
Perturbation-Theory Machine Learning for Multi-Target Drug Discovery in Modern Anticancer Research




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
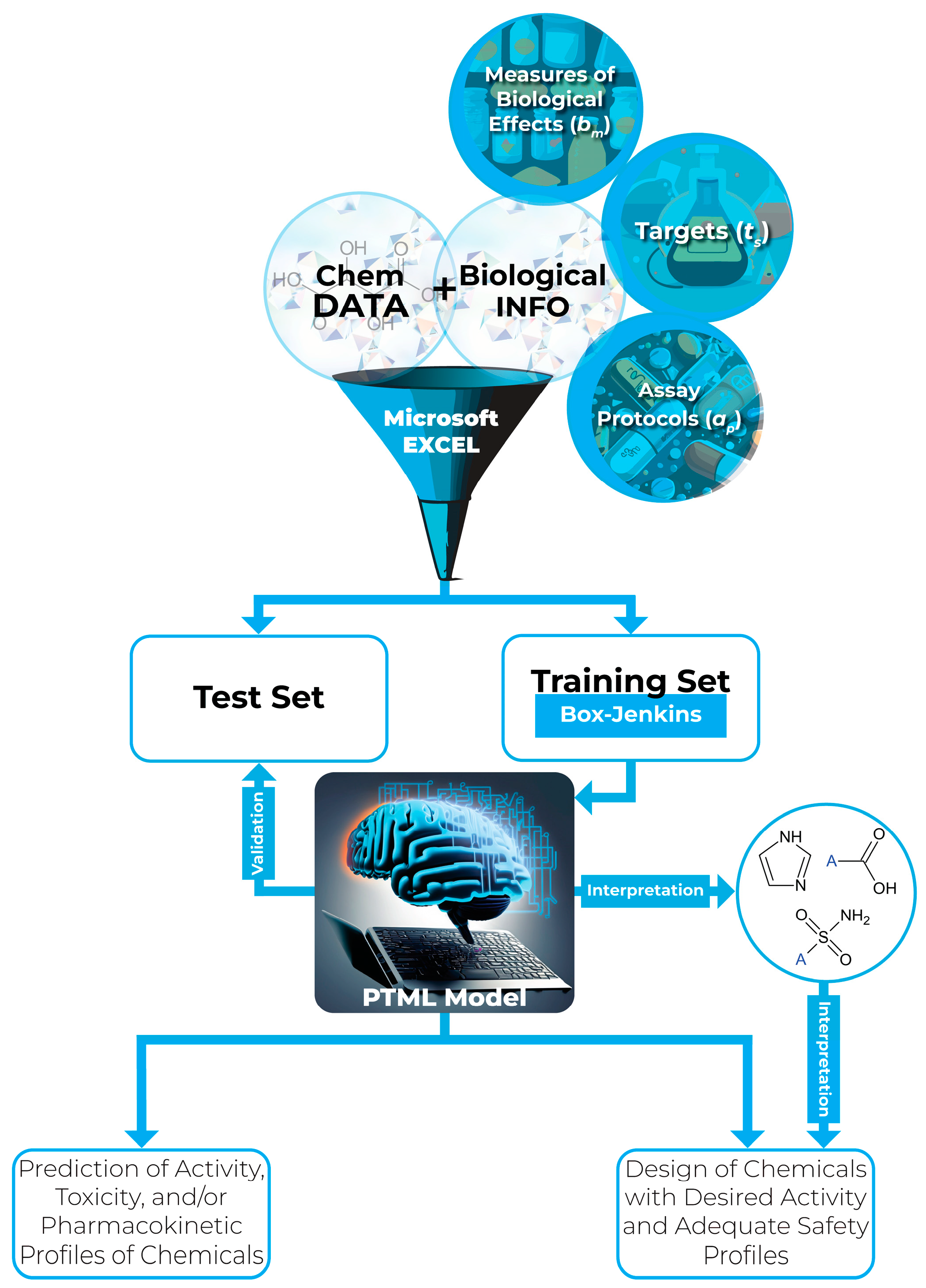
2. An Overview of PTML Modeling
3. PTML Models for MTDD-Based Anticancer Research
3.1. Key Aspects of the Analysis of PTML Modeling for MTDD-Based Anticancer Research
3.2. The PTML Approach for Modeling of Multi-Target Anticancer Activity
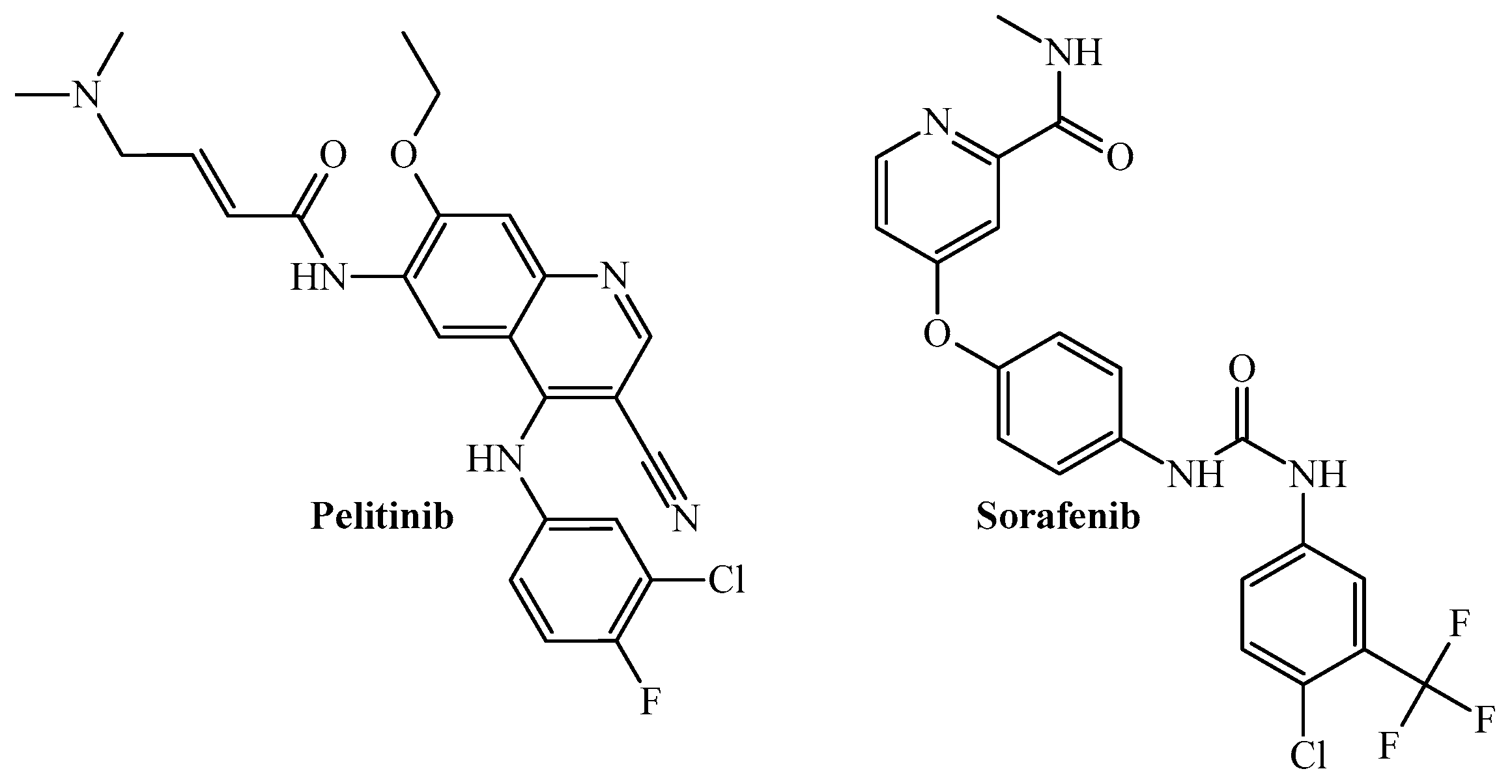
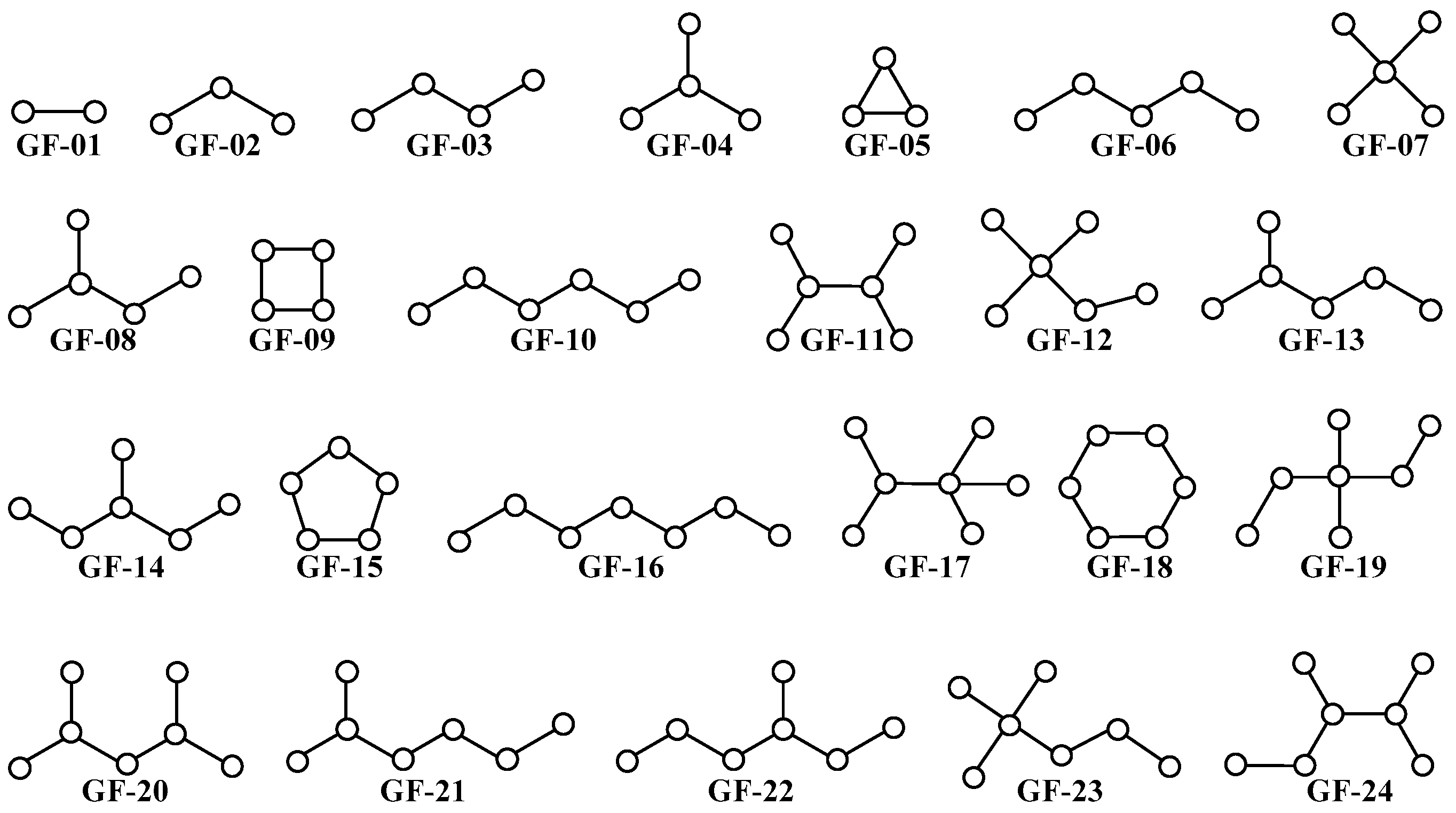
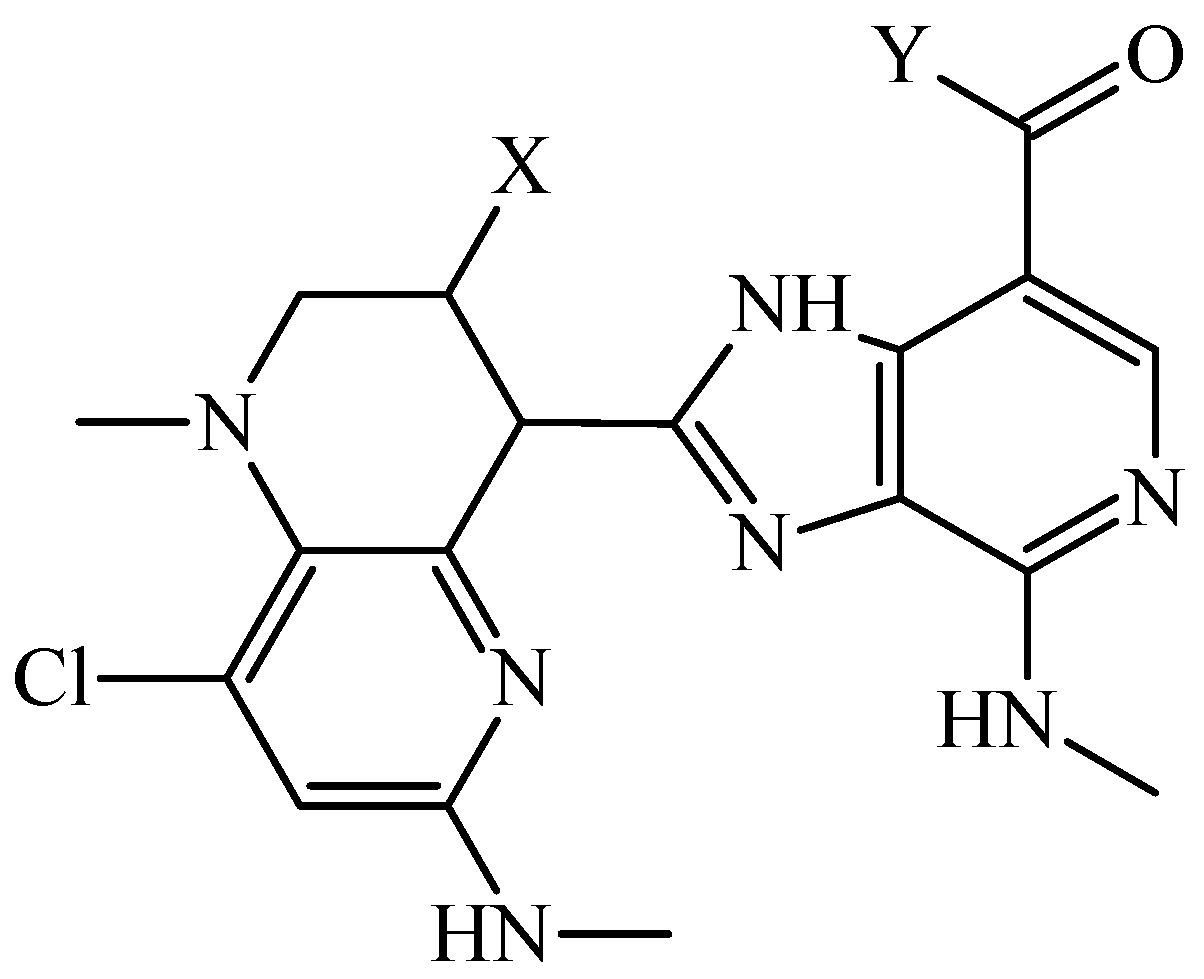
3.3. PTML Modeling for De Novo Drug Design in MTDD-Based Anticancer Research
3.4. Future Perspectives on PTML Modeling
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Acc | Accuracy |
| ANN | Artificial neural networks |
| ap | Assay conditions or protocols |
| avg[X]ej | Average value |
| bm | Biological effects |
| BRD2 | Bromodomain-containing protein 2 |
| BRD3 | Bromodomain-containing protein 3 |
| BRD4 | Bromodomain-containing protein 4 |
| CDK4 | Cyclin-dependent kinase 4 |
| ej | Experimental condition |
| FBTD | Fragment-based topological design |
| GF | Generic fragments |
| HER2 | Human epidermal growth factor receptor 2 |
| IC50 | Half-maximal inhibitory concentration |
| LDA | Linear discriminant analysis |
| logP | Logarithm of the n-octanol/water partition coefficient |
| MLIs | Multi-label indices; these are also denoted as D[X]ej in Equation (2) of this article |
| mt-QSAR | Multi-target QSAR model |
| mt-QSAR-ANN | Multi-target QSAR model based on an artificial neural network |
| mt-QSAR-EL-ANN | Multi-target QSAR model based on an ensemble of artificial neural networks |
| mt-QSAR-LDA | Multi-target QSAR model based on linear discriminant analysis |
| mtc-QSAR | Multi-condition QSAR |
| mtc-QSAR-LDA | Multi-condition QSAR based on linear discriminant analysis |
| MTDD | Multi-target drug discovery |
| mtk-QSBER | Multi-tasking model for quantitative structure-biological effect relationships |
| mtk-QSBER-LDA | Multi-tasking QSBER model based on linear discriminant analysis |
| n(ej) | Number of chemicals that comply with a specific experimental aspect of ej |
| Num | Numerator, which can be the range (difference between the maximum and minimum values of X), the standard deviation of X values, or a value of 1 |
| p(ej) | A priori probability of finding a chemical tested by considering a specific experimental aspect of ej |
| PSA | Polar surface area |
| PTML | Perturbation-theory machine learning |
| PTML-ANN | PTML model based on an artificial neural network |
| PTML-LDA | PTML model based on linear discriminant analysis |
| PTML-MLP | PTML model based on a multilayer perceptron network |
| QSAR | Quantitative structure-activity relationships |
| ts | Targets |
| RF | random forests |
| SMILES | Simplified molecular-input line-entry system |
| Sn | Sensitivity |
| Sp | Specificity |
| SPM | Statistical performance metrics |
| SVM | Support vector machines |
| X | Molecular descriptors |
| vsm | Numerical value for a particular SPM |
| Y | Exponent, which can take the values of −1, −0.5, 0, 0.5, or 1 |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Dominguez-Valentin, M.; Nakken, S.; Tubeuf, H.; Vodak, D.; Ekstrom, P.O.; Nissen, A.M.; Morak, M.; Holinski-Feder, E.; Holth, A.; Capella, G.; et al. Results of multigene panel testing in familial cancer cases without genetic cause demonstrated by single gene testing. Sci. Rep. 2019, 9, 18555. [Google Scholar] [CrossRef] [PubMed]
- Martin-Morales, L.; Rofes, P.; Diaz-Rubio, E.; Llovet, P.; Lorca, V.; Bando, I.; Perez-Segura, P.; de la Hoya, M.; Garre, P.; Garcia-Barberan, V.; et al. Novel genetic mutations detected by multigene panel are associated with hereditary colorectal cancer predisposition. PLoS ONE 2018, 13, e0203885. [Google Scholar] [CrossRef] [PubMed]
- Mezina, A.; Philips, N.; Bogus, Z.; Erez, N.; Xiao, R.; Fan, R.; Olthoff, K.M.; Reddy, K.R.; Samadder, N.J.; Nielsen, S.M.; et al. Multigene Panel Testing in Individuals With Hepatocellular Carcinoma Identifies Pathogenic Germline Variants. JCO Precis. Oncol. 2021, 5, 988–1000. [Google Scholar] [CrossRef]
