
Insulin-like Growth Factor 1 Impact on Alzheimer’s Disease: Role in Inflammation, Stress, and Cognition

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Insulin-like Growth Factor 1
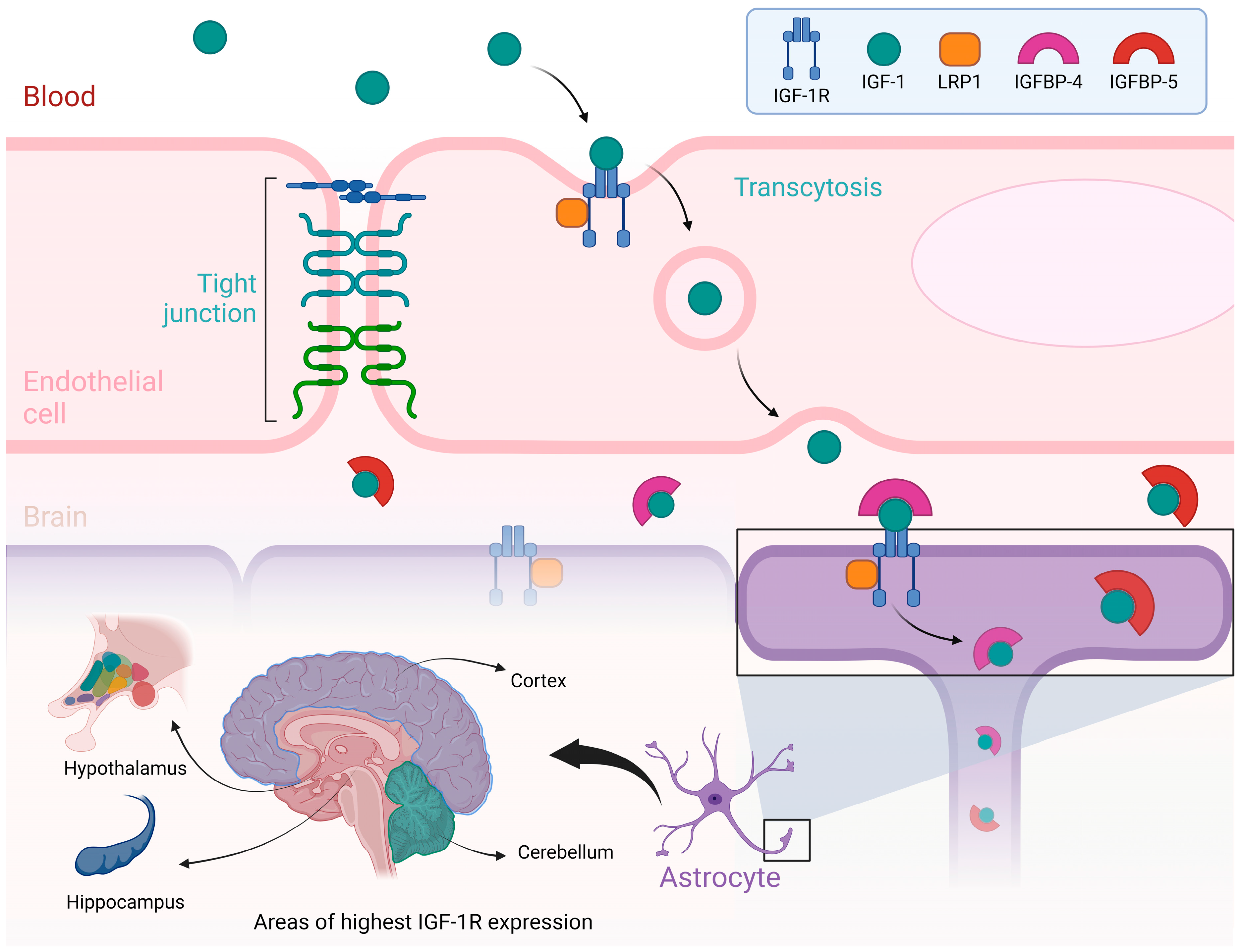
- Note: Figure 1 illustrates the process by which Insulin-like Growth Factor I (IGF-I) is transported from the bloodstream to the brain, emphasizing the role of the blood–brain barrier and the regions of the brain where IGF-IR are most abundantly expressed. At the top of the figure, a blood vessel is depicted as having an endothelial cell, a crucial part of the blood–brain barrier. IGF-I, circulating in the blood, binds to its receptor, IGF-IR, on the endothelial cell membrane. This IGF-I/IGF-IR complex is internalized through a process known as transcytosis, which helps the passage of IGF-I across the blood–brain barrier and into the brain. More proteins, including LRP1 and IGF-binding proteins (IGFBP-4 and IGFBP-5), also play crucial roles in modulating the availability and transport of IGF-I during this process. The lower part of the figure highlights the brain regions with the highest expression of IGF-IR, including the cortex, cerebellum, hypothalamus, and hippocampus. These regions are associated with essential functions such as growth regulation, metabolic control, and cognitive processes. The figure also depicts the interaction of IGF-I with astrocytes, which are glial cells vital for supporting and modulating neuronal function. Illustrations were created using BioRender (www.biorender.com, accessed on 23 August 2024).
1.2. Insulin-like Growth Factor 1 Signaling Mechanisms
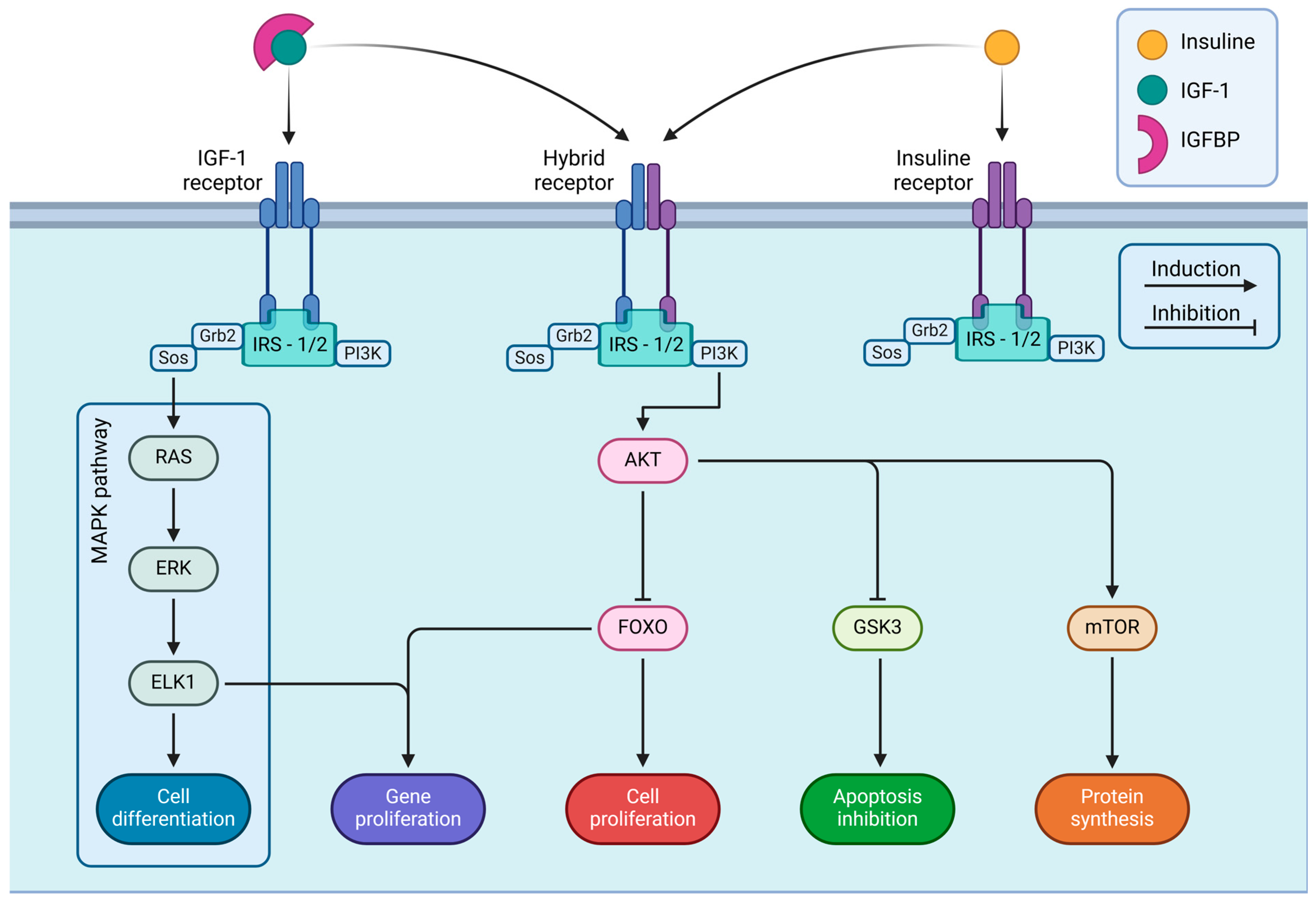
- Note: Figure 2 details the intracellular signaling pathways triggered by IGF-IR and IR. These receptors are embedded in the cell membrane and become activated upon binding with their respective ligands, IGF-I and insulin, starting a series of signaling cascades that regulate various cellular functions. The figure shows three main types of receptors: IGF-IR, IR, and a hybrid receptor combining elements of both. Upon activation, each receptor associates with IRS-1 or IRS-2, which is essential for transmitting signals into the cell. The receptor-IRS complex contains two principal signaling pathways: the MAPK and PI3K/AKT pathways. IGF-I and insulin receptors integrate extracellular signals through these pathways to activate intracellular cascades, which determine cell fate and play critical roles in development, metabolism, and organismal homeostasis. Illustrations were created using BioRender (www.biorender.com, accessed on 3 January 2025).
