, T.; Bi, J. Treadmill exercise decreases Aβ deposition and counteracts cognitive decline in APP/PS1 mice, possibly via hippocampal microglia modifications. Front. Aging Neurosci. 2019, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Ding, Y.; Xu, X. Anti-inflammatory effect of exercise training through reducing inflammasome activation-related inflammatory cytokine levels in overweight/obese populations: A systematic review and meta-analysis. Complement. Ther. Clin. Pract. 2022, 49, 101656. [Google Scholar] [CrossRef]
- Pedersen, B.K. Anti-inflammatory effects of exercise: Role in diabetes and cardiovascular disease. Eur. J. Clin. Investig. 2017, 47, 600–611. [Google Scholar] [CrossRef]
- Zhang, T.; Ding, S.; Wang, R. Research progress of mitochondrial mechanism in NLRP3 inflammasome activation and exercise regulation of NLRP3 Inflammasome. Int. J. Mol. Sci. 2021, 22, 10866. [Google Scholar] [CrossRef]
- Hu, S.; Wan, X.; Li, X.; Wang, X. Aerobic exercise alleviates pyroptosis-related diseases by regulating NLRP3 inflammasome. Front. Physiol. 2022, 13, 965366. [Google Scholar] [CrossRef]
- Liu, W.Y.; He, W.; Li, H. Exhaustive training increases uncoupling protein 2 expression and decreases Bcl-2/Bax ratio in rat skeletal muscle. Oxid. Med. Cell. Longev. 2013, 780719. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Brand, M.D. The regulation and physiology of mitochondrial proton leak. Physiology 2011, 26, 192–205. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef]
- Gleeson, M.; Bishop, N.C.; Stensel, D.J.; Lindley, M.R.; Mastana, S.S.; Nimmo, M.A. The anti-inflammatory effects of exercise: Mechanisms and implications for the prevention and treatment of disease. Nat. Rev. Immunol. 2011, 11, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Lim, S.G.; Suk, K.; Lee, W.H. Mitochondrial dysfunction regulates the JAK-STAT pathway via LKB1-mediated AMPK activation ER-stress-independent manner. Biochem. Cell Biol. 2020, 98, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Serafini, G. Neuroplasticity and major depression, the role of modern antidepressant drugs. World. J. Psychiatr. 2012, 2, 49–57. [Google Scholar] [CrossRef]
- Knaepen, K.; Goekint, M.; Heyman, E.M.; Meeusen, R. Neuroplasticity- exercise-induced response of peripheral brain-derived neurotrophic factor: A systematic review of experimental studies in human subjects. Sports Med. 2010, 40, 765–801. [Google Scholar] [CrossRef]
- Han, Q.; Wang, F. Electroacupuncture at GB20 improves cognitive ability and synaptic plasticity via the CaM-CaMKII-CREB signaling pathway following cerebral ischemia-reperfusion injury in rats. Acupunct. Med. 2024, 42, 23–31. [Google Scholar] [CrossRef]
- Radak, Z.; Hart, N.; Sarga, L.; Koltai, E.; Atalay, M.; Ohno, H.; Boldogh, I. Exercise plays a preventive role against Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, 777–783. [Google Scholar] [CrossRef]
- Almeida, R.D.; Manadas, B.J.; Melo, C.V.; Gomes, J.R.; Mendes, C.S.; Grãos, M.M.; Carvalho, R.F.; Carvalho, A.P.; Duarte, C.B. Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ. 2005, 12, 1329–1343. [Google Scholar] [CrossRef]
