
Ultrasensitive CRISPR/Cas12a-Based System for Detection of BlaOXA-1 Gene in Antibiotic-Resistant Microorganisms

 , ,
, ,  , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Oligonucleotides, Recombinant Proteins, and Buffers
2.2. Selection of Target Sequences in the BlaOXA-1 Antibiotic Resistance Gene to Develop Guide RNAs
2.3. Construction of a Model DNA Matrix Containing the Target blaOXA-1 Gene Fragment
2.4. BlaOXA-1 Target Gene Fragment Pre-Amplification
2.5. BlaOXA-1-Specific RNP Assembly and CRISPR/Cas12a Fluorescence Assay
2.6. Detection of the Target BlaOXA-1 Gene in Real Samples
2.7. Data Processing
3. Results
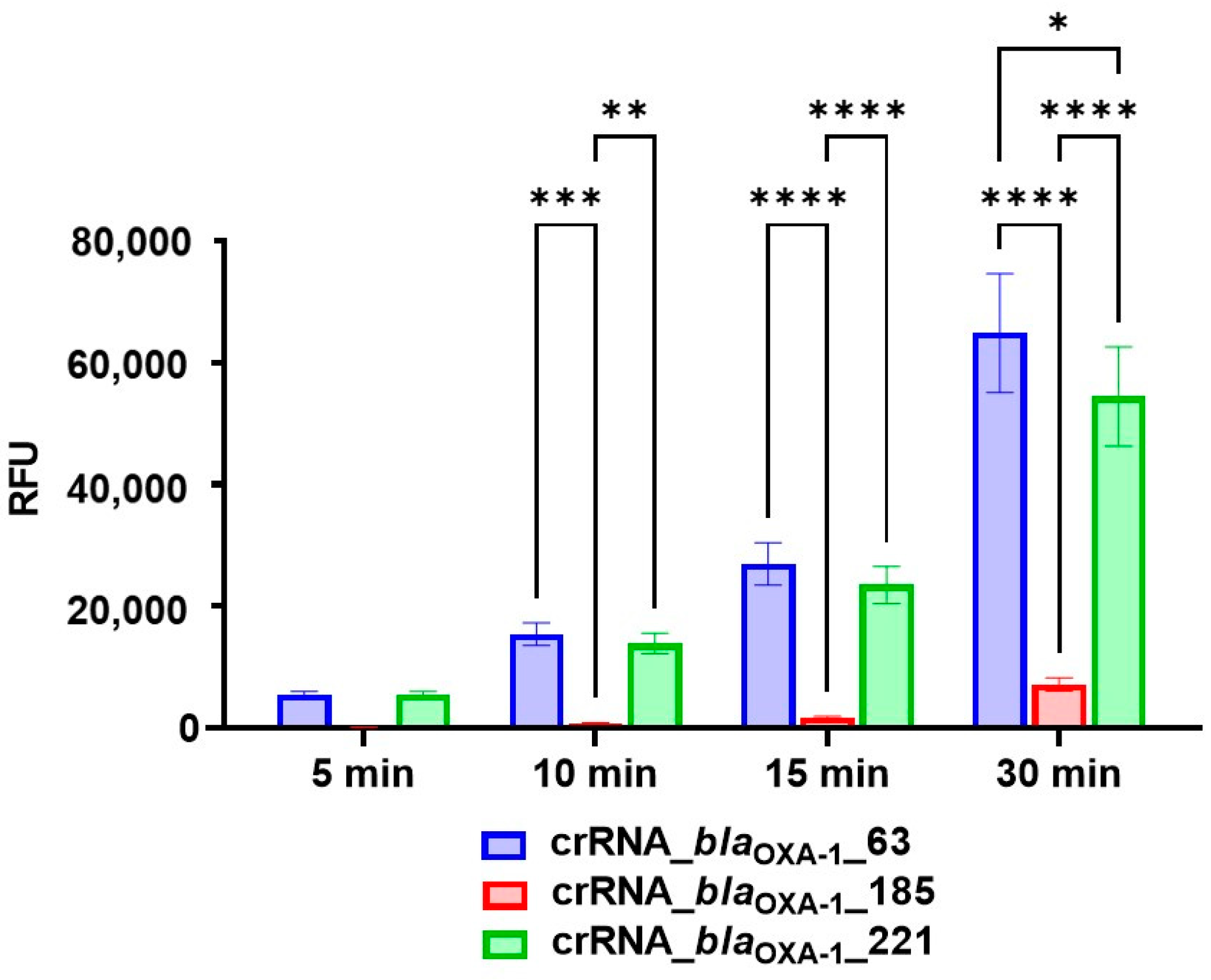
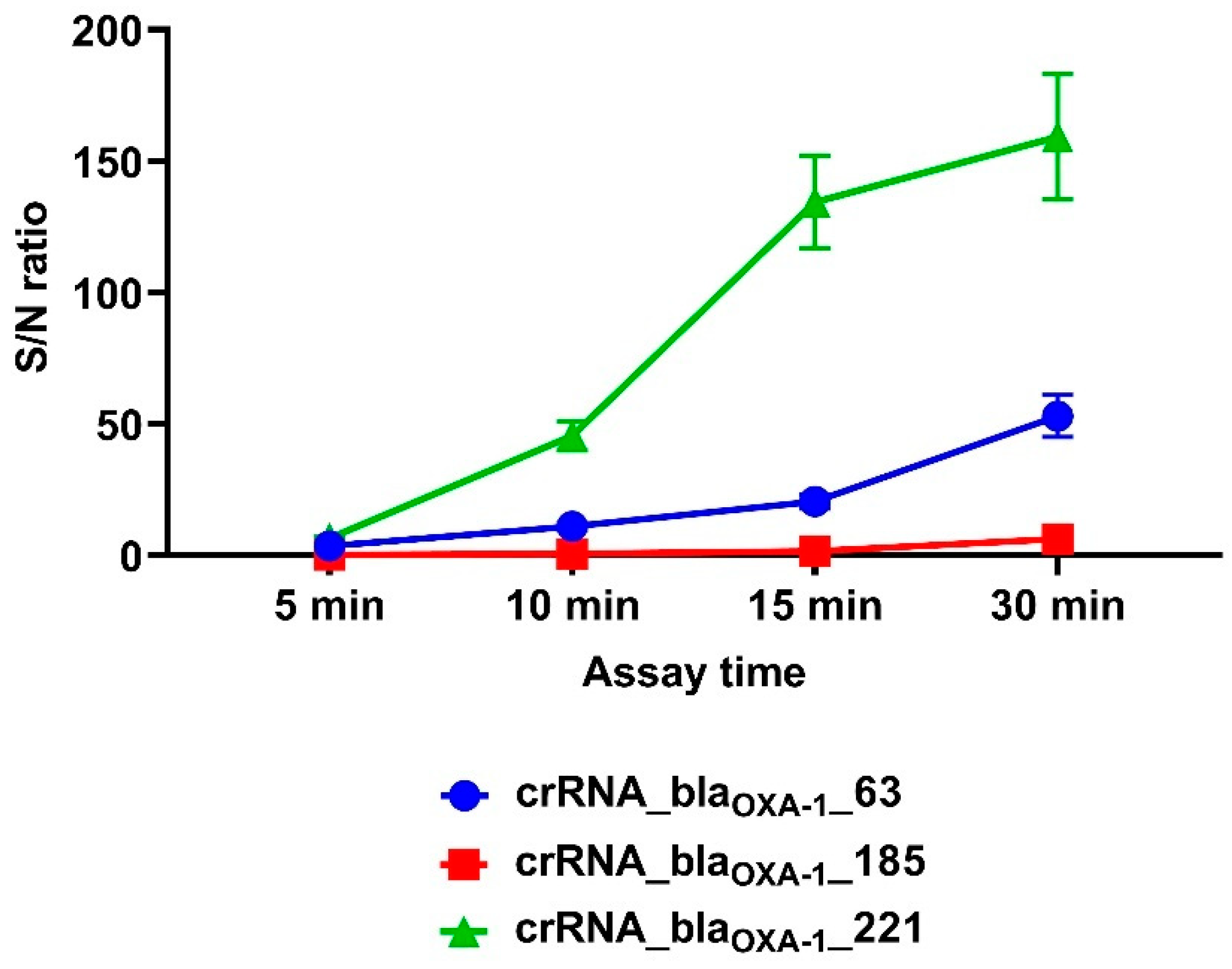
