
Gut Microbiota Changes in Metabolic Dysfunction-Associated Steatohepatitis and Inflammatory Bowel Disease: Common Pathogenic Features

 ,
, 
 , , ,
, , , 

Abstract
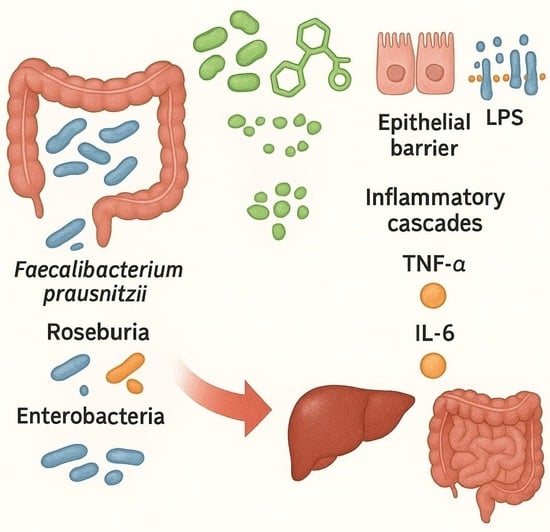
1. Introduction
2. Gut Microbiota and MASH
2.1. Taxonomic Shifts of Gut Microbiota in MASH
2.2. Functional Alterations in Microbial Metabolism in MASH
- Gut microbiota and SCFAs: one of the most consistent findings is the reduced microbial capacity to produce SCFAs, particularly butyrate, which preserves epithelial barrier integrity and exerts anti-inflammatory effects via G-protein–coupled receptor 41/43 (GPR41/43) activation and histone deacetylase (HDAC) inhibition [37,38].
- Bile acid metabolism: MASH-associated dysbiosis is also characterized by disruption of bile acid metabolism. Microbes expressing bile salt hydrolase and 7α-dehydroxylase convert primary bile acids into secondary bile acids, such as deoxycholic acid (DCA). Elevated fecal DCA levels in MASH patients, together with a reduced abundance of Bacteroides, promote activation of hepatic farnesoid X receptor (FXR) and Takeda G protein–coupled receptor 5 (TGR5), enhancing pro-inflammatory and pro-fibrotic signaling [39].
- Microbial translocation and inflammatory mediators: dysbiosis further increases intestinal permeability, as shown by downregulation of tight junction proteins (occludin, claudin-1) in both human MASH and high-fat-diet-fed mouse models. This results in higher circulating levels of lipopolysaccharide (LPS) [40]. LPS activates TLR-4 on Kupffer and hepatic stellate cells, stimulating the release of tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and pro-fibrotic mediators such as transforming growth factor-beta (TGF-β), thereby linking microbial products directly to liver inflammation and fibrogenesis [41].
3. Gut Microbiota and IBD
3.1. Taxonomic Shifts of Gut Microbiota in IBD
3.2. Functional Alterations in Microbial Metabolism in IBD
- Gut microbiota and SCFAs: metagenomic and metabolomic analyses consistently demonstrate reduced production of SCFAs, particularly butyrate and propionate [56,57]. Butyrate, mainly produced by F. prausnitzii, Roseburia, and Eubacterium rectale, serves as the primary energy source for colonocytes and exerts potent anti-inflammatory effects. Its depletion impairs epithelial barrier integrity and exacerbates mucosal inflammation in IBD [58].
- Redox imbalance: IBD microbiota displays increased oxidative stress and preferential use of nitrate and sulfate respiration, creating conditions that favor facultative anaerobes such as Enterobacteriaceae over obligate anaerobes. This shift in redox balance reinforces microbial instability and drives chronic immune activation [59].
- Bile acid metabolism: in healthy individuals, microbial deconjugation and 7α-dehydroxylation convert primary bile acids into secondary ones such as DCA and lithocholic acid. In IBD, these pathways are impaired, resulting in an accumulation of conjugated bile acids that promote barrier dysfunction and mucosal inflammation through FXR and TGR5 signaling [60].