- Hamilton, J.G.; Symecko, H.; Spielman, K.; Breen, K.; Mueller, R.; Catchings, A.; Trottier, M.; Salo-Mullen, E.E.; Shah, I.; Arutyunova, A.; et al. Uptake and acceptability of a mainstreaming model of hereditary cancer multigene panel testing among patients with ovarian, pancreatic, and prostate cancer. Genet. Med. 2021, 23, 2105–2113. [Google Scholar] [CrossRef]
- Hu, C.; LaDuca, H.; Shimelis, H.; Polley, E.C.; Lilyquist, J.; Hart, S.N.; Na, J.; Thomas, A.; Lee, K.Y.; Davis, B.T.; et al. Multigene Hereditary Cancer Panels Reveal High-Risk Pancreatic Cancer Susceptibility Genes. JCO Precis. Oncol. 2018, 2, 1–28. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.J.; Jang, H. Anticancer drug resistance: An update and perspective. Drug Resist. Updat. 2021, 59, 100796. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Hilal, T.; Gonzalez-Velez, M.; Prasad, V. Limitations in Clinical Trials Leading to Anticancer Drug Approvals by the US Food and Drug Administration. JAMA Intern. Med. 2020, 180, 1108–1115. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Schaduangrat, N.; Lampa, S.; Simeon, S.; Gleeson, M.P.; Spjuth, O.; Nantasenamat, C. Towards reproducible computational drug discovery. J. Cheminformatics 2020, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Brogi, S.; Ramalho, T.C.; Kuca, K.; Medina-Franco, J.L.; Valko, M. Editorial: In silico Methods for Drug Design and Discovery. Front. Chem. 2020, 8, 612. [Google Scholar] [CrossRef] [PubMed]
- Pirhadi, S.; Damghani, T.; Avestan, M.S.; Sharifi, S. Dual potent c-Met and ALK inhibitors: From common feature pharmacophore modeling to structure based virtual screening. J. Recept. Signal Transduct. Res. 2020, 40, 357–364. [Google Scholar] [CrossRef]
- He, Q.; Liu, C.; Wang, X.; Rong, K.; Zhu, M.; Duan, L.; Zheng, P.; Mi, Y. Exploring the mechanism of curcumin in the treatment of colon cancer based on network pharmacology and molecular docking. Front. Pharmacol. 2023, 14, 1102581. [Google Scholar] [CrossRef]
- Khalid, H.R.; Aamir, M.; Tabassum, S.; Alghamdi, Y.S.; Alzamami, A.; Ashfaq, U.A. Integrated System Pharmacology Approaches to Elucidate Multi-Target Mechanism of Solanum surattense against Hepatocellular Carcinoma. Molecules 2022, 27, 6220. [Google Scholar] [CrossRef]
- Batool, S.; Javed, M.R.; Aslam, S.; Noor, F.; Javed, H.M.F.; Seemab, R.; Rehman, A.; Aslam, M.F.; Paray, B.A.; Gulnaz, A. Network Pharmacology and Bioinformatics Approach Reveals the Multi-Target Pharmacological Mechanism of Fumaria indica in the Treatment of Liver Cancer. Pharmaceuticals 2022, 15, 654. [Google Scholar] [CrossRef]
- Ahmed, B.; Khan, S.; Nouroz, F.; Farooq, U.; Khalid, S. Exploring multi-target inhibitors using in silico approach targeting cell cycle dysregulator-CDK proteins. J. Biomol. Struct. Dyn. 2022, 40, 8825–8839. [Google Scholar] [CrossRef]
- Al-Khafaji, K.; Taskin Tok, T. Amygdalin as multi-target anticancer drug against targets of cell division cycle: Double docking and molecular dynamics simulation. J. Biomol. Struct. Dyn. 2021, 39, 1965–1974. [Google Scholar] [CrossRef]
- Deng, Z.; Chen, G.; Shi, Y.; Lin, Y.; Ou, J.; Zhu, H.; Wu, J.; Li, G.; Lv, L. Curcumin and its nano-formulations: Defining triple-negative breast cancer targets through network pharmacology, molecular docking, and experimental verification. Front. Pharmacol. 2022, 13, 920514. [Google Scholar] [CrossRef]
- Sharma, A.; Sinha, S.; Rathaur, P.; Vora, J.; Jha, P.C.; Johar, K.; Rawal, R.M.; Shrivastava, N. Reckoning apigenin and kaempferol as a potential multi-targeted inhibitor of EGFR/HER2-MEK pathway of metastatic colorectal cancer identified using rigorous computational workflow. Mol. Divers. 2022, 26, 3337–3356. [Google Scholar] [CrossRef] [PubMed]
- Prabhavathi, H.; Dasegowda, K.R.; Renukananda, K.H.; Karunakar, P.; Lingaraju, K.; Raja Naika, H. Molecular docking and dynamic simulation to identify potential phytocompound inhibitors for EGFR and HER2 as anti-breast cancer agents. J. Biomol. Struct. Dyn. 2022, 40, 4713–4724. [Google Scholar] [CrossRef] [PubMed]