1.3. Insulin-like Growth Factor 1 and Alzheimer’s Disease
1.4. Social Interaction and Insulin-like Growth Factor 1
1.5. Other Mechanisms Involved in Alzheimer’s Disease and Insulin-like Growth Factor 1
2. Link Between Mechanisms Contributing to AD and IGF-I Signaling Pathways
2.1. Alzheimer’s Disease, Neuroinflammation, and Stress
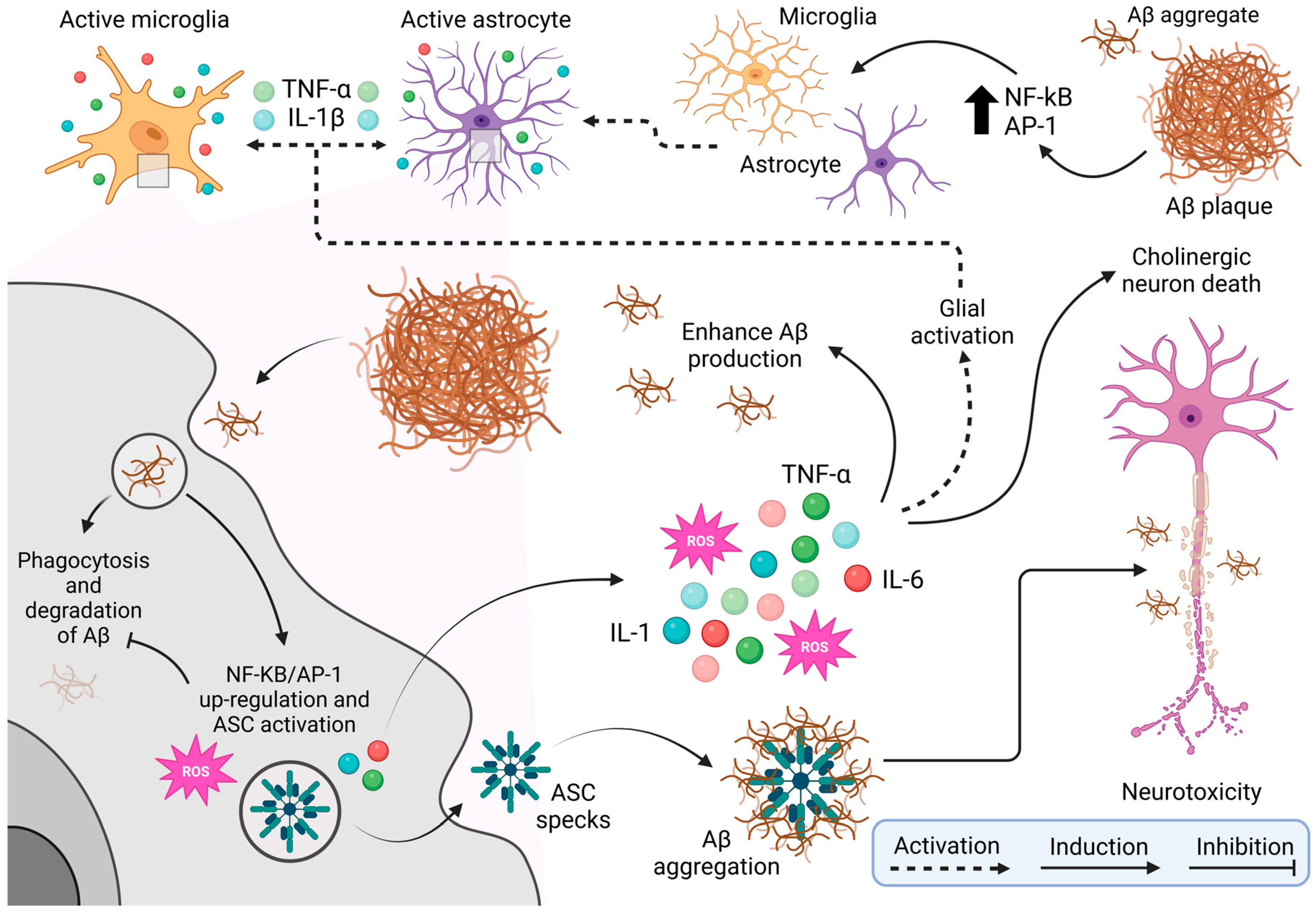
- Note: Figure 3 provides a comprehensive schematic of the cellular and molecular mechanisms underlying neurotoxicity associated with beta-amyloid (Aβ) plaque accumulation, a hallmark of neurodegenerative diseases such as Alzheimer’s disease. The figure begins by showing the activation of microglia, the brain’s resident immune cells, in response to Aβ presence. This activation is mediated by transcription factors such as NF-kB and AP-1, which drive a proinflammatory state. Astrocytes, another type of glial cell, are similarly activated by inflammatory signals, including cytokines like TNF-α (tumor necrosis factor-alpha) and IL-1β (interleukin 1 beta) released by microglia. This glial activation, mediated by NF-kB and AP-1, perpetuates inflammation within brain tissue. Microglia try to clear Aβ plaques through phagocytosis, but this process is often incomplete or ineffective, leading to Aβ accumulation. Persistent interaction with Aβ upregulates NF-kB/AP-1 expression and activates the ASC (apoptosis-associated speck-like protein holding a caspase recruitment domain) complex, which generates reactive oxygen species (ROS) and releases more pro-inflammatory cytokines such as IL-1, TNF-α, and IL-6. The ROS production, combined with ongoing inflammation, worsens oxidative stress, further aggravates Aβ aggregation and promotes a toxic environment detrimental to cholinergic neurons. These neurons, crucial for memory and learning, are especially vulnerable to Alzheimer’s disease. The resulting neurotoxicity and later death of cholinergic neurons drive the cognitive decline characteristic of the disease, creating a vicious cycle of chronic inflammation, neuronal damage, and worsening pathology. Illustrations were created using BioRender (www.biorender.com, accessed on 3 January 2025).

- Note: Figure 4 illustrates IGF-1’s role in oxidative stress regulation. Under normal conditions, IGF-1 enhances antioxidative enzymes—superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx)—converting superoxide radicals (O2−) and hydrogen peroxide (H2O2) into water, reducing reactive oxygen species (ROS) production and preventing neurotoxicity and neurodegeneration in neurons via induction of these enzymes. Conversely, reduced IGF-1 signaling decreases antioxidant capacity, causes mitochondrial dysfunction, impairs ATP production, and elevates mitochondrial ROS, increasing vulnerability to hydrogen peroxide-induced cytotoxicity and exacerbating neuronal damage. The diagram highlights IGF-1’s protective effect, with evidence linking its deficiency to heightened ROS and neurodegeneration, while overexpression mitigates oxidative damage. Illustrations were created using BioRender (www.biorender.com, accessed on 3 January 2025).