- Lee, H.Y.; Kim, Y.K. Plasma brain-derived neurotrophic factor as a peripheral marker for the action mechanism of antidepressants. Neuropsychobiology 2008, 57, 194–199. [Google Scholar] [CrossRef]
- De Sousa, R.A.L.; Rocha-Dias, I.; de Oliveira, L.R.S.; Improta-Caria, A.C.; Monteiro-Junior, R.S.; Cassilhas, R.C. Molecular mechanisms of physical exercise on depression in the elderly: A systematic review. Mol. Biol. Rep. 2021, 48, 3853–3862. [Google Scholar] [CrossRef]
- Seo, J.H.; Park, H.S.; Park, S.S.; Kim, C.J.; Kim, D.H.; Kim, T.W. Physical exercise ameliorates psychiatric disorders and cognitive dysfunctions by hippocampal mitochondrial function and neuroplasticity in post-traumatic stress disorder. Exp. Neurol. 2019, 322, 113043. [Google Scholar] [CrossRef]
- Utpal, B.K.; Roy, S.C.; Zehravi, M.; Sweilam, S.H.; Raja, A.D.; Haque, M.A.; Nayak, C.; Balakrishnan, S.; Singh, L.P.; Panigrahi, S.; et al. Polyphenols as Wnt/β-catenin pathway modulators: A promising strategy in clinical neurodegeneration. Anim. Models Exp. Med. 2025, 8, 266–286. [Google Scholar] [CrossRef]
- Alizadeh Pahlavani, H. Possible role of exercise therapy on depression: Effector neurotransmitters as key players. Behav. Brain Res. 2024, 459, 114791. [Google Scholar] [CrossRef]
- Xu, Y.; Li, Y.; Wang, C.; Han, T.; Wu, Y.; Wang, S.; Wei, J. Clinical value and mechanistic analysis of HIIT on modulating risk and symptoms of depression: A systematic review. Int. J. Clin. Health Psychol. 2024, 24, 100433. [Google Scholar] [CrossRef]
- De Araujo, G.G.; Papoti, M.; Reis, I.G.D.; De Mello, M.A.; Gobatto, C.A. Short and long term effects of high-intensity interval training on hormones, metabolites, antioxidant system, glycogen concentration, and aerobic performance adaptations in rats. Front. Physiol. 2016, 7, 505. [Google Scholar] [CrossRef]
- Blomqvist Mickelsson, T. Modern unexplored martial arts—What can mixed martial arts and Brazilian Jiu-Jitsu do for youth development? Eur. J. Sport Sci. 2020, 20, 386–393. [Google Scholar] [CrossRef]
- Eather, N.; Wade, L.; Pankowiak, A.; Eime, R. The impact of sports participation on mental health and social outcomes in adults: A systematic review and the ‘Mental Health through Sport’ conceptual model. Syst. Rev. 2023, 12, 102. [Google Scholar] [CrossRef]
- Chan, J.S.Y.; Liu, G.; Liang, D.; Deng, K.; Wu, J.; Yan, J.H. Special issue—Therapeutic benefits of physical activity for mood: A systematic review on the effects of exercise intensity, duration, and modality. J. Psychol. 2019, 153, 102–125. [Google Scholar] [CrossRef]
- Lezi, E.; Burns, J.M.; Swerdlow, R.H. Effect of high-intensity exercise on aged mouse brain mitochondria, neurogenesis, and inflammation. Neurobiol. Aging 2014, 35, 2574–2583. [Google Scholar]
- Fiorenza, M.; Gunnarsson, T.P.; Ehlers, T.S.; Bangsbo, J. High-intensity exercise training ameliorates aberrant expression of markers of mitochondrial turnover but not oxidative damage in skeletal muscle of men with essential hypertension. Acta Physiol. 2019, 227, e13344. [Google Scholar] [CrossRef]
- Kang, J.; Wang, Y.; Wang, D. Endurance and resistance training mitigate the negative consequences of depression on synaptic plasticity through different molecular mechanisms. Int. J. Neurosci. 2020, 130, 541–550. [Google Scholar] [CrossRef]
