
Oncogene-Induced Senescence Transcriptomes Signify Premalignant Colorectal Adenomas

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset Acquisition and Preprocessing
2.2. The Senescence-Associated MSigDB Gene Collections
2.3. Single-Sample Gene Set Enrichment Analysis (ssGSEA)
2.4. Statistical Analysis
3. Results
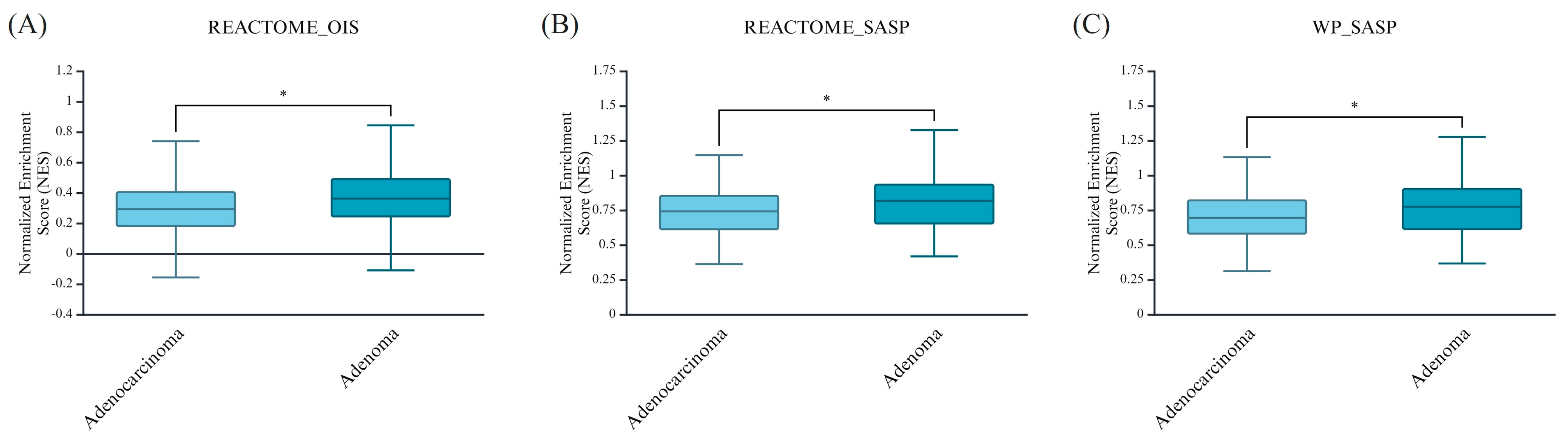
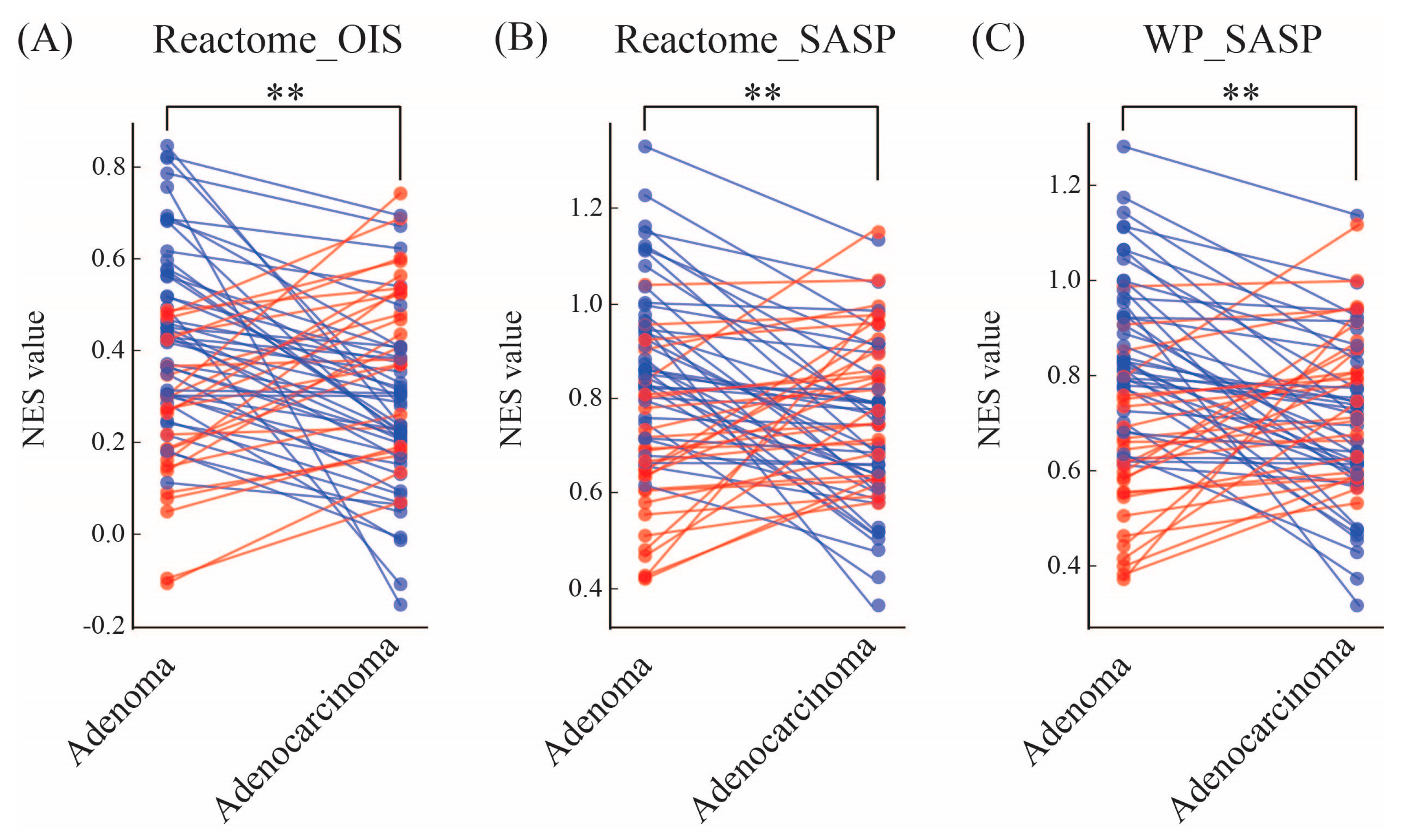
3.1. OIS and SASP Transcriptomic Signatures Predominate in Colorctal Adenomas in Comparison to Their Malignant Counterparts
3.2. Cell Cycle-Related Genes Are Downregulated Through Malignant Progression of Colorectal Lesions
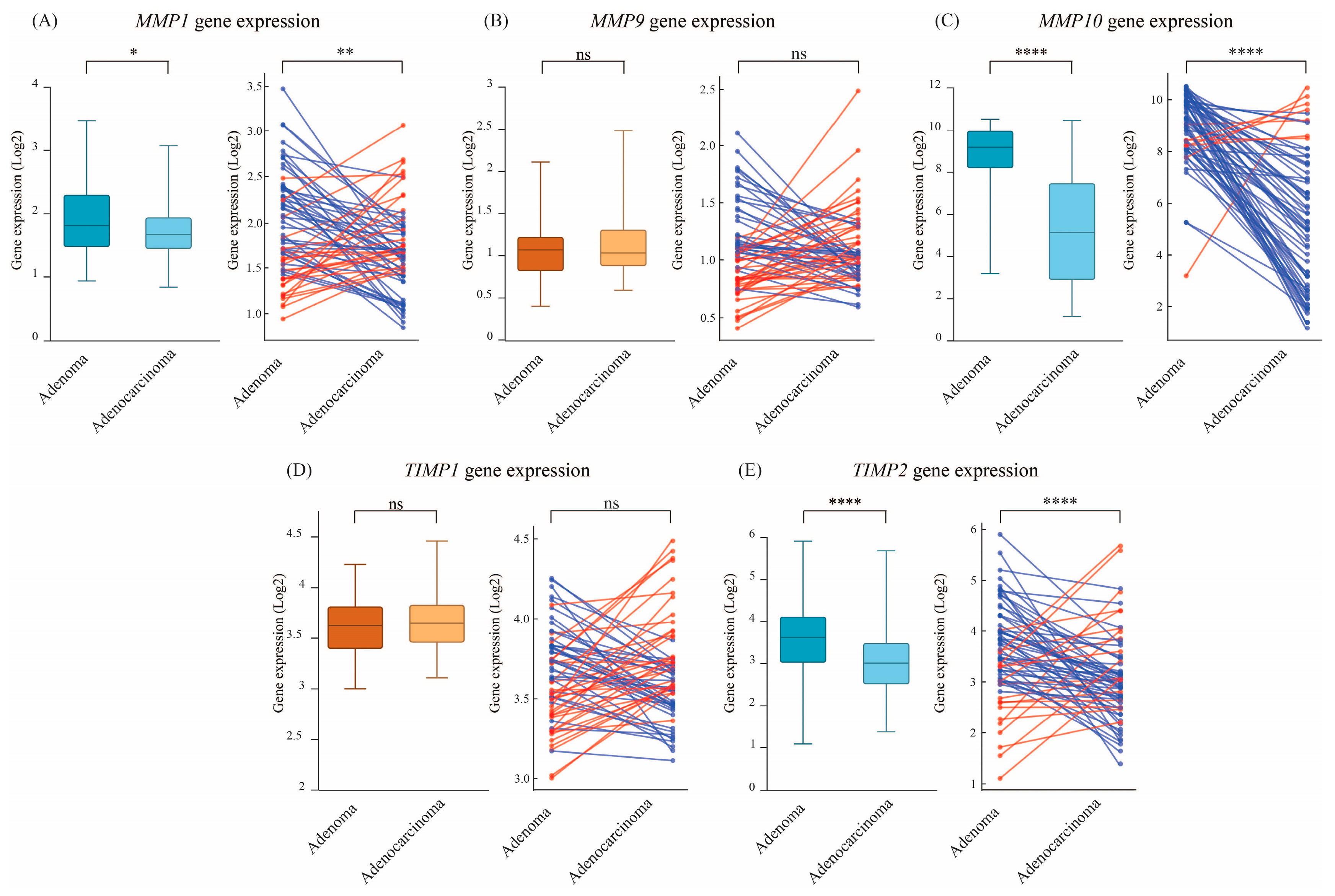
3.3. Senescence-Associated ECM Remodeling Genes Exhibit Lower Expresssion in Colorectal Adenocarcinomas
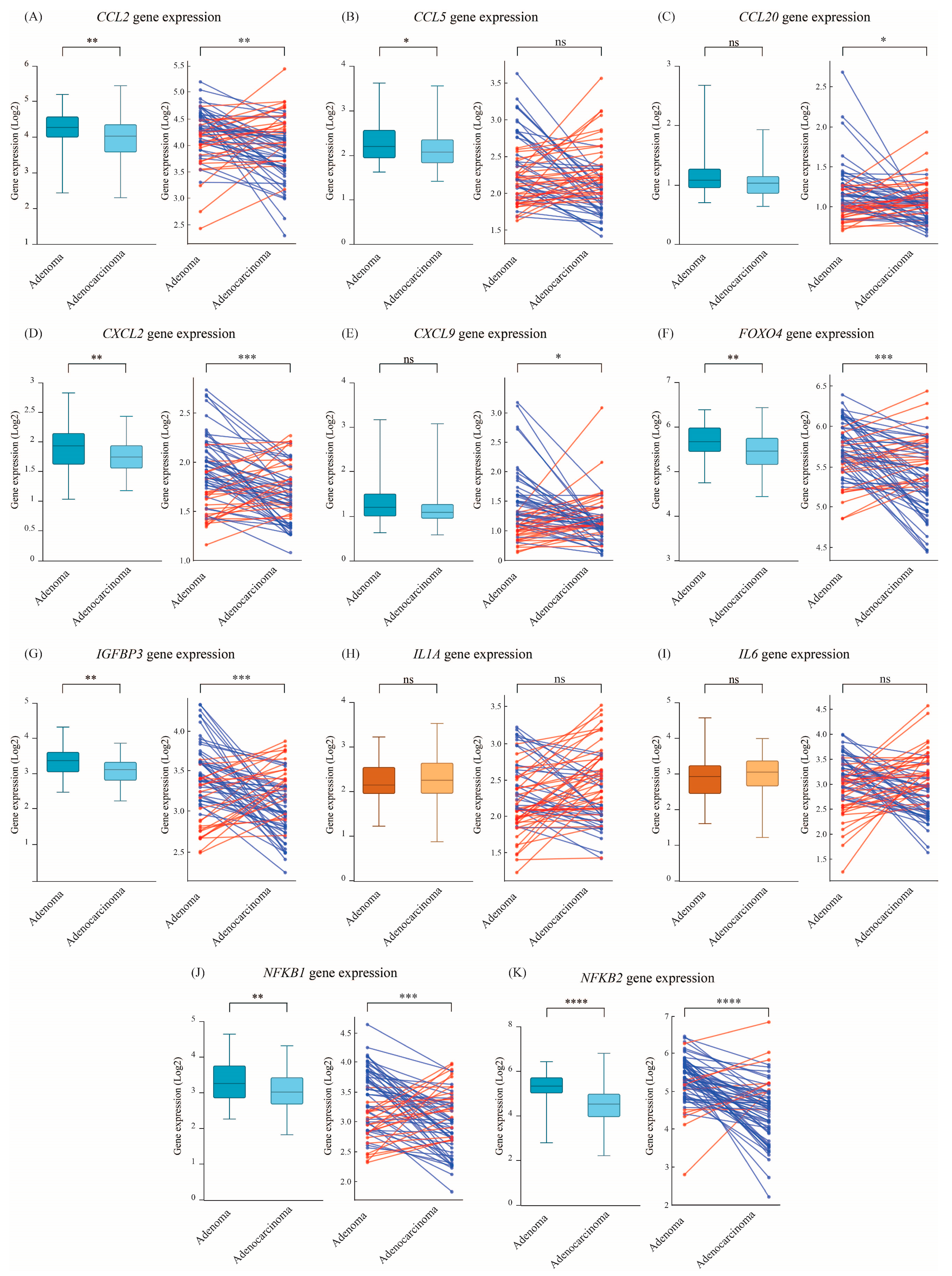
3.4. NF-κB and SASP-Related Cytokines Decline upon Progression from Adenoma to Adenocarcinoma Status
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| APC | Adenomatous polyposis coli |
| CRC | Colorectal cancer |
| CDKI | Cyclin-dependent Kinase Inhibitor |
| GEO | Gene Expression Omnibus |
| MSigDB | Molecular Signatures Database |
| OIS | Oncogene-induced senescence |
| SA-β-gal | Senescence-associated-β-galactosidase |
| SAGA | Senescence-associated growth arrest |
| SASP | Senescence-associated secretory phenotype |
| ssGSEA | Single-sample gene set enrichment analysis |
References
- Gorgoulis, V.; Adams, P.D.D.; Alimonti, A.; Bennett, D.C.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.A.; Sung, J.R.; Yoon, S.O.; Ji, H.P.; Jung, W.L.; Kim, H.P.; Kyung, T.K.; Ik, S.J.; Sang, C.P. Morphological Adjustment of Senescent Cells by Modulating Caveolin-1 Status. J. Biol. Chem. 2004, 279, 42270–42278. [Google Scholar] [CrossRef]
- Son, H.N.; Chi, H.N.Q.; Chung, D.C.; Long, L.T. Morphological Changes during Replicative Senescence in Bovine Ovarian Granulosa Cells. Cell Cycle 2019, 18, 1490. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, J.; Zheng, L.; Meissl, K.; Chaneton, B.; Selivanov, V.A.; MacKay, G.; Van Der Burg, S.H.; Verdegaal, E.M.E.; Cascante, M.; Shlomi, T.; et al. A Key Role for Mitochondrial Gatekeeper Pyruvate Dehydrogenase in Oncogene-Induced Senescence. Nature 2013, 498, 109–112. [Google Scholar] [CrossRef]
- von Zglinicki, T.; Saretzki, G.; Ladhoff, J.; di Fagagna, F.D.A.; Jackson, S.P. Human Cell Senescence as a DNA Damage Response. Mech. Ageing Dev. 2005, 126, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Muñoz, D.P.; Teachenor, R.; Chu, V.; Le, O.; Bhaumik, D.; Coppé, J.P.; Campeau, E.; Beauséjour, C.M.; Kim, S.H.; et al. DNA-SCARS: Distinct Nuclear Structures That Sustain Damage-Induced Senescence Growth Arrest and Inflammatory Cytokine Secretion. J. Cell Sci. 2011, 124, 68–81. [Google Scholar] [CrossRef]