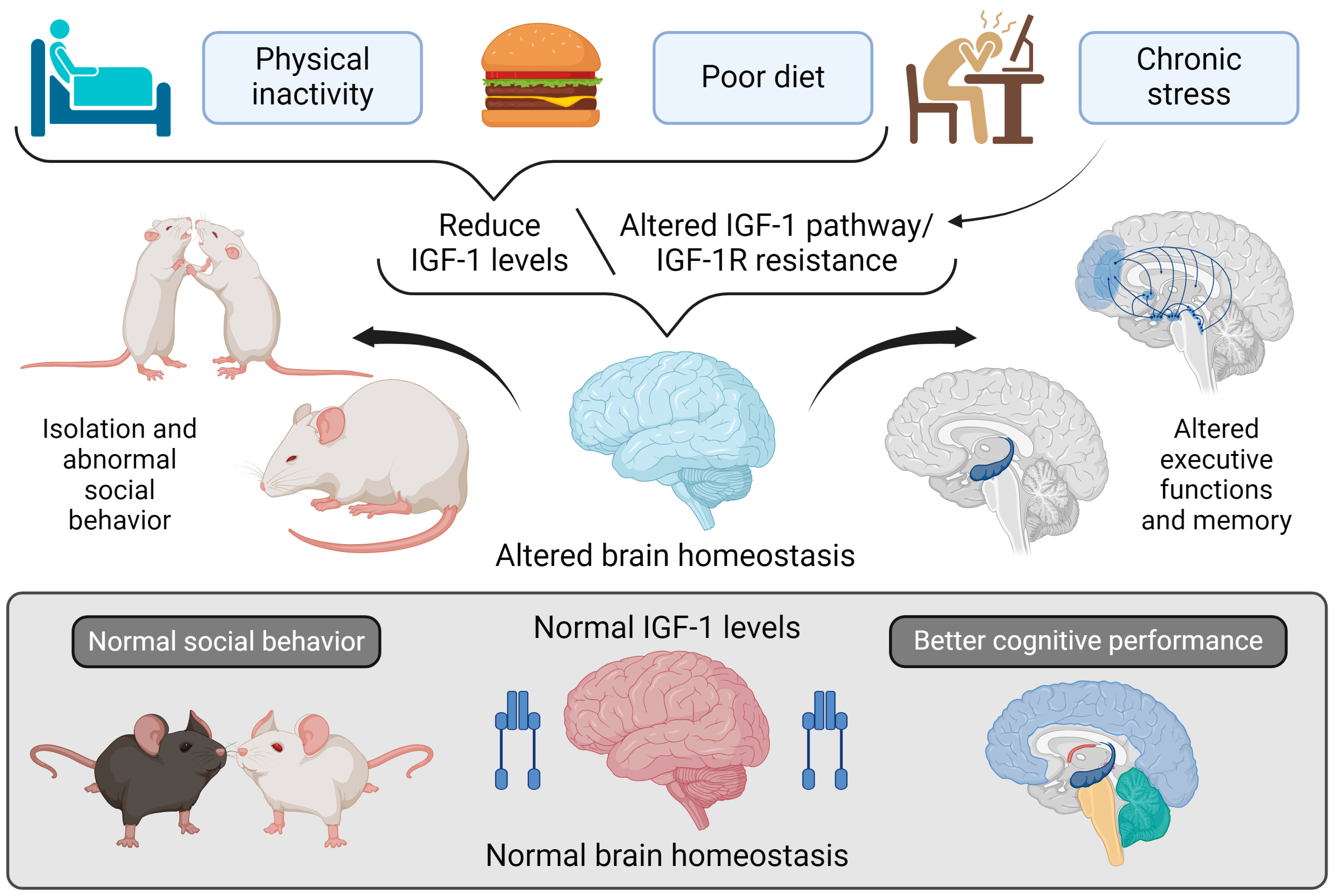
- Note: Figure 5 explores the relationship between lifestyle factors and regulating IGF-I in the brain and its effects on brain homeostasis, social behavior, and cognitive functions. At the top of the figure, three lifestyle factors are shown that can lower IGF-I levels or disrupt its signaling pathway: physical inactivity, poor diet, and chronic stress. These factors contribute to reduced IGF-I levels or IGF-IR resistance, impairing normal IGF-I signaling in the brain. This disruption in IGF-I signaling adversely affects brain homeostasis in two keyways: 1. Altered Social Behavior: Mice with diminished IGF-I levels show social withdrawal and abnormal social interactions, reflecting dysfunction in brain areas responsible for social behavior. 2. Impaired Executive Functions and Memory: Reduced IGF-I signaling, or IGF-IR resistance, also adversely affects executive functions and memory, as depicted by changes in critical brain regions such as the hippocampus and prefrontal cortex.
2.2. Executive Functions (EF) in Alzheimer’s Disease
2.3. Social Cognition in AD
2.4. Diagnostic Tools for Alzheimer’s Disease
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | Amyloid Beta |
| AD | Alzheimer’s Disease |
| AKT | Serine/Threonine Kinase 1 |
| BBB | Blood-Brain Barrier |
| BDNF | Brain-Derived Neurotrophic Factor |
| CSF | Cerebrospinal Fluid |
| EEG | Electroencephalography |
| FOXO | Forkhead Box Protein O |
| GH | Growth Hormone |
| GSK3 | Glycogen Synthase Kinase-3 |
| IGF-I | Insulin-Like Growth Factor 1 |
| IGFBP | Insulin-Like Growth Factor Binding Proteins |
| IR | Insulin Receptor |
| IRS | Insulin Receptor Substrate |
| MAPK | Mitogen-Activated Protein Kinase |
| MCI | Mild Cognitive Impairment |
| MMSE | Mini-Mental State Examination |
| mTOR | Mammalian Target of Rapamycin |
| PET | Positron Emission Tomography |
| PI3K | Phosphatidylinositol-3 Kinase |
| TMT-B | Trail Making Test B |
| TNF-α | Tumor Necrosis Factor-alpha |
References
- Alzheimer’s Association 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 2020, 16, 391–460. [CrossRef]
- Chakrabarti, S.; Khemka, V.K.; Banerjee, A.; Chatterjee, G.; Ganguly, A.; Biswas, A. Metabolic risk factors of sporadic Alzheimer’s disease: Implications in the pathology, pathogenesis and treatment. Aging Dis. 2015, 6, 282–299. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Mattsson, N. Understanding the cause of sporadic Alzheimer’s disease. Expert Rev. Neurother. 2014, 14, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Santi, A.; Torres Aleman, I. Insulin Peptides as Mediators of the Impact of Life Style in Alzheimer’s disease. Brain Plast. 2018, 4, 3–15. [Google Scholar] [CrossRef]
- Scobie, J.; Amos, S.; Beales, S.; Dobbing, C.; Gillam, S.; Knox-Vydmanov, C.; Mihnovits, A.; Mikkonen-Jeanneret, E. Global AgeWatch Index 2015: Insight report; 2015, 1–28. Available online: https://www.helpage.org/silo/files/global-agewatch-index-2015-insight-report.pdf (accessed on 23 August 2024).
- Gauthier, S.; Rosa-Neto, P.; Moraís, J.; Webster, C. World Alzheimer Report 2021. 2021. Available online: https://www.alzint.org/resource/world-alzheimer-report-2021/ (accessed on 18 March 2025).
- WHO. Global Action Plan on the Public Health Response to Dementia 2017–2025; Geneva World Health Organization: Geneva, Switzerland, 2017; 27p. [Google Scholar]
- Ferri, C.P.; Jacob, K.S. Dementia in low-income and middle-income countries: Different realities mandate tailored solutions. PLoS Med. 2017, 14, e1002271. [Google Scholar] [CrossRef]
- Wittenberg, R.; Hu, B.; Barraza-Araiza, L.; Funder, A.R. Projections of Older People Dementia and Costs of Dementia Care in the United Kingdom 2019–2040; CPEC Working Paper 5; Alzheimer’s Society: London, UK, 2019; Available online: www.modem-dementia.org.uk (accessed on 10 July 2024).
- Wong, W. Economic burden of Alzheimer disease and managed care considerations. Am. J. Manag. Care 2020, 26, S177–S183. [Google Scholar] [CrossRef]
- Malaha, A.K.; Thebaut, C.; Achille, D.; Preux, P.-M.; Guerchet, M. Costs of dementia in low-and-middle income countries: A systematic review. J. Alzheimers Dis. 2023, 91, 115–128. [Google Scholar]
- Smailagic, N.; Vacante, M.; Hyde, C.; Martin, S.; Ukoumunne, O.; Sachpekidis, C. 18F-FDG PET for the early diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). In Cochrane Database of Systematic Reviews; John Wiley and Sons Ltd.: Hoboken, NJ, USA, 2015; Volume 2017. [Google Scholar] [CrossRef]
- Grossman, M.; Farmer, J.; Leight, S.; Work, M.; Moore, P.; Van Deerlin, V.; Pratico, D.; Clark, C.M.; Coslett, H.B.; Chatterjee, A.; et al. Cerebrospinal fluid profile in frontotemporal dementia and Alzheimer’s disease. Ann. Neurol. 2005, 57, 721–729. [Google Scholar] [CrossRef]
- Rosso, S.M.; Van Herpen, E.; Pijnenburg, Y.A.L.; Schoonenboom, N.S.M.; Scheltens, P.; Heutink, P.; van Swieten, J.C. Total tau and phosphorylated tau 181 levels in the cerebrospinal fluid of patients with frontotemporal dementia due to P301L and G272V tau mutations. Arch. Neurol. 2003, 60, 1209–1213. [Google Scholar] [CrossRef]
- Gottesman, I.I.; Gould, T.D. The endophenotype concept in psychiatry: Etymology and strategic intentions. Am. J. Psychiatry 2003, 160, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Busiguina, S.; Fernandez, A.M.; Barrios, V.; Clark, R.; Tolbert, D.L.; Berciano, J.; Torres-Aleman, I. Neurodegeneration is associated to changes in serum insulin-like growth factors. Neurobiol. Dis. 2000, 7, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Carro, E.; Trejo, J.L.; Gomez-Isla, T.; LeRoith, D.; Torres-Aleman, I. Serum insulin-like growth factor I regulates brain amyloid-β levels. Nat. Med. 2002, 8, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Burden of Mental Disorders and the Need for a Comprehensive, Coordinated Response from Health and Social Sectors at the Country Level; WHO: Geneva, Switzerland, 2011; Available online: https://apps.who.int/gb/ebwha/pdf_files/EB130/B130_9-en.pdf (accessed on 17 March 2025).