- Yuping, Z.; Tianbi, L.; Wentao, S.; Yun, L.; Guodong, Z. The optimal type and dose of exercise for elevating brain-derived neurotrophic factor levels in patients with depression: A systematic review with pairwise, network, and dose-response meta-analyses. Depress. Anxiety 2024, 2024, 5716755. [Google Scholar] [CrossRef]
- Ross, R.E.; Saladin, M.E.; George, M.S.; Gregory, C.M. High-intensity aerobic exercise acutely increases brain-derived neurotrophic factor. Med. Sci. Sports Exerc. 2019, 51, 1698–1709. [Google Scholar] [CrossRef]
- Kim, H.D.; Hesterman, J.; Call, T.; Magazu, S.; Keeley, E.; Armenta, K.; Kronman, H.; Neve, R.L.; Nestler, E.J.; Ferguson, D. SIRT1 mediates depression-like behaviors in the nucleus accumbens. J. Neurosci. 2016, 36, 8441–8452. [Google Scholar] [CrossRef]
- Abe-Higuchi, N.; Uchida, S.; Yamagata, H.; Higuchi, F.; Hobara, T.; Hara, K.; Kobayashi, A.; Watanabe, Y. Hippocampal sirtuin 1 signaling mediates depression-like behavior. Biol. Psychiatry 2016, 80, 815–826. [Google Scholar] [CrossRef]
- Guo, W.; Xiao, X.; Tian, Y.T.; Yang, J.J. The role and mechanism of SIRT1 gene in depression. Sheng Li Xue Bao 2021, 73, 828–834. [Google Scholar]
- Khalaj, L.; Nejad, S.C.; Mohammadi, M.; Zadeh, S.S.; Pour, M.H.; Ahmadiani, A.; Khodagholi, F.; Ashabi, G.; Alamdary, S.Z.; Samami, E. Gemfibrozil pretreatment proved protection against acute restraint stress-induced changes in the male rats’ hippocampus. Brain Res. 2013, 1527, 117–130. [Google Scholar] [CrossRef]
- Picard, M.; McManus, M.J.; Gray, J.D.; Nasca, C.; Moffat, C.; Kopinski, P.K.; Seifert, E.L.; McEwen, B.S.; Wallace, D.C. Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress. Proc. Natl. Acad. Sci. USA 2015, 112, E6614–E6623. [Google Scholar] [CrossRef]
- Hao, Y.; Ge, H.; Sun, M.; Gao, Y. Selecting an appropriate animal model of depression. Int. J. Mol. Sci. 2019, 20, 4827. [Google Scholar] [CrossRef]
- Disner, S.G.; Beevers, C.G.; Haigh, E.A.; Beck, A.T. Neural mechanisms of the cognitive model of depression. Nat. Rev. Neurosci. 2011, 12, 467–477. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Zhong, C.; Yang, Y.; Hu, J.; Yi, X.; Huang, J.; Li, H.; Liu, X.; Xue, K.; Chen, X. The Role of Mitochondrial Energy Metabolism in the Mechanism of Exercise Improving Depression. Curr. Issues Mol. Biol. 2025, 47, 382. https://doi.org/10.3390/cimb47050382
Liu Y, Zhong C, Yang Y, Hu J, Yi X, Huang J, Li H, Liu X, Xue K, Chen X. The Role of Mitochondrial Energy Metabolism in the Mechanism of Exercise Improving Depression. Current Issues in Molecular Biology. 2025; 47(5):382. https://doi.org/10.3390/cimb47050382
Chicago/Turabian StyleLiu, Yuwei, Chenghao Zhong, Yuxin Yang, Jianbo Hu, Xiaoyan Yi, Jiating Huang, Haonan Li, Xiaojie Liu, Ke Xue, and Xianghe Chen. 2025. "The Role of Mitochondrial Energy Metabolism in the Mechanism of Exercise Improving Depression" Current Issues in Molecular Biology 47, no. 5: 382. https://doi.org/10.3390/cimb47050382
APA StyleLiu, Y., Zhong, C., Yang, Y., Hu, J., Yi, X., Huang, J., Li, H., Liu, X., Xue, K., & Chen, X. (2025). The Role of Mitochondrial Energy Metabolism in the Mechanism of Exercise Improving Depression. Current Issues in Molecular Biology, 47(5), 382. https://doi.org/10.3390/cimb47050382