- Ahmed, E.K.; Rogowska-Wrzesinska, A.; Roepstorff, P.; Bulteau, A.L.; Friguet, B. Protein Modification and Replicative Senescence of WI-38 Human Embryonic Fibroblasts. Aging Cell 2010, 9, 252–272. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular Senescence Drives Age-Dependent Hepatic Steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef]
- Kurz, D.J.D.J.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D.J.D.D. Senescence-Associated (Beta)-Galactosidase Reflects an Increase in Lysosomal Mass during Replicative Ageing of Human Endothelial Cells. J. Cell Sci. 2000, 113 Pt 2, 3613–3622. [Google Scholar] [CrossRef]
- Fridman, A.L.; Tainsky, M.A. Critical Pathways in Cellular Senescence and Immortalization Revealed by Gene Expression Profiling. Oncogene 2008, 27, 5975–5987. [Google Scholar] [CrossRef]
- Casella, G.; Munk, R.; Kim, K.M.M.; Piao, Y.; De, S.; Abdelmohsen, K.; Gorospe, M. Transcriptome Signature of Cellular Senescence. Nucleic Acids Res. 2019, 47, 7294–7305. [Google Scholar] [CrossRef]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the P53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Fridlyanskaya, I.; Alekseenko, L.; Nikolsky, N. Senescence as a General Cellular Response to Stress: A Mini-Review. Exp. Gerontol. 2015, 72, 124–128. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I. Oncogene-Induced Senescence and Tumour Control in Complex Biological Systems. Cell Death Differ. 2018, 25, 1005–1006. [Google Scholar] [CrossRef] [PubMed]
- Mooi, W.J.; Peeper, D.S. Oncogene-Induced Cell Senescence—Halting on the Road to Cancer. N. Engl. J. Med. 2006, 355, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.W.; Yaswen, P.; Campisi, J.; Beausejour, C.M.; Krtolica, A.; Galimi, F.; et al. Reversal of Human Cellular Senescence: Roles of the P53 and P16 Pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Carrire, C.; Gore, A.J.; Norris, A.M.; Gunn, J.R.; Young, A.L.; Longnecker, D.S.; Korc, M. Deletion of Rb Accelerates Pancreatic Carcinogenesis by Oncogenic Kras and Impairs Senescence in Premalignant Lesions. Gastroenterology 2011, 141, 1091–1101. [Google Scholar] [CrossRef]
- Sarkisian, C.J.; Keister, B.A.; Stairs, D.B.; Boxer, R.B.; Moody, S.E.; Chodosh, L.A. Dose-Dependent Oncogene-Induced Senescence in Vivo and Its Evasion during Mammary Tumorigenesis. Nat. Cell Biol. 2007, 9, 493–505. [Google Scholar] [CrossRef]