- Zahr, N.M. Alcohol Use Disorder and Dementia: A Review. Alcohol Res. Curr. Rev. 2024, 44, 3. [Google Scholar] [CrossRef]
- Wang, G.; Li, D.Y.; Vance, D.E.; Li, W. Alcohol Use Disorder as a Risk Factor for Cognitive Impairment. J. Alzheimer’s Dis. 2023, 94, 899–907. [Google Scholar] [CrossRef]
- Morys, F.; Dadar, M.; Dagher, A. Association between midlife obesity and its metabolic consequences, cerebrovascular disease, and cognitive decline. J. Clin. Endocrinol. Metab. 2021, 106, e4260–e4274. [Google Scholar] [CrossRef]
- Patel, V.; Edison, P. Cardiometabolic risk factors and neurodegeneration: A review of the mechanisms underlying diabetes, obesity and hypertension in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2024, 95, 581–589. [Google Scholar] [CrossRef]
- Michailidis, M.; Moraitou, D.; Tata, D.A.; Kalinderi, K.; Papamitsou, T.; Papaliagkas, V. Alzheimer’s Disease as Type 3 Diabetes: Common Pathophysiological Mechanisms between Alzheimer’s Disease and Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 2687. [Google Scholar] [CrossRef]
- Hamzé, R.; Delangre, E.; Tolu, S.; Moreau, M.; Janel, N.; Bailbé, D.; Movassat, J. Type 2 Diabetes Mellitus and Alzheimer’s Disease: Shared Molecular Mechanisms and Potential Common Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 15287. [Google Scholar] [CrossRef]
- Zhong, G.; Wang, Y.; Zhang, Y.; Guo, J.J.; Zhao, Y. Smoking is associated with an increased risk of dementia: A meta-analysis of prospective cohort studies with investigation of potential effect modifiers. PLoS ONE 2015, 10, e0126169. [Google Scholar] [CrossRef]
- Ourry, V.; Binette, A.P.; St-Onge, F.; Strikwerda-Brown, C.; Chagnot, A.; Poirier, J.; Breitner, J.; Arenaza-Urquijo, E.M.; Rabin, J.S.; Buckley, R.; et al. How Do Modifiable Risk Factors Affect Alzheimer’s Disease Pathology or Mitigate Its Effect on Clinical Symptom Expression? Biol. Psychiatry 2024, 95, 1006–1019. [Google Scholar] [CrossRef] [PubMed]
- Shafighi, K.; Villeneuve, S.; Neto, P.R.; Badhwar, A.P.; Poirier, J.; Sharma, V.; Medina, Y.I.; Silveira, P.P.; Dube, L.; Glahn, D.; et al. Social isolation is linked to classical risk factors of Alzheimer’s disease-related dementias. PLoS ONE 2023, 18, e0280471. [Google Scholar] [CrossRef]
- Ren, Y.; Savadlou, A.; Park, S.; Siska, P.; Epp, J.R.; Sargin, D. The impact of loneliness and social isolation on the development of cognitive decline and Alzheimer’s Disease. Front. Neuroendocrinol. 2023, 69, 101061. [Google Scholar] [CrossRef] [PubMed]
- Drinkwater, E.; Davies, C.; Spires-Jones, T.L. Potential neurobiological links between social isolation and Alzheimer’s disease risk. Eur. J. Neurosci. 2022, 56, 5397–5412. [Google Scholar] [CrossRef]
- Lachner, C.; Craver, E.C.; Babulal, G.M.; Lucas, J.A.; Ferman, T.J.; White, R.O.; Graff-Radford, N.R.; Day, G.S. Disparate Dementia Risk Factors Are Associated with Cognitive Impairment and Rates of Decline in African Americans. Ann. Neurol. 2024, 95, 518–529. [Google Scholar] [CrossRef]
- Majoka, M.A.; Schimming, C. Effect of Social Determinants of Health on Cognition and Risk of Alzheimer Disease and Related Dementias. Clin. Ther. 2021, 43, 922–929. [Google Scholar] [CrossRef]
- Torres-aleman, I. Targeting insulin-like growth factor-1 to treat Alzheimer’ s disease. Expert Opin. Ther. Targets 2007, 11, 1535–1542. [Google Scholar]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- De Roeck, E.E.; De Deyn, P.P.; Dierckx, E.; Engelborghs, S. Brief cognitive screening instruments for early detection of Alzheimer’s disease: A systematic review. Alzheimer’s Res. Ther. 2019, 11, 21. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Torres-Alemán, I. The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef]
- Muller, A.P.; Fernandez, A.M.; Haas, C.; Zimmer, E.; Portela, L.V.; Torres-Aleman, I. Reduced brain insulin-like growth factor I function during aging. Mol. Cell. Neurosci. 2012, 49, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Frysak, Z.; Schovanek, J.; Iacobone, M.; Karasek, D. Insulin-like Growth Factors in a clinical setting: Review of IGF-I. Biomed. Pap. 2015, 159, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Rinderknecht, E.; Humbel, R.E. The amino acid sequence of human insulin-like growth factor I and its structural homology with proinsulin. J. Biol. Chem. 1978, 253, 2769–2776. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-L.; Yakar, S.; LeRoith, D. Mice Deficient in Liver Production of Insulin-Like Growth Factor I Display Sexual Dimorphism in Growth Hormone-Stimulated Postnatal Growth. Endocrinology 2000, 141, 4436–4441. [Google Scholar] [CrossRef]
- Trueba-Sáiz, A.; Cavada, C.; Fernandez, A.M.; Leon, T.; González, D.A.; Fortea Ormaechea, J.; Lleó, A.; Del Ser, T.; Nuñez, A.; Torres-Aleman, I. Loss of serum IGF-I input to the brain as an early biomarker of disease onset in Alzheimer mice. Transl. Psychiatry 2013, 3, 2–7. [Google Scholar] [CrossRef]
- Allard, J.B.; Duan, C. IGF-binding proteins: Why do they exist and why are there so many? Front. Endocrinol. 2018, 9, 117. [Google Scholar] [CrossRef]
- Ranke, M.B. Insulin-like growth factor binding-protein-3 (IGFBP-3). Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 701–711. [Google Scholar] [CrossRef]
- Iams, W.T.; Lovly, C.M. Molecular pathways: Clinical applications and future direction of insulin-like growth factor-1 receptor pathway blockade. Clin. Cancer Res. 2015, 21, 4270–4277. [Google Scholar] [CrossRef]
- Rechler, M.M.; Clemmons, D.R. Regulatory actions of insulin-like growth factor-binding proteins. Trends Endocrinol. Metab. 1998, 9, 176–183. [Google Scholar] [CrossRef]
- Khan, S. IGFBP-2 Signaling in the Brain: From Brain Development to Higher Order Brain Functions. Front. Endocrinol. 2019, 10, 822. [Google Scholar] [CrossRef]
- Martinez-Rachadell, L.; Aguilera, A.; Perez-Domper, P.; Pignatelli, J.; Fernandez, A.M.; Torres-Aleman, I. Cell-specific expression of insulin/insulin-like growth factor-I receptor hybrids in the mouse brain. Growth Horm. IGF Res. 2019, 45, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Hakuno, F.; Takahashi, S.I. 40 years of IGF1: IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [PubMed]
- Carro, E.; Torres-Aleman, I. The role of insulin and insulin-like growth factor I in the molecular and cellular mechanisms underlying the pathology of Alzheimer’s disease. Eur. J. Pharmacol. 2004, 490, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A.M.; Bueno, R.; Pedrini, M.T.; Murray, J.M.; Smith, R.J. Insulin-like growth factor I receptor gene structure. J. Biol. Chem. 1992, 267, 10759–10763. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Hernandez-Garzón, E.; Perez-Domper, P.; Perez-Alvarez, A.; Mederos, S.; Matsui, T.; Santi, A.; Trueba-Saiz, A.; García-Guerra, L.; Pose-Utrilla, J.; et al. Insulin regulates astrocytic glucose handling through cooperation with IGF-I. Diabetes 2017, 66, 64–74. [Google Scholar] [CrossRef]
- Campos-Bedolla, P.; Walter, F.R.; Veszelka, S.; Deli, M.A. Role of the Blood-Brain Barrier in the Nutrition of the Central Nervous System. Arch. Med. Res. 2014, 45, 610–638. [Google Scholar] [CrossRef]