3.1. Design of CRISPR/Cas12a Fluorescence Assays and Guide RNA Selection
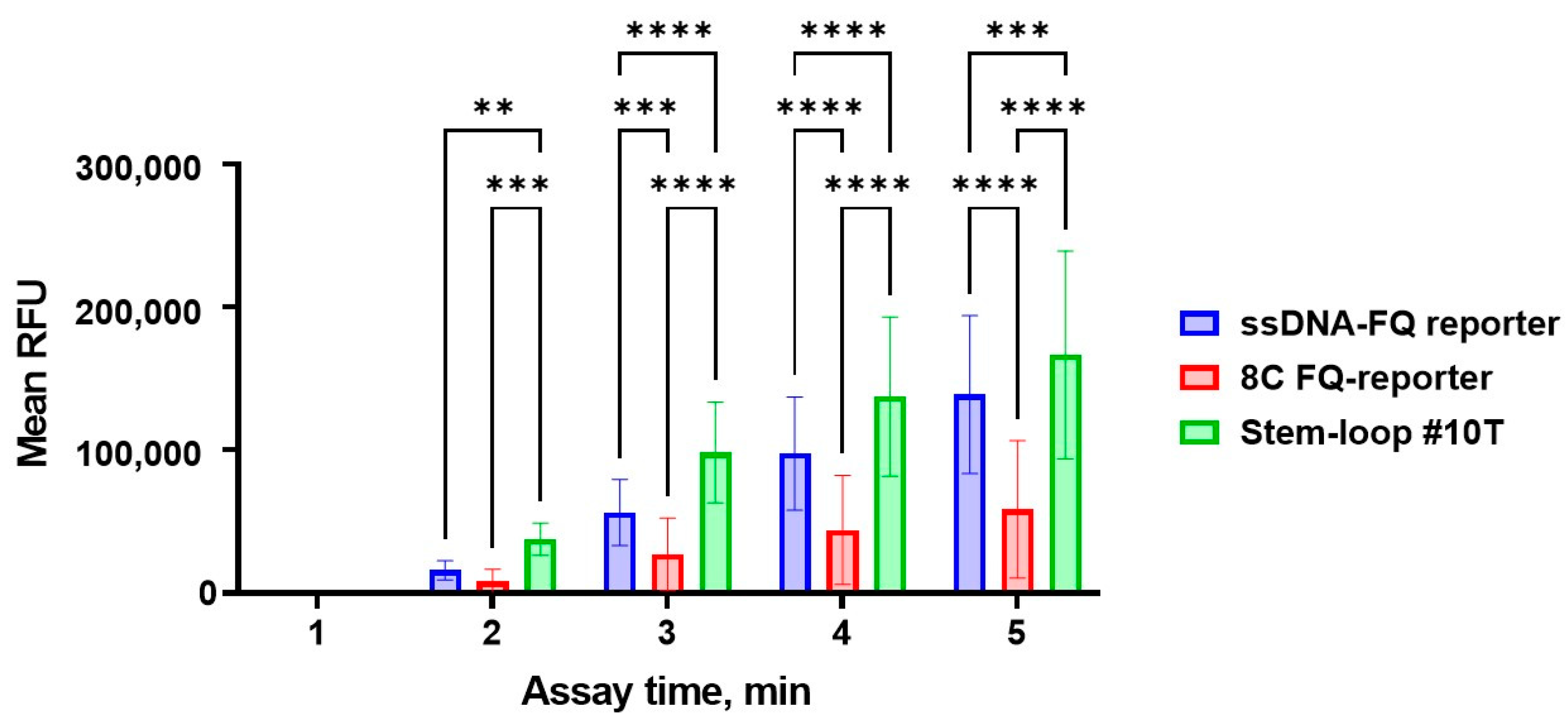
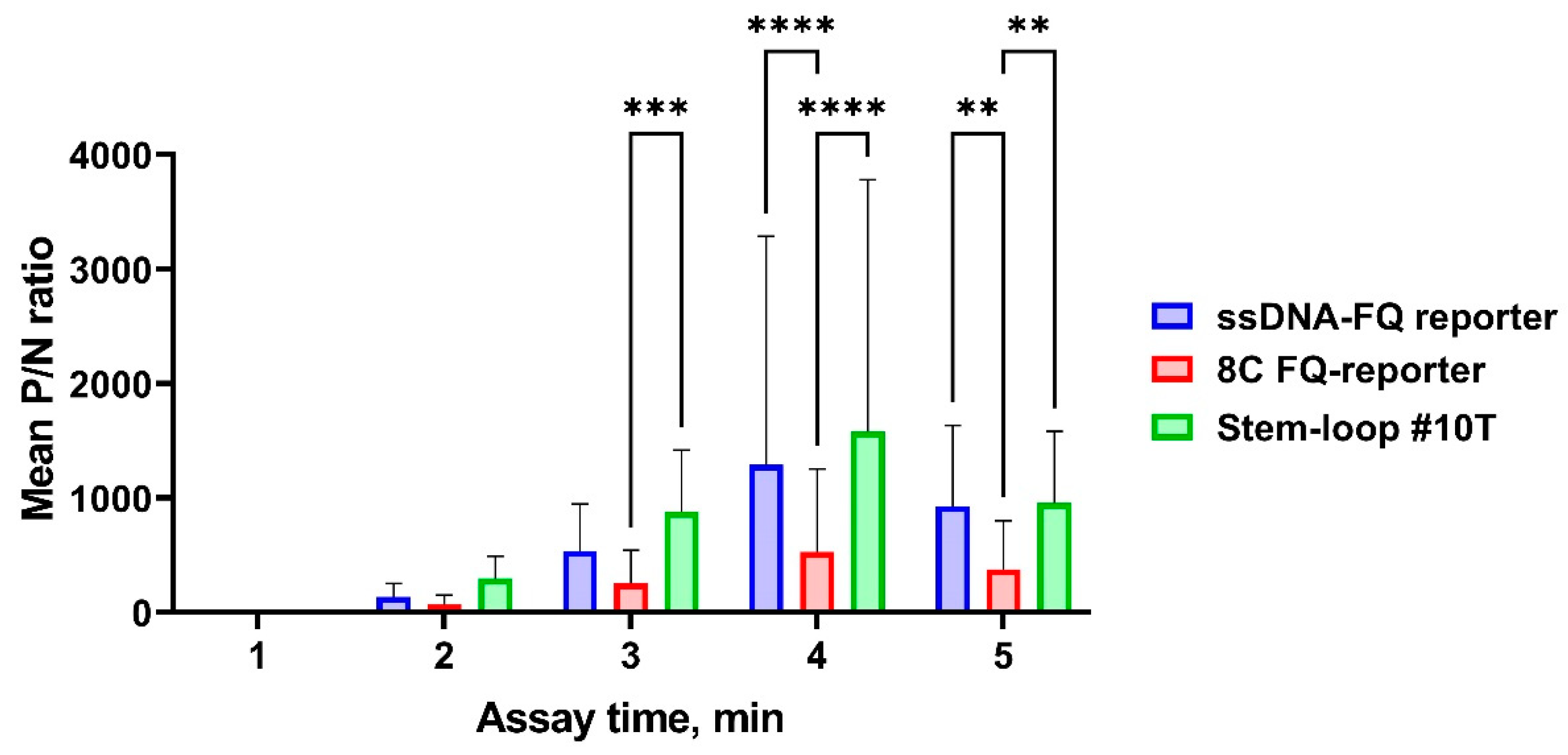
3.2. Performance Analysis of the CRISPR/Cas12a Fluorescence Assay
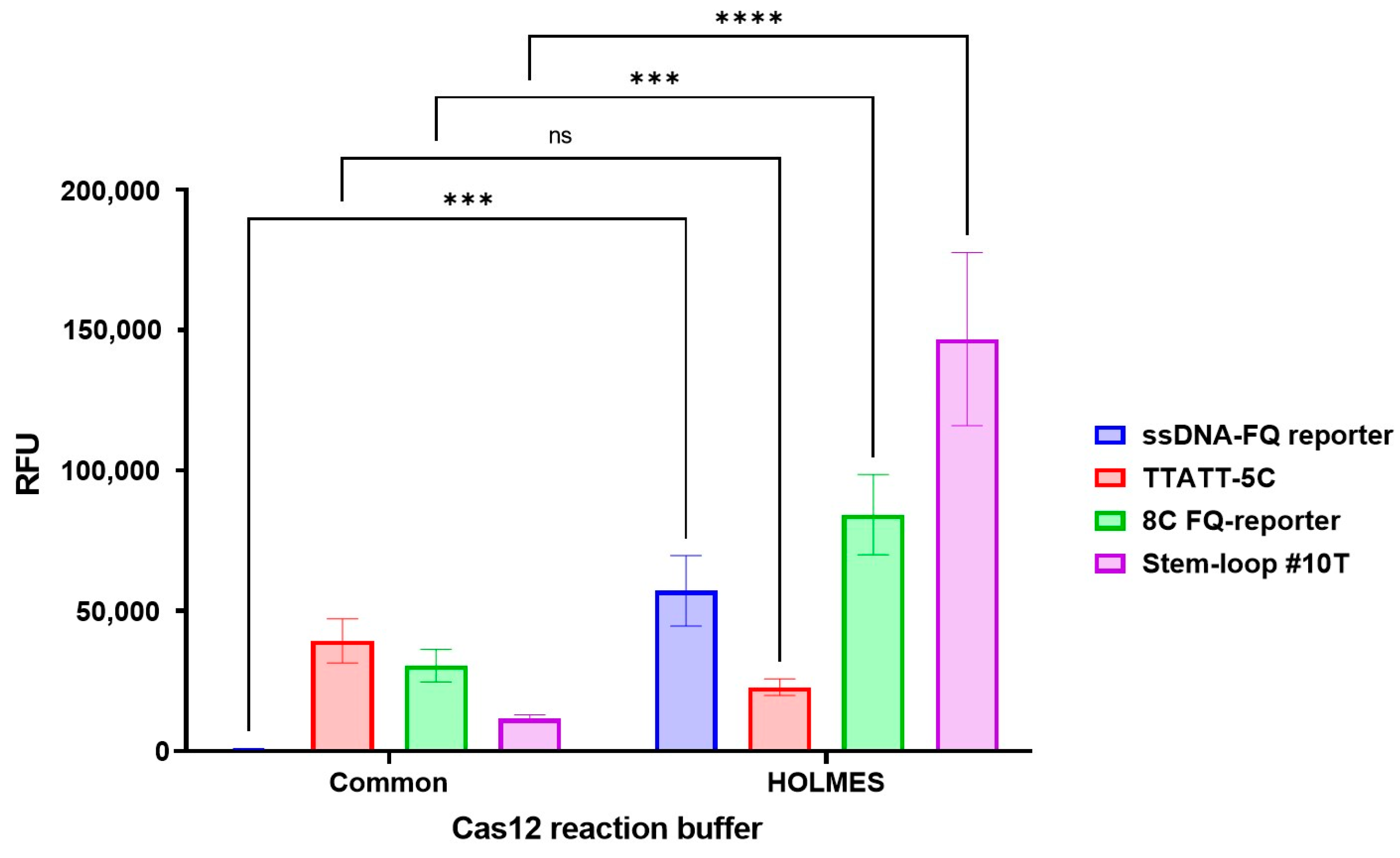