- Elasbali, A.M.; Al-Soud, W.A.; Mousa Elayyan, A.E.; Al-Oanzi, Z.H.; Alhassan, H.H.; Mohamed, B.M.; Alanazi, H.H.; Ashraf, M.S.; Moiz, S.; Patel, M.; et al. Integrating network pharmacology approaches for the investigation of multi-target pharmacological mechanism of 6-shogaol against cervical cancer. J. Biomol. Struct. Dyn. 2023, 41, 14135–14151. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Sardina, D.S.; Gulotta, M.R.; Perricone, U. KUALA: A machine learning-driven framework for kinase inhibitors repositioning. Sci. Rep. 2022, 12, 17877. [Google Scholar] [CrossRef]
- Brindha, G.R.; Rishiikeshwer, B.S.; Santhi, B.; Nakendraprasath, K.; Manikandan, R.; Gandomi, A.H. Precise prediction of multiple anticancer drug efficacy using multi target regression and support vector regression analysis. Comput. Methods Programs Biomed. 2022, 224, 107027. [Google Scholar] [CrossRef]
- Al Taweraqi, N.; King, R.D. Improved prediction of gene expression through integrating cell signalling models with machine learning. BMC Bioinform. 2022, 23, 323. [Google Scholar] [CrossRef]
- Nguyen, L.C.; Naulaerts, S.; Bruna, A.; Ghislat, G.; Ballester, P.J. Predicting Cancer Drug Response In Vivo by Learning an Optimal Feature Selection of Tumour Molecular Profiles. Biomedicines 2021, 9, 1319. [Google Scholar] [CrossRef]
- Simeon, S.; Ghislat, G.; Ballester, P. Characterizing the Relationship Between the Chemical Structures of Drugs and their Activities on Primary Cultures of Pediatric Solid Tumors. Curr. Med. Chem. 2021, 28, 7830–7839. [Google Scholar] [CrossRef]
- Gonzalez-Diaz, H.; Arrasate, S.; Gomez-SanJuan, A.; Sotomayor, N.; Lete, E.; Besada-Porto, L.; Ruso, J.M. General theory for multiple input-output perturbations in complex molecular systems. 1. Linear QSPR electronegativity models in physical, organic, and medicinal chemistry. Curr. Top. Med. Chem. 2013, 13, 1713–1741. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Multitasking models for quantitative structure-biological effect relationships: Current status and future perspectives to speed up drug discovery. Expert Opin. Drug Discov. 2015, 10, 245–256. [Google Scholar] [CrossRef]
- Halder, A.K.; Moura, A.S.; Cordeiro, M.N.D.S. Moving Average-Based Multitasking In Silico Classification Modeling: Where Do We Stand and What Is Next? Int. J. Mol. Sci. 2022, 23, 4937. [Google Scholar] [CrossRef] [PubMed]
- Kleandrova, V.V.; Cordeiro, M.N.D.S.; Speck-Planche, A. Optimizing drug discovery using multitasking models for quantitative structure-biological effect relationships: An update of the literature. Expert Opin. Drug Discov. 2023, 18, 1231–1243. [Google Scholar] [CrossRef] [PubMed]
- Kleandrova, V.V.; Cordeiro, M.N.D.S.; Speck-Planche, A. Current in silico methods for multi-target drug discovery in early anticancer research: The rise of the perturbation-theory machine learning approach. Future Med. Chem. 2023, 15, 1647–1650. [Google Scholar] [CrossRef]
- Kleandrova, V.V.; Cordeiro, M.N.D.S.; Speck-Planche, A. Perturbation-Theory Machine Learning for Multi-Objective Antibacterial Discovery: Current Status and Future Perspectives. Appl. Sci. 2025, 15, 1166. [Google Scholar] [CrossRef]
- Velasquez-Lopez, Y.; Ruiz-Escudero, A.; Arrasate, S.; Gonzalez-Diaz, H. Implementation of IFPTML Computational Models in Drug Discovery Against Flaviviridae Family. J. Chem. Inf. Model. 2024, 64, 1841–1852. [Google Scholar] [CrossRef] [PubMed]
- Dieguez-Santana, K.; Gonzalez-Diaz, H. Machine learning in antibacterial discovery and development: A bibliometric and network analysis of research hotspots and trends. Comput. Biol. Med. 2023, 155, 106638. [Google Scholar] [CrossRef]
- Santiago, C.; Ortega-Tenezaca, B.; Barbolla, I.; Fundora-Ortiz, B.; Arrasate, S.; Dea-Ayuela, M.A.; Gonzalez-Diaz, H.; Sotomayor, N.; Lete, E. Prediction of Antileishmanial Compounds: General Model, Preparation, and Evaluation of 2-Acylpyrrole Derivatives. J. Chem. Inf. Model. 2022, 62, 3928–3940. [Google Scholar] [CrossRef]
- Dieguez-Santana, K.; Casanola-Martin, G.M.; Torres, R.; Rasulev, B.; Green, J.R.; Gonzalez-Diaz, H. Machine Learning Study of Metabolic Networks vs ChEMBL Data of Antibacterial Compounds. Mol. Pharm. 2022, 19, 2151–2163. [Google Scholar] [CrossRef]