- Rane, S.G.; Cosenza, S.C.; Mettus, R.V.; Reddy, E.P. Germ Line Transmission of the Cdk4R24C Mutation Facilitates Tumorigenesis and Escape from Cellular Senescence. Mol. Cell. Biol. 2002, 22, 644–656. [Google Scholar] [CrossRef]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguría, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Tumour Biology: Senescence in Premalignant Tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef]
- Natarajan, E.; Saeb, M.; Crum, C.P.; Woo, S.B.; McKee, P.H.; Rheinwald, J.G. Co-Expression of P16INK4A and Laminin 5 Γ2 by Microinvasive and Superficial Squamous Cell Carcinomas in Vivo and by Migrating Wound and Senescent Keratinocytes in Culture. Am. J. Pathol. 2003, 163, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Gray-Schopfer, V.C.; Cheong, S.C.; Chong, H.; Chow, J.; Moss, T.; Abdel-Malek, Z.A.; Marais, R.; Wynford-Thomas, D.; Bennett, D.C. Cellular Senescence in Naevi and Immortalisation in Melanoma: A Role for P16? Br. J. Cancer 2006, 95, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.K.; Grisanzio, C.; O’Connell, F.; Barry, M.; Brito, J.M.; Xu, Q.; Guney, I.; Berger, R.; Herman, P.; Bikoff, R.; et al. A Prostatic Intraepithelial Neoplasia-Dependent P27 Kip1 Checkpoint Induces Senescence and Inhibits Cell Proliferation and Cancer Progression. Cancer Cell 2008, 14, 146–155. [Google Scholar] [CrossRef]
- Bascones-Martínez, A.; López-Durán, M.; Cano-Sánchez, J.; Sánchez-Verde, L.; Díez-Rodríguez, A.; Aguirre-Echebarría, P.; Alvarez-Fernández, E.; González-Moles, M.A.; Bascones-Ilundain, J.; Muzio, L.L.; et al. Differences in the Expression of Five Senescence Markers in Oral Cancer, Oral Leukoplakia and Control Samples in Humans. Oncol. Lett. 2012, 3, 1319–1325. [Google Scholar] [CrossRef]
- Tsang, C.M.; Yip, Y.L.; Lo, K.W.; Deng, W.; To, K.F.; Hau, P.M.; Lau, V.M.Y.; Takada, K.; Lui, V.W.Y.; Lung, M.L.; et al. Cyclin D1 Overexpression Supports Stable EBV Infection in Nasopharyngeal Epithelial Cells. Proc. Natl. Acad. Sci. USA 2012, 109, E3473–E3482. [Google Scholar] [CrossRef]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.F.M.; Schlegelberger, B.; Stein, H.; Dörken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-Induced Senescence as an Initial Barrier in Lymphoma Development. Nature 2005, 436, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Yu, Q.; Subrahmanyam, R.; Difilippantonio, M.J.; Ried, T.; Sen, J.M. β-Catenin Expression Results in P53-Independent DNA Damage and Oncogene-Induced Senescence in Prelymphomagenic Thymocytes In Vivo. Mol. Cell. Biol. 2008, 28, 1713–1723. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A Genetic Model for Colorectal Tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Oikonomou, E.; Makrodouli, E.; Evagelidou, M.; Joyce, T.; Probert, L.; Pintzas, A. BRAF(V600E) Efficient Transformation and Induction of Microsatellite Instability versus KRAS(G12V) Induction of Senescence Markers in Human Colon Cancer Cells. Neoplasia 2009, 11, 1116–1131. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-Induced Senescence Is Part of the Tumorigenesis Barrier Imposed by DNA Damage Checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Milacic, M.; Beavers, D.; Conley, P.; Gong, C.; Gillespie, M.; Griss, J.; Haw, R.; Jassal, B.; Matthews, L.; May, B.; et al. The Reactome Pathway Knowledgebase 2024. Nucleic Acids Res. 2024, 52, D672–D678. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Balcı, H.; Hanspers, K.; Coort, S.L.; Martens, M.; Slenter, D.N.; Ehrhart, F.; Digles, D.; Waagmeester, A.; Wassink, I.; et al. WikiPathways 2024: Next Generation Pathway Database. Nucleic Acids Res. 2024, 52, D679–D689. [Google Scholar] [CrossRef]
- Savić, R.; Yang, J.; Koplev, S.; An, M.C.; Patel, P.L.; O’Brien, R.N.; Dubose, B.N.; Dodatko, T.; Rogatsky, E.; Sukhavasi, K.; et al. Integration of Transcriptomes of Senescent Cell Models with Multi-Tissue Patient Samples Reveals Reduced COL6A3 as an Inducer of Senescence. Cell Rep. 2023, 42, 113371. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.D.; Wagner, N. The Senescence Markers P16INK4A, P14ARF/P19ARF, and P21 in Organ Development and Homeostasis. Cells 2022, 11, 1966. [Google Scholar] [CrossRef]
- Kriegl, L.; Neumann, J.; Vieth, M.; Greten, F.R.; Reu, S.; Jung, A.; Kirchner, T. Up and Downregulation of P16(Ink4a) Expression in BRAF-Mutated Polyps/Adenomas Indicates a Senescence Barrier in the Serrated Route to Colon Cancer. Mod. Pathol. 2011, 24, 1015–1022. [Google Scholar] [CrossRef]
- Dai, C.Y.; Furth, E.E.; Mick, R.; Koh, J.; Takayama, T.; Niitsu, Y.; Enders, G.H. P16(INK4a) Expression Begins Early in Human Colon Neoplasia and Correlates Inversely with Markers of Cell Proliferation. Gastroenterology 2000, 119, 929–942. [Google Scholar] [CrossRef]
- Guo, Y.; Ayers, J.L.; Carter, K.T.; Wang, T.; Maden, S.K.; Edmond, D.; Newcomb, P.P.; Li, C.; Ulrich, C.; Yu, M.; et al. Senescence-Associated Tissue Microenvironment Promotes Colon Cancer Formation through the Secretory Factor GDF15. Aging Cell 2019, 18, e13013. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.W.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef]