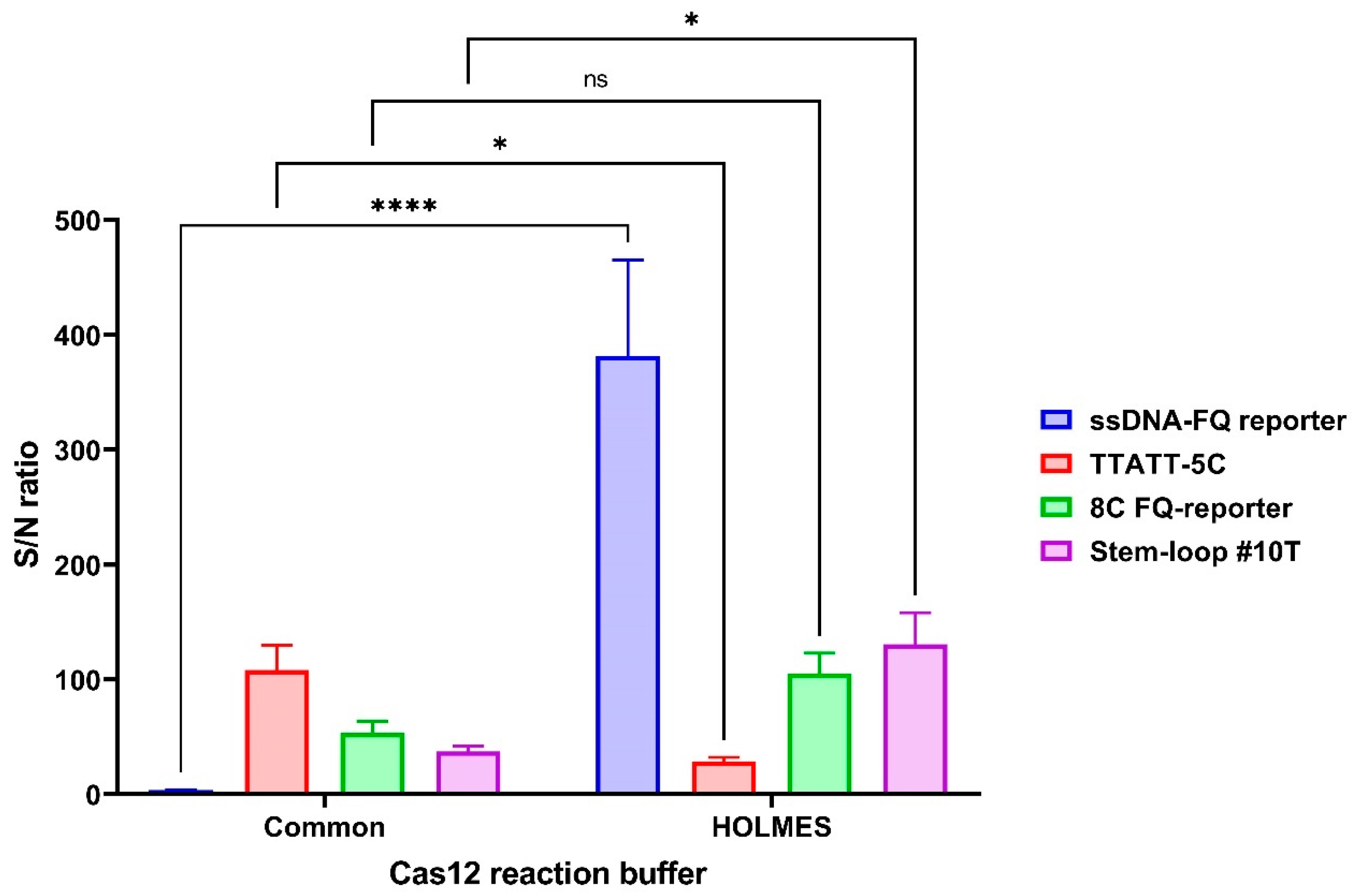
3.3. Assay Optimization
3.4. Detecting BlaOXA-1 Gene in Real Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Antimicrobial Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 20 March 2025).