- Vasquez-Dominguez, E.; Armijos-Jaramillo, V.D.; Tejera, E.; Gonzalez-Diaz, H. Multioutput Perturbation-Theory Machine Learning (PTML) Model of ChEMBL Data for Antiretroviral Compounds. Mol. Pharm. 2019, 16, 4200–4212. [Google Scholar] [CrossRef]
- Nocedo-Mena, D.; Cornelio, C.; Camacho-Corona, M.D.R.; Garza-Gonzalez, E.; Waksman de Torres, N.; Arrasate, S.; Sotomayor, N.; Lete, E.; Gonzalez-Diaz, H. Modeling Antibacterial Activity with Machine Learning and Fusion of Chemical Structure Information with Microorganism Metabolic Networks. J. Chem. Inf. Model. 2019, 59, 1109–1120. [Google Scholar] [CrossRef]
- Quevedo-Tumailli, V.; Ortega-Tenezaca, B.; Gonzalez-Diaz, H. IFPTML Mapping of Drug Graphs with Protein and Chromosome Structural Networks vs. Pre-Clinical Assay Information for Discovery of Antimalarial Compounds. Int. J. Mol. Sci. 2021, 22, 13066. [Google Scholar] [CrossRef]
- Barbolla, I.; Hernandez-Suarez, L.; Quevedo-Tumailli, V.; Nocedo-Mena, D.; Arrasate, S.; Dea-Ayuela, M.A.; Gonzalez-Diaz, H.; Sotomayor, N.; Lete, E. Palladium-mediated synthesis and biological evaluation of C-10b substituted Dihydropyrrolo[1,2-b]isoquinolines as antileishmanial agents. Eur. J. Med. Chem. 2021, 220, 113458. [Google Scholar] [CrossRef]
- Herrera-Ibata, D.M.; Pazos, A.; Orbegozo-Medina, R.A.; Romero-Duran, F.J.; Gonzalez-Diaz, H. Mapping chemical structure-activity information of HAART-drug cocktails over complex networks of AIDS epidemiology and socioeconomic data of U.S. counties. Biosystems 2015, 132–133, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Ibata, D.M.; Orbegozo-Medina, R.A.; Gonzalez-Diaz, H. Multiscale mapping of AIDS in U.S. countries vs anti-HIV drugs activity with complex networks and information indices. Curr. Bioinform. 2015, 10, 639–657. [Google Scholar] [CrossRef]
- Baltasar-Marchueta, M.; Llona, L.; M-Alicante, S.; Barbolla, I.; Ibarluzea, M.G.; Ramis, R.; Salomon, A.M.; Fundora, B.; Araujo, A.; Muguruza-Montero, A.; et al. Identification of Riluzole derivatives as novel calmodulin inhibitors with neuroprotective activity by a joint synthesis, biosensor, and computational guided strategy. Biomed. Pharmacother. 2024, 174, 116602. [Google Scholar] [CrossRef] [PubMed]
- Sampaio-Dias, I.E.; Rodriguez-Borges, J.E.; Yanez-Perez, V.; Arrasate, S.; Llorente, J.; Brea, J.M.; Bediaga, H.; Vina, D.; Loza, M.I.; Caamano, O.; et al. Synthesis, Pharmacological, and Biological Evaluation of 2-Furoyl-Based MIF-1 Peptidomimetics and the Development of a General-Purpose Model for Allosteric Modulators (ALLOPTML). ACS Chem. Neurosci. 2021, 12, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Diez-Alarcia, R.; Yanez-Perez, V.; Muneta-Arrate, I.; Arrasate, S.; Lete, E.; Meana, J.J.; Gonzalez-Diaz, H. Big Data Challenges Targeting Proteins in GPCR Signaling Pathways; Combining PTML-ChEMBL Models and [(35)S]GTPgammaS Binding Assays. ACS Chem. Neurosci. 2019, 10, 4476–4491. [Google Scholar] [CrossRef]
- Ferreira da Costa, J.; Silva, D.; Caamano, O.; Brea, J.M.; Loza, M.I.; Munteanu, C.R.; Pazos, A.; Garcia-Mera, X.; Gonzalez-Diaz, H. Perturbation Theory/Machine Learning Model of ChEMBL Data for Dopamine Targets: Docking, Synthesis, and Assay of New l-Prolyl-l-leucyl-glycinamide Peptidomimetics. ACS Chem. Neurosci. 2018, 9, 2572–2587. [Google Scholar] [CrossRef]
- Abeijon, P.; Garcia-Mera, X.; Caamano, O.; Yanez, M.; Lopez-Castro, E.; Romero-Duran, F.J.; Gonzalez-Diaz, H. Multi-Target Mining of Alzheimer Disease Proteome with Hansch’s QSBR-Perturbation Theory and Experimental-Theoretic Study of New Thiophene Isosters of Rasagiline. Curr. Drug Targets 2017, 18, 511–521. [Google Scholar] [CrossRef]
- Romero-Duran, F.J.; Alonso, N.; Yanez, M.; Caamano, O.; Garcia-Mera, X.; Gonzalez-Diaz, H. Brain-inspired cheminformatics of drug-target brain interactome, synthesis, and assay of TVP1022 derivatives. Neuropharmacology 2016, 103, 270–278. [Google Scholar] [CrossRef]