- Carro, E.; Trejo, J.L.; Spuch, C.; Bohl, D.; Heard, J.M.; Torres-Aleman, I. Blockade of the insulin-like growth factor I receptor in the choroid plexus originates Alzheimer’s-like neuropathology in rodents: New cues into the human disease? Neurobiol. Aging 2006, 27, 1618–1631. [Google Scholar] [CrossRef]
- Mangiola, A.; Vigo, V.; Anile, C.; De Bonis, P.; Marziali, G.; Lofrese, G. Role and Importance of IGF-1 in Traumatic Brain Injuries. Biomed Res. Int. 2015, 2015, 736104. [Google Scholar] [CrossRef]
- Santi, A.; Genis, L.; Torres Aleman, I. A coordinated action of blood-borne and brain insulin-like growth factor I in the response to traumatic brain injury. Cereb. Cortex 2018, 28, 2007–2014. [Google Scholar] [CrossRef]
- Aleman, A.; Torres-Alemán, I. Circulating insulin-like growth factor I and cognitive function: Neuromodulation throughout the lifespan. Prog. Neurobiol. 2009, 89, 256–265. [Google Scholar] [CrossRef]
- Trueba-Saiz, Á. Neurotrophic Uncoupling of IGF-1 in Alzheimer’s Disease: Translation Into Early Diagnosis and Involvement of Lifestyle Risk Factors. Ph.D. Thesis, Universidad Autónoma de Madrid, Madrid, Spain, 2015. [Google Scholar]
- Zegarra-Valdivia, J. Insulin-like growth factor type 1 and its relation with neuropsychiatric disorders. Medwave 2017, 17, e7031. [Google Scholar] [CrossRef] [PubMed]
- Trejo, L.; Carro, E.; Torres-Aleman, I. Circulating Insulin-Like Growth Factor I Mediates Exercise-Induced. J. Neurosci. 2001, 21, 1628–1634. [Google Scholar] [PubMed]
- Trejo, J.L.; LLorens-Martín, M.V.; Torres-Alemán, I. The effects of exercise on spatial learning and anxiety-like behavior are mediated by an IGF-I-dependent mechanism related to hippocampal neurogenesis. Mol. Cell. Neurosci. 2008, 37, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Zegarra-Valdivia, J.A.; Pignatelli, J.; Fernandez de Sevilla, M.E.; Fernandez, A.M.; Munive, V.; Martinez-Rachadell, L.; Nuñez, A.; Aleman, I.T. Insulin-like growth factor I modulates sleep through hypothalamic orexin neurons. FASEB J. 2020, 34, 15975–15990. [Google Scholar] [CrossRef]
- Noriega-Prieto, J.A.; Maglio, L.E.; Zegarra-Valdivia, J.; Pignatelli, J.; Fernandez, A.M.; Martinez-Rachadell, L.; Fernandes, J.; Núñez, Á.; Araque, A.; Alemán, I.T.; et al. IGF-I Governs Cortical Inhibitory Synaptic Plasticity by Astrocyte Activation. bioRxiv 2020. bioRxiv:2020.02.11.942532. [Google Scholar] [CrossRef]
- Zegarra-Valdivia, J.A.; Chaves-Coira, I.; Fernandez de Sevilla, M.E.; Martinez-Rachadell, L.; Esparza, J.; Torres-Aleman, I.; Nuñez, A. Reduced Insulin-Like Growth Factor-I Effects in the Basal Forebrain of Aging Mouse. Front. Aging Neurosci. 2021, 13, 440. [Google Scholar] [CrossRef]
- Lewitt, M.S.; Boyd, G.W. The Role of Insulin-Like Growth Factors and Insulin-Like Growth Factor–Binding Proteins in the Nervous System. Biochem. Insights 2019, 12, 117862641984217. [Google Scholar] [CrossRef]
- Zegarra, J.; Santi, A.; De Sevilla, E.F.; Nuñez, A.; Torres, I. Serum IGF-I deficiency and Alzheimer’s disease: Implications for disease modeling. IBRO Rep. 2019, 6, S486. [Google Scholar] [CrossRef]
- Ostrowski, P.P.; Barszczyk, A.; Forstenpointner, J.; Zheng, W.; Feng, Z.-P. Meta-Analysis of Serum Insulin-Like Growth Factor 1 in Alzheimer’s Disease. PLoS ONE 2016, 11, e0155733. [Google Scholar] [CrossRef]
- Westwood, A.J.; Beiser, A.; DeCarli, C.; Harris, T.B.; Chen, T.C.; He, X.-M.; Roubenoff, R.; Pikula, A.; Au, R.; Braverman, L.E.; et al. Insulin-like growth factor-1 and risk of Alzheimer dementia and brain atrophy. Neurology 2014, 82, 1613–1619. [Google Scholar] [CrossRef]
- Vitale, G.; Pellegrino, G.; Vollery, M.; Hofland, L.J. ROLE of IGF-1 System in the Modulation of Longevity: Controversies and New Insights from a Centenarians’ Perspective. Front. Endocrinol. 2019, 10, 27. [Google Scholar] [CrossRef]
- Kim, B.; Elzinga, S.E.; Henn, R.E.; McGinley, L.M.; Feldman, E.L. The effects of insulin and insulin-like growth factor I on amyloid precursor protein phosphorylation in in vitro and in vivo models of Alzheimer’s disease. Neurobiol. Dis. 2019, 132, 104541. [Google Scholar] [CrossRef] [PubMed]
- Galle, S.A.; Van der Spek, A.; Drent, M.L.; Brugts, M.P.; Scherder, E.J.; Janssen, J.A.; Ikram, M.A.; Van Duijn, C.M. Revisiting the Role of Insulin-Like Growth Factor-I Receptor Stimulating Activity and the Apolipoprotein E in Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- LLorens-Martín, M.; Torres-Alemán, I.; Trejo, J.L. Exercise modulates insulin-like growth factor 1-dependent and -independent effects on adult hippocampal neurogenesis and behaviour. Mol. Cell. Neurosci. 2010, 44, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Roth, A.R.; Apostolova, L.G.; Saykin, A.J.; Perry, B.L. Cognitively stimulating environments and cognitive reserve: The case of personal social networks. Neurobiol. Aging 2022, 112, 197–203. [Google Scholar] [CrossRef]
- Perry, B.L.; McConnell, W.R.; Coleman, M.E.; Roth, A.R.; Peng, S.; Apostolova, L.G. Why the cognitive “fountain of youth” may be upstream: Pathways to dementia risk and resilience through social connectedness. Alzheimer’s Dement. 2022, 18, 934–941. [Google Scholar] [CrossRef]
- Manchella, M.K.; Logan, P.E.; Perry, B.L.; Peng, S.; Risacher, S.L.; Saykin, A.J.; Apostolova, L.G. Associations Between Social Network Characteristics and Brain Structure Among Older Adults. Alzheimer’s Dement. 2024, 20, 1406–1420. [Google Scholar] [CrossRef]
- Angevaare, M.J.; Pieters, J.A.; Twisk, J.W.R.; Van Hout, H.P.J. Social Activity and Cognitive Decline in Older Residents of Long-Term Care Facilities: A Cohort Study. J. Alzheimers Dis. 2024, 98, 433–443. [Google Scholar] [CrossRef]
- Doulames, V.; Lee, S.; Shea, T.B. Environmental enrichment and social interaction improve cognitive function and decrease reactive oxidative species in normal adult mice. Int. J. Neurosci. 2014, 124, 369–376. [Google Scholar] [CrossRef]
- Trueba-Saiz, A.; Fernandez, A.M.; Nishijima, T.; Mecha, M.; Santi, A.; Munive, V.; Aleman, I.T. Circulating Insulin-Like Growth Factor I regulates its receptor in the brain od male mice. Endocrinology 2017, 158, 349–355. [Google Scholar]
- Schoentgen, B.; Gagliardi, G.; Défontaines, B. Environmental and Cognitive Enrichment in Childhood as Protective Factors in the Adult and Aging Brain. Front. Psychol. 2020, 11, 1814. [Google Scholar] [CrossRef]
- Zuelsdorff, M.L.; Koscik, R.L.; Okonkwo, O.C.; Peppard, P.E.; Hermann, B.P.; Sager, M.A.; Johnson, S.C.; Engelman, C.D. Social support and verbal interaction are differentially associated with cognitive function in midlife and older age. Aging Neuropsychol. Cogn. 2019, 26, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Bellar, D.; Glickman, E.L.; Juvancic-Heltzel, J.; Gunstad, J. Serum insulin like growth factor-1 is associated with working memory, executive function and selective attention in a sample of healthy, fit older adults. Neuroscience 2011, 178, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Calvo, D.; Gunstad, J.; Miller, L.A.; Glickman, E.; Spitznagel, M.B. Higher serum insulin-like growth factor-1 is associated with better cognitive performance in persons with mild cognitive impairment. Psychogeriatrics 2013, 13, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Bove, R.M.; Brick, D.J.; Healy, B.C.; Mancuso, S.M.; Gerweck, A.V.; Bredella, M.A.; Sherman, J.; Miller, K. Metabolic and endocrine correlates of cognitive function in healthy young women. Obesity 2013, 21, 1343–1349. [Google Scholar] [CrossRef]