- Tryptophan metabolism and immune regulation: microbial catabolism of tryptophan into indole derivatives is a major source of ligands for the aryl hydrocarbon receptor (AhR), which maintains intestinal immune tolerance and epithelial repair. Loss of this pathway in IBD reduces AhR activation, contributing to chronic inflammation [61]. In addition, multi-omics evaluations reveal that strain-level variation modulates functional outputs. For example, Ruminococcus gnavus, commonly enriched in IBD, may exert either pro- or anti-inflammatory effects depending on capsular polysaccharide expression [62].
4. Shared Pathogenic Mechanisms of Gut Dysbiosis in MASH and IBD
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 16S rRNA | 16S ribosomal RNA |
| AhR | Aryl hydrocarbon Receptor |
| AUC | Area Under the Curve |
| BMI | Body mass index |
| CD | Crohn’s disease |
| DCA | Deoxycholic Acid |
| E. coli | Escherichia coli |
| F/B | Firmicutes/Bacteroidetes ratio |
| FXR | Farnesoid X Receptor |
| GPR41/43 | G-protein–coupled Receptor 41/43 |
| HCC | Hepatocellular carcinoma |
| HDAC | Histone Deacetylase |
| IBD | Inflammatory Bowel Disease |
| IL | Interleukin |
| LPS | Lipopolysaccharide |
| MAMPs | Microbe-Associated Molecular Patterns |
| MASH | Metabolic dysfunction-Associated Steatohepatitis |
| MASLD | Metabolic dysfunction-Associated Steatotic Liver Disease |
| SCFA | Short-Chain Fatty Acid |
| T2DM | Type 2 diabetes mellitus |
| TGF-β | Transforming Growth Factor-beta |
| TGR5 | Takeda G protein-coupled Receptor 5 |
| TLR | Toll-Like Receptor |
| TNF-α | Tumor Necrosis Factor-alpha |
| UC | Ulcerative Colitis |
References
- Hou, K.; Wu, Z.X.; Chen, X.Y.; Wang, J.Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in health and diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Shen, Y.; Fan, N.; Ma, S.X.; Cheng, X.; Yang, X.; Wang, G. Gut Microbiota Dysbiosis: Pathogenesis, Diseases, Prevention, and Therapy. MedComm (2020) 2025, 6, e70168. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Román, A.; Pagán-Zayas, N.; Velázquez-Rivera, L.I.; Torres-Ventura, A.C.; Godoy-Vitorino, F. Insights into Gut Dysbiosis: Inflammatory Diseases, Obesity, and Restoration Approaches. Int. J. Mol. Sci. 2024, 25, 9715. [Google Scholar] [CrossRef] [PubMed]
- Bandopadhyay, P.; Ganguly, D. Gut dysbiosis and metabolic diseases. Prog. Mol. Biol. Transl. Sci. 2022, 191, 153–174. [Google Scholar] [PubMed]
- Scarlata, G.G.M.; Abenavoli, L. Gut microbiota: The pathogenetic bridge between inflammatory bowel disease and metabolic-associated steatotic liver disease. Expert. Rev. Gastroenterol. Hepatol. 2025, 19, 85–88. [Google Scholar]
- Ohtani, N.; Kamiya, T.; Kawada, N. Recent updates on the role of the gut-liver axis in the pathogenesis of NAFLD/NASH, HCC, and beyond. Hepatol. Commun. 2023, 7, e0241. [Google Scholar]
- Rinella, M.E.; Sookoian, S. From NAFLD to MASLD: Updated naming and diagnosis criteria for fatty liver disease. J. Lipid Res. 2024, 65, 100485. [Google Scholar]