- He, S.; Segura Abarrategi, J.; Bediaga, H.; Arrasate, S.; Gonzalez-Diaz, H. On the additive artificial intelligence-based discovery of nanoparticle neurodegenerative disease drug delivery systems. Beilstein J. Nanotechnol. 2024, 15, 535–555. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Nader, K.; Abarrategi, J.S.; Bediaga, H.; Nocedo-Mena, D.; Ascencio, E.; Casanola-Martin, G.M.; Castellanos-Rubio, I.; Insausti, M.; Rasulev, B.; et al. NANO.PTML model for read-across prediction of nanosystems in neurosciences. computational model and experimental case of study. J. Nanobiotechnol. 2024, 22, 435. [Google Scholar] [CrossRef]
- Diéguez-Santana, K.; Rasulev, B.; González-Díaz, H. Towards rational nanomaterial design by predicting drug–nanoparticle system interaction vs. bacterial metabolic networks. Environ. Sci. Nano 2022, 9, 1391–1413. [Google Scholar] [CrossRef]
- Ortega-Tenezaca, B.; Gonzalez-Diaz, H. IFPTML mapping of nanoparticle antibacterial activity vs. pathogen metabolic networks. Nanoscale 2021, 13, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, C.R.; Gutierrez-Asorey, P.; Blanes-Rodriguez, M.; Hidalgo-Delgado, I.; Blanco Liverio, M.J.; Castineiras Galdo, B.; Porto-Pazos, A.B.; Gestal, M.; Arrasate, S.; Gonzalez-Diaz, H. Prediction of Anti-Glioblastoma Drug-Decorated Nanoparticle Delivery Systems Using Molecular Descriptors and Machine Learning. Int. J. Mol. Sci. 2021, 22, 11519. [Google Scholar] [CrossRef]
- Dieguez-Santana, K.; Gonzalez-Diaz, H. Towards machine learning discovery of dual antibacterial drug-nanoparticle systems. Nanoscale 2021, 13, 17854–17870. [Google Scholar] [CrossRef] [PubMed]
- Urista, D.V.; Carrue, D.B.; Otero, I.; Arrasate, S.; Quevedo-Tumailli, V.F.; Gestal, M.; Gonzalez-Diaz, H.; Munteanu, C.R. Prediction of Antimalarial Drug-Decorated Nanoparticle Delivery Systems with Random Forest Models. Biology 2020, 9, 198. [Google Scholar] [CrossRef]
- Santana, R.; Zuluaga, R.; Ganan, P.; Arrasate, S.; Onieva, E.; Montemore, M.M.; Gonzalez-Diaz, H. PTML Model for Selection of Nanoparticles, Anticancer Drugs, and Vitamins in the Design of Drug-Vitamin Nanoparticle Release Systems for Cancer Cotherapy. Mol. Pharm. 2020, 17, 2612–2627. [Google Scholar] [CrossRef]
- Santana, R.; Zuluaga, R.; Ganan, P.; Arrasate, S.; Onieva, E.; Gonzalez-Diaz, H. Predicting coated-nanoparticle drug release systems with perturbation-theory machine learning (PTML) models. Nanoscale 2020, 12, 13471–13483. [Google Scholar] [CrossRef]
- Tenorio-Borroto, E.; Castanedo, N.; Garcia-Mera, X.; Rivadeneira, K.; Vazquez Chagoyan, J.C.; Barbabosa Pliego, A.; Munteanu, C.R.; Gonzalez-Diaz, H. Perturbation Theory Machine Learning Modeling of Immunotoxicity for Drugs Targeting Inflammatory Cytokines and Study of the Antimicrobial G1 Using Cytometric Bead Arrays. Chem. Res. Toxicol. 2019, 32, 1811–1823. [Google Scholar] [CrossRef]
- Vazquez-Prieto, S.; Paniagua, E.; Solana, H.; Ubeira, F.M.; Gonzalez-Diaz, H. A study of the Immune Epitope Database for some fungi species using network topological indices. Mol. Divers. 2017, 21, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Arzate, S.G.; Tenorio-Borroto, E.; Barbabosa Pliego, A.; Diaz-Albiter, H.M.; Vazquez-Chagoyan, J.C.; Gonzalez-Diaz, H. PTML Model for Proteome Mining of B-Cell Epitopes and Theoretical-Experimental Study of Bm86 Protein Sequences from Colima, Mexico. J. Proteome Res. 2017, 16, 4093–4103. [Google Scholar] [CrossRef]
- Tenorio-Borroto, E.; Penuelas-Rivas, C.G.; Vasquez-Chagoyan, J.C.; Castanedo, N.; Prado-Prado, F.J.; Garcia-Mera, X.; Gonzalez-Diaz, H. Model for high-throughput screening of drug immunotoxicity—Study of the anti-microbial G1 over peritoneal macrophages using flow cytometry. Eur. J. Med. Chem. 2014, 72, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Daghighi, A.; Casanola-Martin, G.M.; Iduoku, K.; Kusic, H.; Gonzalez-Diaz, H.; Rasulev, B. Multi-Endpoint Acute Toxicity Assessment of Organic Compounds Using Large-Scale Machine Learning Modeling. Environ. Sci. Technol. 2024, 58, 10116–10127. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. (Eds.) Molecular Descriptors for Chemoinformatics; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; Volume I–II. [Google Scholar]
- Estrada, E.; Gutiérrez, Y. MODESLAB, Santiago de Compostela, Spain, 2004; v1.5.