- Torres-Aleman, I. Toward a comprehensive neurobiology of IGF-I. Dev. Neurobiol. 2010, 70, 384–396. [Google Scholar] [CrossRef]
- Dobolyi, A.; Lékó, A.H. The insulin-like growth factor-1 system in the adult mammalian brain and its implications in central maternal adaptation. Front. Neuroendocrinol. 2019, 52, 181–194. [Google Scholar] [CrossRef]
- Zegarra-Valdivia, J.; Fernandez, A.M.; Martinez-Rachadell, L.; Herrero-Labrador, R.; Fernandes, J.; Torres Aleman, I. Insulin and insulin-like growth factor-I receptors in astrocytes exert different effects on behavior and Alzheimers-like pathology. F1000Research 2022, 11, 663. [Google Scholar] [CrossRef]
- Yue, S.; Wang, Y.; Wang, Z.J. Insulin-like growth factor 1 regulates excitatory synaptic transmission in pyramidal neurons from adult prefrontal cortex. Neuropharmacology 2022, 217, 109204. [Google Scholar] [CrossRef]
- Nieto-Estévez, V.; Defterali, Ç.; Vicario-Abejón, C. IGF-I: A key growth factor that regulates neurogenesis and synaptogenesis from embryonic to adult stages of the brain. Front. Neurosci. 2016, 10, 52. [Google Scholar] [CrossRef]
- Skuse, D.; Lawrence, K.; Tang, J. Measuring social-cognitive functions in children with somatotropic axis dysfunction. Horm. Res. 2005, 64, 73–82. [Google Scholar] [CrossRef]
- Valtorta, N.; Hanratty, B. Loneliness, isolation and the health of older adults: Do we need a new research agenda? J. R. Soc. Med. 2012, 105, 518. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, A.; Zegarra-Valdivia, J.; Fernandez de Sevilla, D.; Pignatelli, J.; Torres Aleman, I. The neurobiology of insulin-like growth factor I: From neuroprotection to modulation of brain states. Mol. Psychiatry 2023, 28, 3220–3230. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, A.B.; Stern, S.A.; Kohtz, A.S.; Descalzi, G.; Alberini, C.M. Insulin-Like Growth Factor II Targets the mTOR Pathway to Reverse Autism-Like Phenotypes in Mice. J. Neurosci. 2018, 38, 1015–1029. [Google Scholar] [CrossRef] [PubMed]
- Riikonen, R. Treatment of autistic spectrum disorder with insulin-like growth factors. Eur. J. Paediatr. Neurol. 2016, 20, 816–823. [Google Scholar] [CrossRef]
- Bhalla, S.; Mehan, S.; Khan, A.; Rehman, M.U. Protective role of IGF-1 and GLP-1 signaling activation in neurological dysfunctions. Neurosci. Biobehav. Rev. 2022, 142, 104896. [Google Scholar] [CrossRef]
- Doré, S.; Satyabrata, K.; Quirion, R. Rediscovering an old friend, IFG-I: Potential use in the treatment of neurodegenerative diseases. Trends Neurosci. 1997, 20, 326–331. [Google Scholar] [CrossRef]
- Makkar, R.; Behl, T.; Sehgal, A.; Singh, S.; Sharma, N.; Makeen, H.A.; Albratty, M.; Alhazmi, H.A.; Meraya, A.M. Targeting Insulin-Like Growth Factor-I in Management of Neurological Disorders. Neurotox. Res. 2022, 40, 874–883. [Google Scholar] [CrossRef]
- Fitzgerald, G.S.; Chuchta, T.G.; McNay, E.C. Insulin-like growth factor-2 is a promising candidate for the treatment and prevention of Alzheimer’s disease. CNS Neurosci. Ther. 2023, 29, 1449–1469. [Google Scholar] [CrossRef]
- Kimura, A.; Namekata, K.; Guo, X.; Harada, C.; Harada, T. Neuroprotection, growth factors and BDNF-TRKB signalling in retinal degeneration. Int. J. Mol. Sci. 2016, 17, 1584. [Google Scholar] [CrossRef]
- Aguirre, V.; Uchida, T.; Yenush, L.; Davis, R.; White, M.F. The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser307. J. Biol. Chem. 2000, 275, 9047–9054. [Google Scholar] [CrossRef]
- Strandberg, R.B.; Graue, M.; Wentzel-Larsen, T.; Peyrot, M.; Rokne, B. Relationships of diabetes-specific emotional distress, depression, anxiety, and overall well-being with HbA1c in adult persons with type 1 diabetes. J. Psychosom. Res. 2014, 77, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Basta-Kaim, A.; Szczesny, E.; Glombik, K.; Stachowicz, K.; Slusarczyk, J.; Nalepa, I.; Molik, A.Z.; Zablocka, K.R.; Budziszewska, B.; Kubera, M.; et al. Prenatal stress affects insulin-like growth factor-1 (IGF-1) level and IGF-1 receptor phosphorylation in the brain of adult rats. Eur. Neuropsychopharmacol. 2014, 24, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Zegarra-Valdivia, J.A.; Chino-Vilca, B.N. Neurobiología del trastorno de estrés postraumático. Rev. Mex. Neurocienc. 2019, 20, 21–28. [Google Scholar] [CrossRef]
- Solas, M.; Aisa, B.; Mugueta, M.C.; Del Río, J.; Tordera, R.M.; Ramírez, M.J. Interactions between age, stress and insulin on cognition: Implications for alzheimer’s disease. Neuropsychopharmacology 2010, 35, 1664–1673. [Google Scholar] [CrossRef]
- Ahmadi, N.; Arora, R.; Vaidya, N.; Yehuda, R.; Ebrahimi, R. Post-traumatic stress disorder is associated with increased incidence of insulin resistance and metabolic syndrome. J. Am. Coll. Cardiol. 2013, 61, E1347. [Google Scholar] [CrossRef]
- Li, L.; Li, X.; Zhou, W.; Messina, J.L. Acute psychological stress results in the rapid development of insulin resistance. J. Endocrinol. 2013, 217, 175–184. [Google Scholar] [CrossRef]
- Kelly, Á.M. Exercise-Induced Modulation of Neuroinflammation in Models of Alzheimer’s Disease. Brain Plast. 2018, 4, 81–94. [Google Scholar] [CrossRef]
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 2016, 136, 457–474. [Google Scholar] [CrossRef]
- Finneran, D.J.; Nash, K.R. Neuroinflammation and fractalkine signaling in Alzheimer’s disease. J. Neuroinflammation 2019, 16, 30. [Google Scholar] [CrossRef]
- Pak, V.M.; Onen, S.H.; Bliwise, D.L.; Kutner, N.G.; Russell, K.L.; Onen, F. Sleep Disturbances in MCI and AD: Neuroinflammation as a Possible Mediating Pathway. Front. Aging Neurosci. 2020, 12, 69. [Google Scholar] [CrossRef]
- Park, J.C.; Han, S.H.; Mook-Jung, I. Peripheral inflammatory biomarkers in Alzheimer’s disease: A brief review. BMB Rep. 2020, 53, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Madore, C.; Yin, Z.; Leibowitz, J.; Butovsky, O. Microglia, Lifestyle Stress, and Neurodegeneration. Immunity 2020, 52, 222–240. [Google Scholar] [CrossRef] [PubMed]
- Munoz, U.; Castilla-Cortazar, I. Protection Against Oxidative Stress and “IGF-I Deficiency Conditions”. In Antioxidant Enzyme; IntechOpen: London, UK, 2012. [Google Scholar]
- Logan, S.; Pharaoh, G.A.; Marlin, M.C.; Masser, D.R.; Matsuzaki, S.; Wronowski, B.; Yeganeh, A.; Parks, E.E.; Premkumar, P.; Farley, J.A.; et al. Insulin-like growth factor receptor signaling regulates working memory, mitochondrial metabolism, and amyloid-β uptake in astrocytes. Mol. Metab. 2018, 9, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Calixto, A.; Jara, J.S.; Court, F.A. Diapause Formation and Downregulation of Insulin-Like Signaling via DAF-16/FOXO Delays Axonal Degeneration and Neuronal Loss. PLoS Genet. 2012, 8, e1003141. [Google Scholar] [CrossRef]
- Sadagurski, M.; Cheng, Z.; Rozzo, A.; Palazzolo, I.; Kelley, G.R.; Dong, X.; Krainc, D.; White, M.F. IRS2 increases mitochondrial dysfunction and oxidative stress in a mouse model of Huntington disease. J. Clin. Investig. 2011, 121, 4070–4081. [Google Scholar] [CrossRef]