- Guo, Z.; Wu, D.; Mao, R.; Yao, Z.; Wu, Q.; Lv, W. Global burden of MAFLD, MAFLD related cirrhosis and MASH related liver cancer from 1990 to 2021. Sci. Rep. 2025, 15, 7083. [Google Scholar] [CrossRef]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology 2023, 78, 1966–1986. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Newsome, P.N. NAFLD vs MASLD (Metabolic Dysfunction-Associated Steatotic Liver Disease)-Why the Need for a Change of Nomenclature? J. Clin. Endocrinol. Metab. 2025, 110, e2407–e2410. [Google Scholar]
- Sergi, C.M. NAFLD (MASLD)/NASH (MASH): Does It Bother to Label at All? A Comprehensive Narrative Review. Int. J. Mol. Sci. 2024, 25, 8462. [Google Scholar] [CrossRef] [PubMed]
- Jegodzinski, L.; Rudolph, L.; Castven, D.; Sayk, F.; Rout, A.K.; Föh, B.; Hölzen, L.; Meyhöfer, S.; Schenk, A.; Weber, S.N.; et al. PNPLA3 I148M variant links to adverse metabolic traits in MASLD during fasting and feeding. JHEP Rep. 2025, 7, 101450. [Google Scholar] [CrossRef] [PubMed]
- Armandi, A.; Rosso, C.; Caviglia, G.P.; Bugianesi, E. An updated overview on hepatocellular carcinoma in patients with Metabolic dysfunction-Associated Steatotic Liver Disease: Trends, pathophysiology and risk-based surveillance. Metabolism 2025, 162, 156080. [Google Scholar] [CrossRef] [PubMed]
- Calvez, V.; Puca, P.; Di Vincenzo, F.; Del Gaudio, A.; Bartocci, B.; Murgiano, M.; Iaccarino, J.; Parand, E.; Napolitano, D.; Pugliese, D.; et al. Novel Insights into the Pathogenesis of Inflammatory Bowel Diseases. Biomedicines 2025, 13, 305. [Google Scholar] [CrossRef]
- Cao, Y.; Shen, J.; Ran, Z.H. Association between Faecalibacterium prausnitzii Reduction and Inflammatory Bowel Disease: A Meta-Analysis and Systematic Review of the Literature. Gastroenterol. Res. Pract. 2014, 2014, 872725. [Google Scholar]
- Zheng, M.; Han, R.; Yuan, Y.; Xing, Y.; Zhang, W.; Sun, Z.; Liu, Y.; Li, J.; Mao, T. The role of Akkermansia muciniphila in inflammatory bowel disease: Current knowledge and perspectives. Front. Immunol. 2023, 13, 1089600. [Google Scholar] [CrossRef]
- Mirsepasi-Lauridsen, H.C.; Vallance, B.A.; Krogfelt, K.A.; Petersen, A.M. Escherichia coli Pathobionts Associated with Inflammatory Bowel Disease. Clin. Microbiol. Rev. 2019, 32, e00060-18. [Google Scholar] [CrossRef]
- Weiss, G.A.; Hennet, T. Mechanisms and consequences of intestinal dysbiosis. Cell Mol. Life Sci. 2017, 74, 2959–2977. [Google Scholar] [CrossRef]
- Peterson, D.; Weidenmaier, C.; Timberlake, S.; Gura Sadovsky, R. Depletion of key gut bacteria predicts disrupted bile acid metabolism in inflammatory bowel disease. Microbiol. Spectr. 2025, 13, e0199924. [Google Scholar] [CrossRef]
- Camilleri, M. What is the leaky gut? Clinical considerations in humans. Curr. Opin. Clin. Nutr. Metab. Care 2021, 24, 473–482. [Google Scholar] [CrossRef]
- Hsu, C.L.; Schnabl, B. The gut-liver axis and gut microbiota in health and liver disease. Nat. Rev. Microbiol. 2023, 21, 719–733. [Google Scholar] [CrossRef]
- Dawson, S.L.; Todd, E.; Ward, A.C. The Interplay of Nutrition, the Gut Microbiota and Immunity and Its Contribution to Human Disease. Biomedicines 2025, 13, 329. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Whitley, C.S.; Haribabu, B.; Jala, V.R. Regulation of Intestinal Barrier Function by Microbial Metabolites. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 1463–1482. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, L.; Hua, H.; Liu, L.; Mao, Y.; Wang, R. Interactions between toll-like receptors signaling pathway and gut microbiota in host homeostasis. Immun. Inflamm. Dis. 2024, 12, e1356. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, S.G.; van der Merwe, S.; Krag, A.; Wiest, R. Gut-liver axis: Pathophysiological concepts and medical perspective in chronic liver diseases. Semin. Immunol. 2024, 71, 101859. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar]