- Todeschini, R.; Consonni, V.; Mauri, A.; Pavan, M. DRAGON for Windows (Software for Molecular Descriptor Calculations), Milano Chemometrics and QSAR Research Group: Milano, Italy, 2005; v5.3.
- Valdés-Martini, J.R.; García-Jacas, C.R.; Marrero-Ponce, Y.; Silveira Vaz ‘d Almeida, Y.; Morell, C. QUBILs-MAS: Free Software for Molecular Descriptors Calculator from Quadratic, Bilinear and Linear Maps Based on Graph-Theoretic Electronic-Density Matrices and Atomic Weightings, v1.0. CAMD-BIR Unit, CENDA Registration Number: 2373-2012: Villa Clara, Cuba, 2012. Available online: https://tomocomd.com/ (accessed on 15 February 2025).
- Medina Marrero, R.; Marrero-Ponce, Y.; Barigye, S.J.; Echeverria Diaz, Y.; Acevedo-Barrios, R.; Casanola-Martin, G.M.; Garcia Bernal, M.; Torrens, F.; Perez-Gimenez, F. QuBiLs-MAS method in early drug discovery and rational drug identification of antifungal agents. SAR QSAR Environ. Res. 2015, 26, 943–958. [Google Scholar] [CrossRef]
- Valdes-Martini, J.R.; Marrero-Ponce, Y.; Garcia-Jacas, C.R.; Martinez-Mayorga, K.; Barigye, S.J.; Vaz d’Almeida, Y.S.; Pham-The, H.; Perez-Gimenez, F.; Morell, C.A. QuBiLS-MAS, open source multi-platform software for atom- and bond-based topological (2D) and chiral (2.5D) algebraic molecular descriptors computations. J. Cheminformatics 2017, 9, 35. [Google Scholar] [CrossRef]
- TIBCO-Software-Inc. STATISTICA (Data Analysis Software System), TIBCO-Software-Inc.: Palo Alto, CA, USA, 2018; v13.5.0.17.
- Marzaro, G.; Chilin, A.; Guiotto, A.; Uriarte, E.; Brun, P.; Castagliuolo, I.; Tonus, F.; Gonzalez-Diaz, H. Using the TOPS-MODE approach to fit multi-target QSAR models for tyrosine kinases inhibitors. Eur. J. Med. Chem. 2011, 46, 2185–2192. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Fragment-based QSAR model toward the selection of versatile anti-sarcoma leads. Eur. J. Med. Chem. 2011, 46, 5910–5916. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Cordeiro, M.N.D.S. In Bladder Cancer: Risk Factors, Emerging Treatment Strategies and Challenges; Haggerty, S., Ed.; Multi-tasking chemoinformatic model for the efficient discovery of potent and safer anti-bladder cancer agents. Nova Science Publishers, Inc.: New York, NY, USA, 2014; pp. 71–94. [Google Scholar]
- Pelon, M.; Krzeminski, P.; Tracz-Gaszewska, Z.; Misiewicz-Krzeminska, I. Factors determining the sensitivity to proteasome inhibitors of multiple myeloma cells. Front. Pharmacol. 2024, 15, 1351565. [Google Scholar] [CrossRef]
- Velez, B.; Razi, A.; Hubbard, R.D.; Walsh, R.; Rawson, S.; Tian, G.; Finley, D.; Hanna, J. Rational design of proteasome inhibitors based on the structure of the endogenous inhibitor PI31/Fub1. Proc. Natl. Acad. Sci. USA 2023, 120, e2308417120. [Google Scholar] [CrossRef]
- Bennett, M.K.; Li, M.; Tea, M.N.; Pitman, M.R.; Toubia, J.; Wang, P.P.; Anderson, D.; Creek, D.J.; Orlowski, R.Z.; Gliddon, B.L.; et al. Resensitising proteasome inhibitor-resistant myeloma with sphingosine kinase 2 inhibition. Neoplasia 2022, 24, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Casanola-Martin, G.M.; Le-Thi-Thu, H.; Perez-Gimenez, F.; Marrero-Ponce, Y.; Merino-Sanjuan, M.; Abad, C.; Gonzalez-Diaz, H. Multi-output model with Box-Jenkins operators of linear indices to predict multi-target inhibitors of ubiquitin-proteasome pathway. Mol. Divers. 2015, 19, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Bediaga, H.; Arrasate, S.; Gonzalez-Diaz, H. PTML Combinatorial Model of ChEMBL Compounds Assays for Multiple Types of Cancer. ACS Comb. Sci. 2018, 20, 621–632. [Google Scholar] [CrossRef]
- Cabrera-Andrade, A.; Lopez-Cortes, A.; Munteanu, C.R.; Pazos, A.; Perez-Castillo, Y.; Tejera, E.; Arrasate, S.; Gonzalez-Diaz, H. Perturbation-Theory Machine Learning (PTML) Multilabel Model of the ChEMBL Dataset of Preclinical Assays for Antisarcoma Compounds. ACS Omega 2020, 5, 27211–27220. [Google Scholar] [CrossRef] [PubMed]
- Atz, K.; Cotos, L.; Isert, C.; Hakansson, M.; Focht, D.; Hilleke, M.; Nippa, D.F.; Iff, M.; Ledergerber, J.; Schiebroek, C.C.G.; et al. Prospective de novo drug design with deep interactome learning. Nat. Commun. 2024, 15, 3408. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Z.; Sun, H.; Wang, J.; Shen, C.; Weng, G.; Chai, X.; Li, H.; Cao, D.; Hou, T. Deep learning approaches for de novo drug design: An overview. Curr. Opin. Struct. Biol. 2022, 72, 135–144. [Google Scholar] [CrossRef]