- Chen, W.; Wang, X.; Wei, G.; Huang, Y.; Shi, Y.; Li, D.; Qiu, S.; Zhou, B.; Cao, J.; Chen, M.; et al. Single-Cell Transcriptome Analysis Reveals Six Subpopulations Reflecting Distinct Cellular Fates in Senescent Mouse Embryonic Fibroblasts. Front. Genet. 2020, 11, 867. [Google Scholar] [CrossRef]
- Tang, H.; Geng, A.; Zhang, T.; Wang, C.; Jiang, Y.; Mao, Z. Single Senescent Cell Sequencing Reveals Heterogeneity in Senescent Cells Induced by Telomere Erosion. Protein Cell 2019, 10, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Almanzar, N.; Antony, J.; Baghel, A.S.; Bakerman, I.; Bansal, I.; Barres, B.A.; Beachy, P.A.; Berdnik, D.; Bilen, B.; Brownfield, D.; et al. A Single-Cell Transcriptomic Atlas Characterizes Ageing Tissues in the Mouse. Nature 2020, 583, 590–595. [Google Scholar] [CrossRef]
- Yildiz, G.; Arslan-Ergul, A.; Bagislar, S.; Konu, O.; Yuzugullu, H.; Gursoy-Yuzugullu, O.; Ozturk, N.; Ozen, C.; Ozdag, H.; Erdal, E.; et al. Genome-Wide Transcriptional Reorganization Associated with Senescence-to-Immortality Switch during Human Hepatocellular Carcinogenesis. PLoS ONE 2013, 8, e64016. [Google Scholar] [CrossRef]
- Lafferty-Whyte, K.; Cairney, C.J.; Jamieson, N.B.; Oien, K.A.; Keith, W.N. Pathway Analysis of Senescence-Associated MiRNA Targets Reveals Common Processes to Different Senescence Induction Mechanisms. Biochim. Biophys. Acta-Mol. Basis Dis. 2009, 1792, 341–352. [Google Scholar]
- Wang, L.; Leite de Oliveira, R.; Wang, C.; Fernandes Neto, J.M.; Mainardi, S.; Evers, B.; Lieftink, C.; Morris, B.; Jochems, F.; Willemsen, L.; et al. High-Throughput Functional Genetic and Compound Screens Identify Targets for Senescence Induction in Cancer. Cell Rep. 2017, 21, 773–783. [Google Scholar] [CrossRef]
- Gala, M.K.; Mizukami, Y.; Le, L.P.; Moriichi, K.; Austin, T.; Yamamoto, M.; Lauwers, G.Y.; Bardeesy, N.; Chung, D.C. Germline Mutations in Oncogene-Induced Senescence Pathways Are Associated with Multiple Sessile Serrated Adenomas. Gastroenterology 2014, 146, 520–529. [Google Scholar] [CrossRef]
- Khasawneh, A.I.; Al Shboul, S.; Himsawi, N.; Al Rousan, A.; Shahin, N.A.; El-Sadoni, M.; Alhesa, A.; Abu Ghalioun, A.; Khawaldeh, S.; Shawish, B.; et al. Resolution of Oncogene-Induced Senescence Markers in HPV-Infected Cervical Cancer Tissue. BMC Cancer 2025, 25, 111. [Google Scholar] [CrossRef]