- van Belkum, A.; Bachmann, T.T.; Lüdke, G.; Lisby, J.G.; Kahlmeter, G.; Mohess, A.; Becker, K.; Hays, J.P.; Woodford, N.; Mitsakakis, K.; et al. Innovative and Rapid Antimicrobial Susceptibility Testing Systems. Nat. Rev. Microbiol. 2019, 17, 51–62. [Google Scholar] [CrossRef]
- Strich, J.R.; Chertow, D.S. CRISPR-Cas Biology and Its Application to Infectious Diseases. J. Clin. Microbiol. 2021, 59, e01307-18. [Google Scholar]
- Lerminiaux, N.A.; Cameron, A.D.S. Horizontal Transfer of Antibiotic Resistance Genes in Clinical Environments. Can. J. Microbiol. 2019, 65, 34–44. [Google Scholar] [PubMed]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Review on Antimicrobial Resistance; Wellcome Trust: London, UK, 2016; Available online: https://amr-review.org/ (accessed on 20 March 2025).
- Ben-Ami, R.; Rodríguez-Baño, J.; Arslan, H.; Pitout, J.D.D.; Quentin, C.; Calbo, E.S.; Azap, O.K.; Arpin, C.; Pascual, A.; Livermore, D.M.; et al. A multinational survey of risk factors for infection with extended-spectrum beta-lactamase-producing Enterobacteriaceae in nonhospitalized patients. Clin. Infect. Dis. 2009, 49, 682–690. [Google Scholar]
- Karaiskos, I.; Giamarellou, H. Carbapenem-sparing strategies for ESBL producers: When and how. Antibiotics 2020, 9, 61. [Google Scholar] [CrossRef]
- Ouellette, M.; Bissonnette, L.; Roy, P.H. Precise insertion of antibiotic resistance determinants into Tn21-like transposons: Nucleotide sequence of the OXA-1 beta-lactamase gene. Proc. Natl. Acad. Sci. USA 1987, 84, 7378–7382. [Google Scholar] [PubMed]
- Bradford, P.A. Extended-spectrum beta-lactamases in the 21st century: Characterization, epidemiology, and detection of this important resistance threat. Clin. Microbiol. Rev. 2001, 14, 933–951. [Google Scholar]
- Paterson, D.L.; Bonomo, R.A. Extended-spectrum beta-lactamases: A clinical update. Clin. Microbiol. Rev. 2005, 18, 657–686. [Google Scholar] [CrossRef]
- Wiegand, I.; Geiss, H.K.; Mack, D.; Stürenburg, E.; Seifert, H. Detection of extended-spectrum beta-lactamases among Enterobacteriaceae by use of semiautomated microbiology systems and manual detection procedures. J. Clin. Microbiol. 2007, 45, 1167–1174. [Google Scholar]
- Tyumentseva, M.; Tyumentsev, A.; Akimkin, V. CRISPR/Cas9 landscape: Current state and future perspectives. Int. J. Mol. Sci. 2023, 24, 16077. [Google Scholar] [CrossRef]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef]
- Mikhaylova, Y.V.; Nagornykh, A.M.; Petrov, V.V.; Sud’ina, A.E.; Tyumentsev, A.I.; Tyumentseva, M.A.; Shelenkov, A.A. Genetic Technologies; Akimkin, V.G., Ed.; Central Research Institute for Epidemiology: Moscow, Russia, 2020; ISBN 9785604528631. [Google Scholar]
- Lv, H.; Wang, J.; Zhang, J.; Chen, Y.; Yin, L.; Jin, D.; Gu, D.; Zhao, H.; Xu, Y.; Wang, J. Definition of CRISPR Cas12a trans-cleavage units to facilitate CRISPR diagnostics. Front. Microbiol. 2021, 12, 766464. [Google Scholar] [CrossRef]
- Lee, S.; Nam, D.; Park, J.S.; Kim, S.; Lee, E.S.; Cha, B.S.; Park, K.S. Highly efficient DNA reporter for CRISPR/Cas12a-based specific and sensitive biosensor. Biochip J. 2022, 16, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, M.; Merlo, R.; Bagheri, N.; Moscone, D.; Valenti, A.; Saha, A.; Arantes, P.R.; Ippodrino, R.; Ricci, F.; Treglia, I.; et al. Enhancement of CRISPR/Cas12a trans-cleavage activity using hairpin DNA reporters. Nucleic Acids Res. 2022, 50, 8377–8391. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Tyumentseva, M.; Mikhaylova, Y.; Prelovskaya, A.; Karbyshev, K.; Tyumentsev, A.; Petrova, L.; Mironova, A.; Zamyatin, M.; Shelenkov, A.; Akimkin, V. CRISPR element patterns vs. pathoadaptability of clinical Pseudomonas aeruginosa isolates from a medical center in Moscow, Russia. Antibiotics 2021, 10, 1301. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.; Jinek, M. In vitro enzymology of Cas9. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 546, pp. 1–20. [Google Scholar]