- Herrera, M.L.; Champarini, L.G.; Oliveros, A.L.; José, M.; Hereñú, C.B. Exploration of Neuroprotective Therapy Potentialities of IGF-1 for regulating oxidative stress in neuroinflammation and neurodegeneration: Theoretical review. Explor. Neuroprot. Ther. 2024, 4, 442–458. [Google Scholar] [CrossRef]
- Alvarez, A.; Sampedro, C.; Cacabelos, R.; Linares, C.; Aleixandre, M.; García-Fantini, M.; Moessler, H. Reduced TNF- and increased IGF-I levels in the serum of Alzheimer’s disease patients treated with the neurotrophic agent Cerebrolysin. Int. J. Neuropsychopharmacol. 2009, 12, 867–872. [Google Scholar] [CrossRef]
- Rutters, F.; Pilz, S.; Koopman, A.D.; Rauh, S.P.; Te Velde, S.J.; Stehouwer, C.D.; Elders, P.J.; Nijpels, G.; Dekker, J.M. The association between psychosocial stress and mortality is mediated by lifestyle and chronic diseases: The Hoorn Study. Soc. Sci. Med. 2014, 118, 166–172. [Google Scholar] [CrossRef]
- Justice, N.J. The relationship between stress and Alzheimer’s disease. Neurobiol. Stress 2018, 8, 127–133. [Google Scholar] [CrossRef]
- Cortés, N.; Andrade, V.; Maccioni, R.B. Behavioral and Neuropsychiatric Disorders in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 63, 899–910. [Google Scholar] [CrossRef]
- Dietlin, S.; Soto, M.; Kiyasova, V.; Pueyo, M.; De Mauleon, A.; Delrieu, J.; Ousset, P.J.; Vellas, B. Neuropsychiatric Symptoms and Risk of Progression to Alzheimer’s Disease Among Mild Cognitive Impairment Subjects. J. Alzheimer’s Dis. 2019, 70, 25–34. [Google Scholar] [CrossRef]
- Geda, Y.E.; Schneider, L.S.; Gitlin, L.N.; Miller, D.S.; Smith, G.S.; Bell, J.; Evans, J.; Lee, M.; Porsteinsson, A.; Lanctôt, K.L.; et al. Neuropsychiatric symptoms in Alzheimer’s disease: Past progress and anticipation of the future. Alzheimer’s Dement. 2013, 9, 602–608. [Google Scholar] [CrossRef]
- Hallikainen, I.; Hongisto, K.; Välimäki, T.; Hänninen, T.; Martikainen, J.; Koivisto, A.M. The progression of neuropsychiatric symptoms in Alzheimer’s disease during a five-year follow-up: Kuopio ALSOVA study. J. Alzheimer’s Dis. 2018, 61, 1367–1376. [Google Scholar] [CrossRef]
- Lanctôt, K.L.; Agüera-Ortiz, L.; Brodaty, H.; Francis, P.T.; Geda, Y.E.; Ismail, Z.; Marshall, G.A.; Mortby, M.E.; Onyike, C.U.; Padala, P.R.; et al. Apathy associated with neurocognitive disorders: Recent progress and future directions. Alzheimer’s Dement. 2017, 13, 84–100. [Google Scholar] [CrossRef]
- Van Vliet, D.; De Vugt, M.E.; Aalten, P.; Bakker, C.; Pijnenburg, Y.A.L.; Vernooij-Dassen, M.J.F.J.; Koopmans, R.T.; Verhey, F.R. Prevalence of neuropsychiatric symptoms in young-onset compared to late-onset alzheimer’s disease-part 1: Findings of the two-year longitudinal needYD-study. Dement. Geriatr. Cogn. Disord. 2013, 34, 319–327. [Google Scholar] [CrossRef]
- Santi, A.; Bot, M.; Aleman, A.; Penninx, B.W.J.H.; Aleman, I.T. Circulating insulin-like growth factor I modulates mood and is a biomarker of vulnerability to stress: From mouse to man. Transl. Psychiatry 2018, 8, 142. [Google Scholar] [CrossRef]
- Blair, L.J.; Nordhues, B.A.; Hill, S.E.; Scaglione, K.M.; O’Leary, J.C.; Fontaine, S.N.; Breydo, L.; Zhang, B.; Li, P.; Wang, L.; et al. Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Investig. 2013, 123, 4158–4169. [Google Scholar] [CrossRef]
- Sabbagh, J.J.; O’Leary, J.C.; Blair, L.J.; Klengel, T.; Nordhues, B.A.; Fontaine, S.N.; Binder, E.B.; Dickey, C.A. Age-Associated Epigenetic Upregulation of the FKBP5 Gene Selectively Impairs Stress Resiliency. PLoS ONE 2014, 9, e107241. [Google Scholar] [CrossRef]
- Zannas, A.S.; Jia, M.; Hafner, K.; Baumert, J.; Wiechmann, T.; Pape, J.C.; Arloth, J.; Ködel, M.; Martinelli, S.; Roitman, M.; et al. Epigenetic upregulation of FKBP5 by aging and stress contributes to NF-κB-driven inflammation and cardiovascular risk. Proc. Natl. Acad. Sci. USA 2019, 166, 11370–11379. [Google Scholar] [CrossRef]
- Zegarra-Valdivia, J.A. Funcionamiento ejecutivo: Modelos conceptuales. Rev. Psicol. 2014, 16, 108–119. [Google Scholar]
- Martyr, A.; Clare, L. Executive Function and Activities of Daily Living in Alzheimer’s Disease: A Correlational Meta-Analysis. Dement. Geriatr. Cogn. Disord. 2012, 33, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Royall, D.R.; Chiodo, L.K.; Polk, M.J. Correlates of Disability Among Elderly Retirees with “Subclinical” Cognitive Impairment. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2000, 55, M541–M546. [Google Scholar] [CrossRef]
- Stopford, C.L.; Thompson, J.C.; Neary, D.; Richardson, A.M.T.; Snowden, J.S. Working memory, attention, and executive function in Alzheimer’s disease and frontotemporal dementia. Cortex 2012, 48, 429–446. [Google Scholar] [CrossRef] [PubMed]
- Chino, B.; Torres-Simón, L.; Żelwetro, A.; Rodríguez-Rojo, I.C.; Carnes-Vendrell, A.; Piñol-Ripoll, G.; Yubero, R.; Paúl, N.; Maestú, F. Understanding the Episodic Memory and Executive Functioning Axis Impairment in MCI Patients: A Multicenter Study in Comparison with CSF Biomarkers. Biomedicines 2023, 11, 3147. [Google Scholar] [CrossRef]
- Abellán-Martínez, M.; Castellanos López, M.Á.; Delgado-Losada, M.L.; Yubero, R.; Paúl, N.; Unturbe, F.M. Executive Control on Memory Test Performance across Life: Test of Memory Strategies. Span. J. Psychol. 2019, 22, E50. [Google Scholar] [CrossRef]
- Van Der Vlies, A.E.; Verwey, N.A.; Bouwman, F.H.; Blankenstein, M.A.; Klein, M.; Scheltens, P.; van der Flier, W.M. CSF biomarkers in relationship to cognitive profiles in Alzheimer disease. Neurology 2009, 72, 1056–1061. [Google Scholar] [CrossRef]
- Kirova, A.M.; Bays, R.B.; Lagalwar, S. Working Memory and Executive Function Decline across Normal Aging, Mild Cognitive Impairment, and Alzheimer’s Disease. BioMed Res. Int. 2015, 2015, 748212. [Google Scholar] [CrossRef]
- Horvath, A.; Quinlan, P.; Eckerström, C.; Aberg, N.D.; Wallin, A.; Svensson, J. The Associations Between Serum Insulin-like Growth Factor-I, Brain White Matter Volumes, and Cognition in Mild Cognitive Impairment and Alzheimer’s Disease. J. Alzheimer’s Dis. 2024, 99, 609–622. [Google Scholar] [CrossRef]
- Huang, R.; Wang, P.; Han, J.; Xia, W.; Cai, R.; Sun, H.; Sun, J.; Wang, S. Decreased Serum IGF-1/IGFBP-3 Molar Ratio is Associated with Executive Function Behaviors in Type 2 Diabetic Patients with Mild Cognitive Impairment. J. Alzheimer’s Dis. 2015, 47, 85–94. [Google Scholar] [CrossRef]
- Al-Delaimy, W.K.; Von Muhlen, D.; Barrett-Connor, E. Insulinlike growth factor-1, insulinlike growth factor binding protein-1, and cognitive function in older men and women. J. Am. Geriatr. Soc. 2009, 57, 1441–1446. [Google Scholar] [CrossRef]
- Zegarra-Valdivia, J.A.; Chino-Vilca, B. Mentalización y teoría de la mente. Rev. Neuropsiquiatr. 2017, 80, 189–199. [Google Scholar] [CrossRef]