- Saleh, T.; Himsawi, N.; Al Rousan, A.; Alhesa, A.; El-Sadoni, M.; Khawaldeh, S.; Shahin, N.A.; Ghalioun, A.A.; Shawish, B.; Friehat, K.; et al. Variable Expression of Oncogene-Induced Senescence/SASP Surrogates in HPV-Associated Precancerous Cervical Tissue. Curr. Issues Mol. Biol. 2024, 46, 13696–13712. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 Loss Is a Senescence-Associated Biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Saleh, T.; Alhesa, A.; El-Sadoni, M.; Abu Shahin, N.; Alsharaiah, E.; Al Shboul, S.; Awad, H.; Bloukh, S.; Al-Balas, M.; Alsalem, M.; et al. The Expression of the Senescence-Associated Biomarker Lamin B1 in Human Breast Cancer. Diagnostics 2022, 12, 609. [Google Scholar] [CrossRef]
- Kolodkin-Gal, D.; Roitman, L.; Ovadya, Y.; Azazmeh, N.; Assouline, B.; Schlesinger, Y.; Kalifa, R.; Horwitz, S.; Khalatnik, Y.; Hochner-Ger, A.; et al. Senolytic Elimination of Cox2-Expressing Senescent Cells Inhibits the Growth of Premalignant Pancreatic Lesions. Gut 2022, 71, 345–355. [Google Scholar] [CrossRef]
- Scanlan, R.L.; Pease, L.; O’Keefe, H.; Martinez-Guimera, A.; Rasmussen, L.; Wordsworth, J.; Shanley, D. Systematic Transcriptomic Analysis and Temporal Modelling of Human Fibroblast Senescence. Front. Aging 2024, 5, 1448543. [Google Scholar] [CrossRef]
- Yasuda, T.; Baba, H.; Ishimoto, T. Cellular Senescence in the Tumor Microenvironment and Context-Specific Cancer Treatment Strategies. FEBS J. 2023, 290, 1290–1302. [Google Scholar] [CrossRef]
- Hoenicke, L.; Zender, L. Immune Surveillance of Senescent Cells--Biological Significance in Cancer- and Non-Cancer Pathologies. Carcinogenesis 2012, 33, 1123–1126. [Google Scholar] [CrossRef]
- Prata, L.G.P.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent Cell Clearance by the Immune System: Emerging Therapeutic Opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef]
- Han, Y.; Micklem, G.; Kim, S.Y. Transcriptional Landscape of Oncogene-Induced Senescence: A Machine Learning-Based Meta-Analytic Approach. Ageing Res. Rev. 2023, 85, 101849. [Google Scholar] [CrossRef]
- Saleh, T.; Bloukh, S.; Hasan, M.; Al Shboul, S. Therapy-Induced Senescence as a Component of Tumor Biology: Evidence from Clinical Cancer. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188994. [Google Scholar] [CrossRef] [PubMed]
- te Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joel, S.P. DNA Damage Is Able to Induce Senescence in Tumor Cells in Vitro and in Vivo. Cancer Res. 2002, 62, 1876–1883. [Google Scholar] [PubMed]
- González-Gualda, E.; Baker, A.G.; Fruk, L.; Muñoz-Espín, D.; González-Gualda, E.; Baker, A.G.; Fruk, L.; Muñoz-Espín, D. A Guide to Assessing Cellular Senescence in Vitro and in Vivo. FEBS J. 2021, 288, 56–80. [Google Scholar] [CrossRef]