- Shin, J.; Kim, S.R.; Xie, Z.; Jin, Y.-S.; Wang, Y.-C. A CRISPR/Cas12a-based system for sensitive detection of antimicrobial-resistant genes in carbapenem-resistant Enterobacterales. Biosensors 2024, 14, 194. [Google Scholar] [CrossRef]
- Akimkin, V.G.; Tiumentsev, A.I.; Tiumentseva, M.A. Crispr/Cas System for Detecting Proviral HIV DNA. World Patent WO2021118409A1, 17 June 2021. [Google Scholar]
- Akimkin, V.G.; Tiumentsev, A.I.; Tiumentseva, M.A. Crispr/Cas System for Detecting an Antibiotic Resistance Gene. World Patent WO2021211012A1, 21 October 2021. [Google Scholar]
- Tiumentsev, A.I.; Tiumentseva, M.A.; Akimkin, V.G.; Prelovskaya, A.N. Crispr-cas14 System for Detecting SARS-CoV-2 Virus RNA at Ultra-Low Concentrations. World Patent WO2023055255A1, 6 April 2023. [Google Scholar]
- Shariq, M.; Khan, M.F.; Raj, R.; Ahsan, N.; Singh, R.; Kumar, P. CRISPR-based diagnostic approaches: Implications for rapid management of future pandemics (Review). Mol. Med. Rep. 2023, 27, 13005. [Google Scholar] [CrossRef]
- Colom, K.; Pérez, J.; Alonso, R.; Fernández-Aranguiz, A.; Lariño, E.; Cisterna, R. Simple and reliable multiplex PCR assay for detection of blaTEM, blaSHV and blaOXA-1 genes in Enterobacteriaceae. FEMS Microbiol. Lett. 2003, 223, 147–151. [Google Scholar] [CrossRef]
- Ogutu, J.O.; Zhang, Q.; Huang, Y.; Yan, H.; Su, L.; Gao, B.; Zhang, W.; Zhao, J.; Cai, W.; Li, W.; et al. Development of a multiplex PCR system and its application in detection of blaSHV, blaTEM, blaCTX-M-1, blaCTX-M-9 and blaOXA-1 group genes in clinical Klebsiella pneumoniae and Escherichia coli strains. J. Antibiot. 2015, 68, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Probst, K.; Boutin, S.; Bandilla, M.; Heeg, K.; Dalpke, A.H. Fast and automated detection of common carbapenemase genes using multiplex real-time PCR on the BD MAXTM system. J. Microbiol. Methods 2021, 185, 106224. [Google Scholar] [CrossRef]
- Cohen, S.S. A Guide to Polyamines; Oxford University Press: New York, NY, USA, 1997; ISBN 9780195110647. [Google Scholar]
- Krasnow, M.A.; Cozzarelli, N.R. Catenation of DNA rings by topoisomerases. Mechanism of control by spermidine. J. Biol. Chem. 1982, 257, 2687–2693. [Google Scholar] [CrossRef] [PubMed]
- Plateau, P.; Moch, C.; Blanquet, S. Spermidine strongly increases the fidelity of Escherichia coli CRISPR Cas1-Cas2 integrase. J. Biol. Chem. 2019, 294, 11311–11322. [Google Scholar] [CrossRef] [PubMed]
- Akabayov, B.; Akabayov, S.R.; Lee, S.-J.; Wagner, G.; Richardson, C.C. Impact of macromolecular crowding on DNA replication. Nat. Commun. 2013, 4, 1615. [Google Scholar] [CrossRef]
- Minton, A.P. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J. Biol. Chem. 2001, 276, 10577–10580. [Google Scholar] [CrossRef]
- Zimmerman, S.B.; Minton, A.P. Macromolecular crowding: Biochemical, biophysical, and physiological consequences. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 27–65. [Google Scholar] [CrossRef]
- Wang, Q.; Liang, K.-C.; Czader, A.; Waxham, M.N.; Cheung, M.S. The effect of macromolecular crowding, ionic strength and calcium binding on calmodulin dynamics. PLoS Comput. Biol. 2011, 7, e1002114. [Google Scholar] [CrossRef]