- Moret, M.; Pachon Angona, I.; Cotos, L.; Yan, S.; Atz, K.; Brunner, C.; Baumgartner, M.; Grisoni, F.; Schneider, G. Leveraging molecular structure and bioactivity with chemical language models for de novo drug design. Nat. Commun. 2023, 14, 114. [Google Scholar] [CrossRef]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in de Novo Drug Design: From Conventional to Machine Learning Methods. Int. J. Mol. Sci. 2021, 22, 1676. [Google Scholar] [CrossRef]
- Kleandrova, V.V.; Speck-Planche, A. The QSAR Paradigm in Fragment-Based Drug Discovery: From the Virtual Generation of Target Inhibitors to Multi-Scale Modeling. Mini Rev. Med. Chem. 2020, 20, 1357–1374. [Google Scholar] [CrossRef]
- Estrada, E.; Molina, E.; Perdomo-Lopez, I. Can 3D structural parameters be predicted from 2D (topological) molecular descriptors? J. Chem. Inf. Comput. Sci. 2001, 41, 1015–1021. [Google Scholar] [CrossRef]
- Estrada, E. Physicochemical Interpretation of Molecular Connectivity Indices. J. Phys. Chem. A 2002, 106, 9085–9091. [Google Scholar] [CrossRef]
- Estrada, E. Edge adjacency relationship and a novel topological index related to molecular volume. J. Chem. Inf. Comput. Sci. 1995, 35, 31–33. [Google Scholar] [CrossRef]
- Estrada, E. Spectral moments of the edge adjacency matrix in molecular graphs. 1. Definition and applications for the prediction of physical properties of alkanes. J. Chem. Inf. Comput. Sci. 1996, 36, 844–849. [Google Scholar] [CrossRef]
- Estrada, E. Spectral moments of the edge adjacency matrix in molecular graphs. 2. Molecules containing heteroatoms and QSAR applications. J. Chem. Inf. Comput. Sci. 1997, 37, 320–328. [Google Scholar] [CrossRef]
- Estrada, E. Spectral moments of the edge adjacency matrix in molecular graphs. 3. Molecules containing cycles. J. Chem. Inf. Comput. Sci. 1998, 38, 23–27. [Google Scholar] [CrossRef]
- Estrada, E.; Pena, A.; Garcia-Domenech, R. Designing sedative/hypnotic compounds from a novel substructural graph-theoretical approach. J. Comput. Aided Mol. Des. 1998, 12, 583–595. [Google Scholar] [CrossRef]
- Kier, L.B.; Hall, L.H. Molecular Connectivity in Structure-Activity Analysis; John Wiley & Sons: New York, NY, USA, 1986. [Google Scholar]
- Baskin, I.I.; Skvortsova, M.I.; Stankevich, I.V.; Zefirov, N.S. On the basis of invariants of labeled molecular graphs. J. Chem. Inf. Comput. Sci. 1995, 35, 527–531. [Google Scholar] [CrossRef]
- Baskin, I.; Varnek, A. In Chemoinformatics Approaches to Virtual Screening; Varnek, A., Tropsha, A., Eds.; Fragment descriptors in SAR/QSAR/QSPR studies, molecular similarity analysis and in virtual screening. Royal Society of Chemistry: Cambridge, UK, 2008; pp. 1–43. [Google Scholar]
- Kleandrova, V.V.; Cordeiro, M.N.D.S.; Speck-Planche, A. In Silico Approach for Antibacterial Discovery: PTML Modeling of Virtual Multi-Strain Inhibitors Against Staphylococcus aureus. Pharmaceuticals 2025, 18, 196. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Multi-target drug discovery in anti-cancer therapy: Fragment-based approach toward the design of potent and versatile anti-prostate cancer agents. Bioorg. Med. Chem. 2011, 19, 6239–6244. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Chemoinformatics in anti-cancer chemotherapy: Multi-target QSAR model for the in silico discovery of anti-breast cancer agents. Eur. J. Pharm. Sci. 2012, 47, 273–279. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Chemoinformatics in multi-target drug discovery for anti-cancer therapy: In silico design of potent and versatile anti-brain tumor agents. Anticancer Agents Med. Chem. 2012, 12, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Rational drug design for anti-cancer chemotherapy: Multi-target QSAR models for the in silico discovery of anti-colorectal cancer agents. Bioorg. Med. Chem. 2012, 20, 4848–4855. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Unified multi-target approach for the rational in silico design of anti-bladder cancer agents. Anticancer Agents Med. Chem. 2013, 13, 791–800. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Cordeiro, M. Fragment-based in silico modeling of multi-target inhibitors against breast cancer-related proteins. Mol. Divers. 2017, 21, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Kleandrova, V.V.; Scotti, M.T.; Scotti, L.; Speck-Planche, A. Multi-Target Drug Discovery Via PTML Modeling: Applications to the Design of Virtual Dual Inhibitors of CDK4 and HER2. Curr. Top. Med. Chem. 2021, 21, 661–675. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Scotti, M.T. BET bromodomain inhibitors: Fragment-based in silico design using multi-target QSAR models. Mol. Divers. 2019, 23, 555–572. [Google Scholar] [CrossRef]