- López-Vicente, L.; Armengol, G.; Pons, B.; Coch, L.; Argelaguet, E.; Lleonart, M.; Hernández-Losa, J.; de Torres, I.; Cajal, S.R.y. Regulation of Replicative and Stress-Induced Senescence by RSK4, Which Is Down-Regulated in Human Tumors. Clin. Cancer Res. 2009, 15, 4546–4553. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, C. Transcriptomic Analysis of Cellular Senescence: One Step Closer to Senescence Atlas. Mol. Cells 2021, 44, 136–145. [Google Scholar] [CrossRef]
- Ding, C.; Xu, X.; Zhang, X.; Zhang, E.; Li, S.; Fan, X.; Ma, J.; Yang, X.; Zang, L. Investigating the Role of Senescence Biomarkers in Colorectal Cancer Heterogeneity by Bulk and Single-Cell RNA Sequencing. Sci. Rep. 2024, 14, 20083. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Park, S.S.; Lee, Y.-K.; Choi, Y.W.; Bin Lim, S.; Park, S.H.; Kim, H.K.; Shin, J.S.; Kim, Y.H.; Lee, D.H.; Kim, J.-H.; et al. Cellular Senescence Is Associated with the Spatial Evolution toward a Higher Metastatic Phenotype in Colorectal Cancer. Cell Rep. 2024, 43, 113912. [Google Scholar] [CrossRef]
- Nakao, T.; Shimada, M.; Yoshikawa, K.; Tokunaga, T.; Nishi, M.; Kashihara, H.; Takasu, C.; Wada, Y.; Yoshimoto, T. Prognostic Impact of the Combination of P16INK4a, P21 and Immunoscore in Rectal Cancer. Int. J. Clin. Oncol. 2024, 29, 1152–1160. [Google Scholar] [CrossRef]
- Hu, X.; Zhu, X.; Chen, Y.; Zhang, W.; Li, L.; Liang, H.; Usmanov, B.B.; Donadon, M.; Yusupbekov, A.A.; Zheng, Y. Senescence-Related Signatures Predict Prognosis and Response to Immunotherapy in Colon Cancer. J. Gastrointest. Oncol. 2024, 15, 1020–1034. [Google Scholar] [CrossRef]
- Kellers, F.; Fernandez, A.; Konukiewitz, B.; Schindeldecker, M.; Tagscherer, K.E.; Heintz, A.; Jesinghaus, M.; Roth, W.; Foersch, S. Senescence-Associated Molecules and Tumor-Immune-Interactions as Prognostic Biomarkers in Colorectal Cancer. Front. Med. 2022, 9, 865230. [Google Scholar] [CrossRef] [PubMed]
- Park, S.S.; Lee, Y.K.; Park, S.H.; Bin Lim, S.; Choi, Y.W.; Shin, J.S.; Kim, Y.H.; Kim, J.H.; Park, T.J. P15INK4B Is an Alternative Marker of Senescent Tumor Cells in Colorectal Cancer. Heliyon 2023, 9, e13170. [Google Scholar] [CrossRef] [PubMed]
- Al Shboul, S.; Abu Al Karsaneh, O.; Alrjoub, M.; Al-Qudah, M.; El-Sadoni, M.; Alhesa, A.; Ramadan, M.; Barukba, M.; Al-Quran, E.F.; Masaadeh, A.; et al. Dissociation between the Expression of CGAS/STING and a Senescence-Associated Signature in Colon Cancer. Int. J. Immunopathol. Pharmacol. 2025, 39, 3946320251324821. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Carpenter, V.J. Potential Use of Senolytics for Pharmacological Targeting of Precancerous Lesions. Mol. Pharmacol. 2021, 100, 580–587. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Higher in Adenoma% (n) | Higher in Adenocarcinoma% (n) | |