- Ramanan, S.; de Souza, L.C.; Moreau, N.; Sarazin, M.; Teixeira, A.L.; Allen, Z.; Guimarães, H.C.; Caramelli, P.; Dubois, B.; Hornberger, M.; et al. Determinants of theory of mind performance in Alzheimer’s disease: A data-mining study. Cortex 2017, 88, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Friedler, B.; Crapser, J.; McCullough, L. One is the deadliest number: The detrimental effects of social isolation on cerebrovascular diseases and cognition. Acta Neuropathol. 2015, 129, 493–509. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.H.; Chang, C.H.; Gean, P.W. Impact of social relationships on Alzheimer’s memory impairment: Mechanistic studies. J. Biomed. Sci. 2018, 25, 3. [Google Scholar] [CrossRef]
- Wilson, R.S.; Krueger, K.R.; Arnold, S.E.; Schneider, J.A.; Kelly, J.F.; Barnes, L.L.; Tang, Y.; Bennett, D.A. Loneliness and risk of Alzheimer disease. Arch. Gen. Psychiatry 2007, 64, 234–240. [Google Scholar] [CrossRef]
- Porcelli, S.; Van Der Wee, N.; van der Werff, S.; Aghajani, M.; Glennon, J.C.; van Heukelum, S.; Mogavero, F.; Lobo, A.; Olivera, F.J.; Lobo, E.; et al. Social brain, social dysfunction and social withdrawal. Neurosci. Biobehav. Rev. 2019, 97, 10–33. [Google Scholar] [CrossRef]
- Dermody, N.; Wong, S.; Ahmed, R.; Piguet, O.; Hodges, J.R.; Irish, M. Uncovering the Neural Bases of Cognitive and Affective Empathy Deficits in Alzheimer’s Disease and the Behavioral-Variant of Frontotemporal Dementia. J. Alzheimer’s Dis. 2016, 53, 801–816. [Google Scholar] [CrossRef]
- Cadieux, N.L.; Greve, K.W. Emotion processing in Alzheimer’s disease. J. Int. Neuropsychol. Soc. 1997, 3, 411–419. [Google Scholar] [CrossRef]
- Cosentino, S.; Zahodne, L.B.; Brandt, J.; Blacker, D.; Albert, M.; Dubois, B.; Stern, Y. Social cognition in Alzheimer’s disease: A separate construct contributing to dependence. Alzheimer’s Dement. 2014, 10, 818–826. [Google Scholar] [CrossRef]
- Zegarra-Valdivia, J.A.; Shany-Ur, T.; Rijpma, M.G.; Callahan, P.; Poorzand, P.; Grossman, S.; McEachen, B.; Kramer, J.H.; Miller, B.L.; Rankin, K.P. Validation of the Cognitive-Emotional Perspective Taking test in patients with neurodegeneration. J. Alzheimers. Dis. 2025, 3, 13872877251317683. [Google Scholar] [CrossRef]
- Multani, N.; Taghdiri, F.; Anor, C.J.; Varriano, B.; Misquitta, K.; Tang-Wai, D.F.; Keren, R.; Fox, S.; Lang, A.E.; Vijverman, A.C.; et al. Association Between Social Cognition Changes and Resting State Functional Connectivity in Frontotemporal Dementia, Alzheimer’s Disease, Parkinson’s Disease, and Healthy Controls. Front. Neurosci. 2019, 13, 1259. [Google Scholar] [CrossRef]
- Levada, O.A.; Troyan, A.S. Insulin-like growth factor-1: A possible marker for emotional and cognitive disturbances, and treatment effectiveness in major depressive disorder. Ann. Gen. Psychiatry 2017, 16, 38. [Google Scholar] [CrossRef] [PubMed]
- Zegarra-Valdivia, J.; Khan, M.; Putzolu, A.; Cipriani, R.; Pignatelli, J.; Torres Aleman, I. Influencie of Lifestyle on Brain sensitivity to circulating insuling-like growth factor 1. bioRxiv 2025. bioRxiv:2025.02.27.640579. [Google Scholar] [CrossRef]
- Smith, P.J.; Mabe, S.M.; Sherwood, A.; Doraiswamy, P.M.; Welsh-Bohmer, K.A.; Burke, J.R.; Kraus, W.E.; Lin, P.-H.; Browndyke, J.N.; Babyak, M.A.; et al. Metabolic and Neurocognitive Changes Following Lifestyle Modification: Examination of Biomarkers from the ENLIGHTEN Randomized Clinical Trial. J. Alzheimer’s. Dis. 2020, 77, 1793. [Google Scholar] [CrossRef]
- Salzmann, A.; James, S.N.; Williams, D.M.; Richards, M.; Cadar, D.; Schott, J.M.; Coath, W.; Sudre, C.H.; Chaturvedi, N.; Garfield, V. Investigating the Relationship between IGF-I, IGF-II, and IGFBP-3 Concentrations and Later-Life Cognition and Brain Volume. J. Clin. Endocrinol. Metab. 2021, 106, 1617–1629. [Google Scholar] [CrossRef]
- Ryoo, S.W.; Anita, N.Z.; Perlman, G.; Xiong, L.Y.; Wu, C.Y.; Wood, M.; Rabin, J.S.; Mitchell, J.; Swardfager, W. Insulin-like growth factor-1 and cognition in normoglycemia, prediabetes, and type 2 diabetes mellitus. Psychoneuroendocrinology 2024, 161, 106946. [Google Scholar] [CrossRef]
- Halloway, S.; Volgman, A.S.; Barnes, L.L.; Schoeny, M.E.; Wilbur, J.E.; Pressler, S.J.; Laddu, D.; Phillips, S.A.; Vispute, S.; Hall, G.; et al. The MindMoves Trial: Cross-Sectional Analyses of Baseline Vascular Risk and Cognition in Older Women with Cardiovascular Disease. J. Alzheimer’s Dis. 2024, 100, 1407–1416. [Google Scholar] [CrossRef]
- Gubbi, S.; Quipildor, G.F.; Barzilai, N.; Huffman, D.M.; Milman, S. 40 years of IGF1: IGF1: The Jekyll and Hyde of the aging brain. J. Mol. Endocrinol. 2018, 61, T171–T185. [Google Scholar] [CrossRef]
- Pharaoh, G.; Owen, D.; Yeganeh, A.; Premkumar, P.; Farley, J.; Bhaskaran, S.; Ashpole, N.; Kinter, M.; Van Remmen, H.; Logan, S. Disparate Central and Peripheral Effects of Circulating IGF-1 Deficiency on Tissue Mitochondrial Function. Mol. Neurobiol. 2020, 57, 1317–1331. [Google Scholar] [CrossRef]
- Cao, Z.; Min, J.; Tan, Q.; Si, K.; Yang, H.; Xu, C. Circulating insulin-like growth factor-1 and brain health: Evidence from 369,711 participants in the UK Biobank. Alzheimer’s Res. Ther. 2023, 15, 140. [Google Scholar] [CrossRef]
- Chino, B.; Lucas, J.D.F.; Antón-toro, L.F.; Susi, G. M/EEG Hallmarks of Healthy and Pathological Aging. In Psychiatry and Neuroscience Update; Springer: Berlin/Heidelberg, Germany, 2024; pp. 507–529. ISBN 9783031722196. [Google Scholar]
- Zegarra-Valdivia, J.A.; Fernandes, J.; Fernandez de Sevilla, M.E.; Trueba-Saiz, A.; Pignatelli, J.; Suda, K.; Martinez-Rachadell, L.; Fernandez, A.M.; Esparza, J.; Vega, M.; et al. Insulin-like growth factor I sensitization rejuvenates sleep patterns in old mice. Geroscience 2022, 44, 2243–2257. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zegarra-Valdivia, J.; Arana-Nombera, H.; Perez-Fernandez, L.; del Rocío Casimiro, M.; Gallegos-Manayay, V.; del Rosario Oliva-Piscoya, M.; Alamo-Medina, R.; Abanto-Saldaña, E.; Cruz-Ordinola, M.C.; Paredes-Manrique, C.; et al. Insulin-like Growth Factor 1 Impact on Alzheimer’s Disease: Role in Inflammation, Stress, and Cognition. Curr. Issues Mol. Biol. 2025, 47, 233. https://doi.org/10.3390/cimb47040233
Zegarra-Valdivia J, Arana-Nombera H, Perez-Fernandez L, del Rocío Casimiro M, Gallegos-Manayay V, del Rosario Oliva-Piscoya M, Alamo-Medina R, Abanto-Saldaña E, Cruz-Ordinola MC, Paredes-Manrique C, et al. Insulin-like Growth Factor 1 Impact on Alzheimer’s Disease: Role in Inflammation, Stress, and Cognition. Current Issues in Molecular Biology. 2025; 47(4):233. https://doi.org/10.3390/cimb47040233
Chicago/Turabian StyleZegarra-Valdivia, Jonathan, Harold Arana-Nombera, Leandro Perez-Fernandez, Milagros del Rocío Casimiro, Viviana Gallegos-Manayay, María del Rosario Oliva-Piscoya, Reyna Alamo-Medina, Eduardo Abanto-Saldaña, María Celinda Cruz-Ordinola, Carmen Paredes-Manrique, and et al. 2025. "Insulin-like Growth Factor 1 Impact on Alzheimer’s Disease: Role in Inflammation, Stress, and Cognition" Current Issues in Molecular Biology 47, no. 4: 233. https://doi.org/10.3390/cimb47040233
APA StyleZegarra-Valdivia, J., Arana-Nombera, H., Perez-Fernandez, L., del Rocío Casimiro, M., Gallegos-Manayay, V., del Rosario Oliva-Piscoya, M., Alamo-Medina, R., Abanto-Saldaña, E., Cruz-Ordinola, M. C., Paredes-Manrique, C., & Chino-Vilca, B. (2025). Insulin-like Growth Factor 1 Impact on Alzheimer’s Disease: Role in Inflammation, Stress, and Cognition. Current Issues in Molecular Biology, 47(4), 233. https://doi.org/10.3390/cimb47040233