- van den Berg, B.; Wain, R.; Dobson, C.M.; Ellis, R.J. Macromolecular crowding perturbs protein refolding kinetics: Implications for folding inside the cell. EMBO J. 2000, 19, 3870–3875. [Google Scholar] [CrossRef]
- Sikorav, J.L.; Church, G.M. Complementary recognition in condensed DNA: Accelerated DNA renaturation. J. Mol. Biol. 1991, 222, 1085–1108. [Google Scholar] [CrossRef]
- Akimkin, V.G.; Tiumentsev, A.I.; Tiumentseva, M.A.; Shagin, D.A. Method for Producing a Preparation of Highly-Purified Recombinant Cas Nuclease. World Patent WO2020197436A1, 1 October 2020. [Google Scholar]
- Qiu, M.; Zhou, X.-M.; Liu, L. Improved strategies for CRISPR-Cas12-based nucleic acids detection. J. Anal. Test. 2022, 6, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Salehian, M.; Emamzadeh, R.; Nazari, M.; Oliayi, M. Glycine as a stabilizing osmolyte for Renilla luciferase: A kinetic and molecular dynamics analysis. Biocatal. Biotransform. 2024, 43, 1–10. [Google Scholar] [CrossRef]
- Tyumentseva, M.A.; Tyumentsev, A.I.; Prelovskaya, A.N.; Akimkin, V.G. Optimization of a method for detecting single copies of hepatitis B virus DNA using CRISPR/Cas systems. Epidemiol. Vakcinoprofil. 2025, 23, 114–128. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Function | Oligonucleotide Sequence | Reference |
|---|---|---|---|
| crRNA_blaOXA-1_63 | LbCpf1 crRNA | 5′ AAUUUCUACUAAGUGUAGAUGGUUAUUUCUUGCGAAACCC 3′ | This study |
| crRNA_blaOXA-1_185 | LbCpf1 crRNA | 5′ AAUUUCUACUAAGUGUAGAUAAGCUACUUUCGAGCCAUGC 3′ | This study |
| crRNA_blaOXA-1_221 | LbCpf1 crRNA | 5′ AAUUUCUACUAAGUGUAGAUCGCAGGAAUUGAAUUUGUUC 3′ | This study |
| OXA-1_for_45 | PCR primer | 5′ GGAATGGAGATCTGGAACAGCAATCATACACC 3′ | This study |
| OXA-1_rev_48 | PCR primer | 5′ ATCCAGATCTTGTAGATACATGTTCTCTATGG 3′ | This study |
| OXA-1_for_2 | PCR primer | 5′ AGCAATCATACACCAAAGACG 3′ | This study |
| OXA-1_rev_1 | PCR primer | 5′ TGGCTGAGTTTTTAACTGGG 3′ | This study |
| ssDNA-FQ reporter | ssDNA reporter | 5′ FAM-TTATT-BHQ1 3′ | [13] |
| 8C FQ-reporter | ssDNA reporter | 5′ FAM-CCCCCCCC-BHQ1 3′ | [16] |
| TTATT-5C | ssDNA reporter | 5′ FAM-TTATTCCCCC-BHQ1 3′ | [17] |
| Stem-loop #10T | ssDNA reporter | 5′ FAM-CTCTCATTTTTTTTTTAGAGAG-BHQ1 3′ | [18] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyumentseva, M.; Tyumentsev, A.; Prelovskaya, A.; Akinin, A.; Mikhailova, Y.; Shelenkov, A.; Panevina, A.; Akimkin, V. Ultrasensitive CRISPR/Cas12a-Based System for Detection of BlaOXA-1 Gene in Antibiotic-Resistant Microorganisms. Curr. Issues Mol. Biol. 2025, 47, 238. https://doi.org/10.3390/cimb47040238
Tyumentseva M, Tyumentsev A, Prelovskaya A, Akinin A, Mikhailova Y, Shelenkov A, Panevina A, Akimkin V. Ultrasensitive CRISPR/Cas12a-Based System for Detection of BlaOXA-1 Gene in Antibiotic-Resistant Microorganisms. Current Issues in Molecular Biology. 2025; 47(4):238. https://doi.org/10.3390/cimb47040238
Chicago/Turabian StyleTyumentseva, Marina, Aleksandr Tyumentsev, Anna Prelovskaya, Andrey Akinin, Yulia Mikhailova, Andrey Shelenkov, Anna Panevina, and Vasiliy Akimkin. 2025. "Ultrasensitive CRISPR/Cas12a-Based System for Detection of BlaOXA-1 Gene in Antibiotic-Resistant Microorganisms" Current Issues in Molecular Biology 47, no. 4: 238. https://doi.org/10.3390/cimb47040238
APA StyleTyumentseva, M., Tyumentsev, A., Prelovskaya, A., Akinin, A., Mikhailova, Y., Shelenkov, A., Panevina, A., & Akimkin, V. (2025). Ultrasensitive CRISPR/Cas12a-Based System for Detection of BlaOXA-1 Gene in Antibiotic-Resistant Microorganisms. Current Issues in Molecular Biology, 47(4), 238. https://doi.org/10.3390/cimb47040238