|---|---|---|
| REACTOME_ONCOGENE_INDUCED_SENESCENCE (NES values) | 65% (43) | 35% (23) |
| REACTOME_SENESCENCE_ASSOCIATED_SECRETORY_PHENOTYPE_SASP (NES values) | 59% (39) | 41% (27) |
| WP_SENESCENCEASSOCIATED_SECRETORY_PHENOTYPE_SASP (NES values) | 61% (40) | 39% (26) |
| Higher in Adenoma% (n) | Higher in Adenocarcinoma% (n) | Fold Change | ||
|---|---|---|---|---|
| Cell cycle-related genes | CDKN1A | 61% (40) | 39% (26) | 1.127 |
| CDKN2B | 65% (43) | 35% (23) | 1.143 | |
| E2F3 | 73% (48) | 27% (18) | 1.254 | |
| Senescence-associated ECM remodeling genes | MMP1 | 53% (35) | 47% (31) | 1.188 |
| MMP9 | 48% (32) | 52% (34) | 0.992 | |
| MMP10 | 89% (59) | 11% (7) | 13.372 | |
| TIMP1 | 53% (35) | 47% (31) | 0.986 | |
| TIMP2 | 64% (42) | 36% (24) | 1.436 | |
| NF-κB and SASP-related cytokines | CCL2 | 64% (42) | 36% (24) | 1.181 |
| CCL5 | 53% (35) | 47% (31) | 1.105 | |
| CCL20 | 59% (39) | 41% (27) | 1.078 | |
| CXCL2 | 67% (44) | 33% (22) | 1.142 | |
| CXCL9 | 59% (39) | 41% (27) | 1.136 | |
| FOXO4 | 68% (45) | 32% (21) | 1.242 | |
| IGFBP3 | 67% (44) | 33% (22) | 1.207 | |
| IL1A | 42% (28) | 58% (38) | 0.925 | |
| IL6 | 55% (36) | 45% (30) | 1.066 | |
| NFKB1 | 68% (45) | 32% (21) | 1.256 | |
| NFKB2 | 86% (57) | 14% (9) | 1.803 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Shboul, S.; Awad, H.; Abu-Humaidan, A.; Ababneh, N.A.; Khasawneh, A.I.; Saleh, T. Oncogene-Induced Senescence Transcriptomes Signify Premalignant Colorectal Adenomas. Curr. Issues Mol. Biol. 2025, 47, 221. https://doi.org/10.3390/cimb47040221
Al Shboul S, Awad H, Abu-Humaidan A, Ababneh NA, Khasawneh AI, Saleh T. Oncogene-Induced Senescence Transcriptomes Signify Premalignant Colorectal Adenomas. Current Issues in Molecular Biology. 2025; 47(4):221. https://doi.org/10.3390/cimb47040221
Chicago/Turabian StyleAl Shboul, Sofian, Heyam Awad, Anas Abu-Humaidan, Nidaa A. Ababneh, Ashraf I. Khasawneh, and Tareq Saleh. 2025. "Oncogene-Induced Senescence Transcriptomes Signify Premalignant Colorectal Adenomas" Current Issues in Molecular Biology 47, no. 4: 221. https://doi.org/10.3390/cimb47040221
APA StyleAl Shboul, S., Awad, H., Abu-Humaidan, A., Ababneh, N. A., Khasawneh, A. I., & Saleh, T. (2025). Oncogene-Induced Senescence Transcriptomes Signify Premalignant Colorectal Adenomas. Current Issues in Molecular Biology, 47(4), 221. https://doi.org/10.3390/cimb47040221