- Kleandrova, V.V.; Scotti, M.T.; Scotti, L.; Nayarisseri, A.; Speck-Planche, A. Cell-based multi-target QSAR model for design of virtual versatile inhibitors of liver cancer cell lines. SAR QSAR Environ. Res. 2020, 31, 815–836. [Google Scholar] [CrossRef]
- Speck-Planche, A. Multicellular Target QSAR Model for Simultaneous Prediction and Design of Anti-Pancreatic Cancer Agents. ACS Omega 2019, 4, 3122–3132. [Google Scholar] [CrossRef]
- Kleandrova, V.V.; Speck-Planche, A. PTML Modeling for Pancreatic Cancer Research: In Silico Design of Simultaneous Multi-Protein and Multi-Cell Inhibitors. Biomedicines 2022, 10, 491. [Google Scholar] [CrossRef]
- Kleandrova, V.V.; Cordeiro, M.N.D.S.; Speck-Planche, A. Perturbation Theory Machine Learning Model for Phenotypic Early Antineoplastic Drug Discovery: Design of Virtual Anti-Lung-Cancer Agents. Appl. Sci. 2024, 14, 9344. [Google Scholar] [CrossRef]
- Budczies, J.; Kazdal, D.; Menzel, M.; Beck, S.; Kluck, K.; Altbürger, C.; Schwab, C.; Allgäuer, M.; Ahadova, A.; Kloor, M.; et al. Tumour mutational burden: Clinical utility, challenges and emerging improvements. Nat. Rev. Clin. Oncol. 2024, 21, 725–742. [Google Scholar] [CrossRef]
- Lagunin, A.A.; Rudik, A.V.; Pogodin, P.V.; Savosina, P.I.; Tarasova, O.A.; Dmitriev, A.V.; Ivanov, S.M.; Biziukova, N.Y.; Druzhilovskiy, D.S.; Filimonov, D.A.; et al. CLC-Pred 2.0: A Freely Available Web Application for In Silico Prediction of Human Cell Line Cytotoxicity and Molecular Mechanisms of Action for Druglike Compounds. Int. J. Mol. Sci. 2023, 24, 1689. [Google Scholar] [CrossRef]
- Cieplinski, T.; Danel, T.; Podlewska, S.; Jastrzebski, S. Generative Models Should at Least Be Able to Design Molecules That Dock Well: A New Benchmark. J. Chem. Inf. Model. 2023, 63, 3238–3247. [Google Scholar] [CrossRef] [PubMed]
- Kleandrova, V.V.; Scotti, L.; Bezerra Mendonça Junior, F.J.; Muratov, E.; Scotti, M.T.; Speck-Planche, A. QSAR Modeling for Multi-Target Drug Discovery: Designing Simultaneous Inhibitors of Proteins in Diverse Pathogenic Parasites. Front. Chem. 2021, 9, 634663. [Google Scholar] [CrossRef] [PubMed]
- Chinnadurai, R.K.; Khan, N.; Meghwanshi, G.K.; Ponne, S.; Althobiti, M.; Kumar, R. Current research status of anti-cancer peptides: Mechanism of action, production, and clinical applications. Biomed. Pharmacother. 2023, 164, 114996. [Google Scholar] [CrossRef] [PubMed]
- Gurbuz, N.; Ozpolat, B. MicroRNA-based Targeted Therapeutics in Pancreatic Cancer. Anticancer Res. 2019, 39, 529–532. [Google Scholar] [CrossRef]
- Venkatesan, S.; Chanda, K.; Balamurali, M.M. Recent Advancements of Aptamers in Cancer Therapy. ACS Omega 2023, 8, 32231–32243. [Google Scholar] [CrossRef]
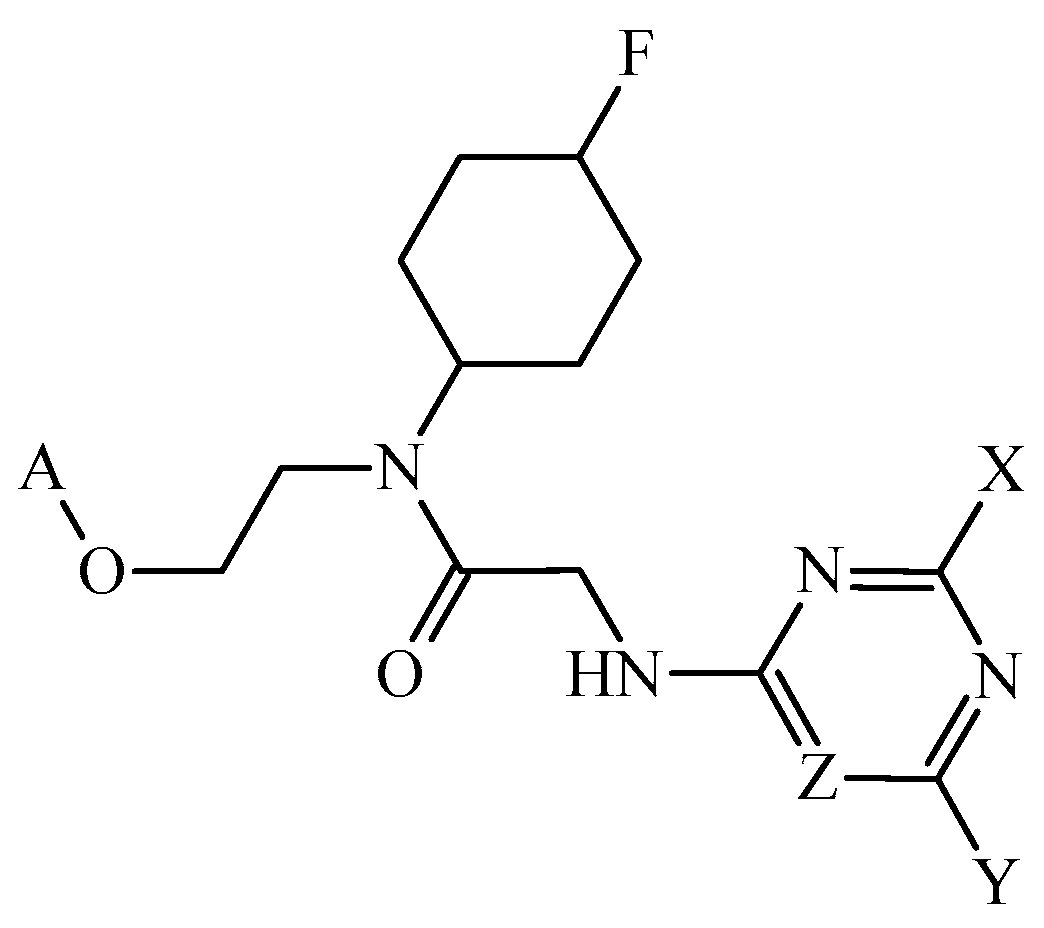
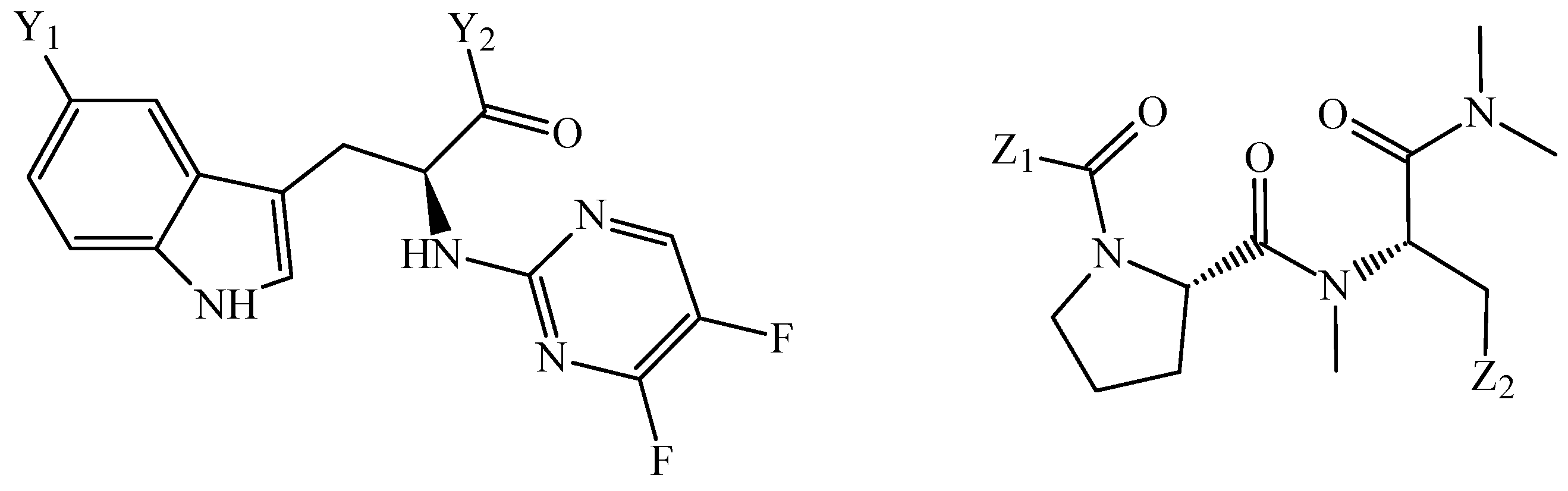
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleandrova, V.V.; Cordeiro, M.N.D.S.; Speck-Planche, A. Perturbation-Theory Machine Learning for Multi-Target Drug Discovery in Modern Anticancer Research. Curr. Issues Mol. Biol. 2025, 47, 301. https://doi.org/10.3390/cimb47050301
Kleandrova VV, Cordeiro MNDS, Speck-Planche A. Perturbation-Theory Machine Learning for Multi-Target Drug Discovery in Modern Anticancer Research. Current Issues in Molecular Biology. 2025; 47(5):301. https://doi.org/10.3390/cimb47050301
Chicago/Turabian StyleKleandrova, Valeria V., M. Natália D. S. Cordeiro, and Alejandro Speck-Planche. 2025. "Perturbation-Theory Machine Learning for Multi-Target Drug Discovery in Modern Anticancer Research" Current Issues in Molecular Biology 47, no. 5: 301. https://doi.org/10.3390/cimb47050301
APA StyleKleandrova, V. V., Cordeiro, M. N. D. S., & Speck-Planche, A. (2025). Perturbation-Theory Machine Learning for Multi-Target Drug Discovery in Modern Anticancer Research. Current Issues in Molecular Biology, 47(5), 301. https://doi.org/10.3390/cimb47050301