- Del Chierico, F.; Nobili, V.; Vernocchi, P.; Russo, A.; De Stefanis, C.; Gnani, D.; Furlanello, C.; Zandonà, A.; Paci, P.; Capuani, G.; et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 2017, 65, 451–464. [Google Scholar] [CrossRef]
- Haslam, D.B. Nonalcoholic steatohepatitis and the intestinal microbiota. Hepatology 2017, 65, 401–403. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5. [Google Scholar] [CrossRef]
- Duarte, S.M.B.; Stefano, J.T.; Miele, L.; Ponziani, F.R.; Souza-Basqueira, M.; Okada, L.S.R.R.; de Barros Costa, F.G.; Toda, K.; Mazo, D.F.C.; Sabino, E.C.; et al. Gut microbiome composition in lean patients with NASH is associated with liver damage independent of caloric intake: A prospective pilot study. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 369–384. [Google Scholar] [CrossRef]
- Caussy, C.; Tripathi, A.; Humphrey, G.; Bassirian, S.; Singh, S.; Faulkner, C.; Bettencourt, R.; Rizo, E.; Richards, L.; Xu, Z.Z.; et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat. Commun. 2019, 10, 1406. [Google Scholar] [CrossRef]
- van Kleef, L.A.; Pustjens, J.; Savas, M.; Ayada, I.; Li, P.; Pan, Q.; van Rossum, E.F.C.; Janssen, H.L.A.; Brouwer, W.P. MASLD, At-Risk MASH and Increased Liver Stiffness Are Associated with Young Adulthood Obesity Without Residual Risk After Losing Obesity. Liver Int. 2025, 45, e16169. [Google Scholar] [CrossRef]
- Bhattacharya, I.; Maity, D.K.; Kumar, A.; Sarkar, S.; Bhattacharya, T.; Sahu, A.; Sreedhar, R.; Arumugam, S. Beyond obesity: Lean metabolic dysfunction-associated steatohepatitis from unveiling molecular pathogenesis to therapeutic advancement. Naunyn Schmiedebergs Arch. Pharmacol. 2025, 398, 13647–13665. [Google Scholar] [CrossRef]
- Sookoian, S.; Salatino, A.; Castaño, G.O.; Landa, M.S.; Fijalkowky, C.; Garaycoechea, M.; Pirola, C.J. Intrahepatic bacterial metataxonomic signature in non-alcoholic fatty liver disease. Gut 2020, 69, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Lyu, B.; Xie, F.; Li, F.; Xing, Y.; Han, Z.; Lai, J.; Ma, J.; Zou, Y.; Zeng, H.; et al. From gut to liver: Unveiling the differences of intestinal microbiota in NAFL and NASH patients. Front. Microbiol. 2024, 15, 1366744. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shi, M.; Zhao, C.; Liang, G.; Li, C.; Ge, X.; Pei, C.; Kong, Y.; Li, D.; Yang, W.; et al. Role of intestinal flora in the development of nonalcoholic fatty liver disease in children. Microbiol. Spectr. 2024, 12, e0100623. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Covián, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; de Los Reyes-Gavilán, C.G.; Salazar, N. Intestinal Short Chain Fatty Acids and their Link with Diet and Human Health. Front. Microbiol. 2016, 7, 185. [Google Scholar] [CrossRef]
- Du, Y.; He, C.; An, Y.; Huang, Y.; Zhang, H.; Fu, W.; Wang, M.; Shan, Z.; Xie, J.; Yang, Y.; et al. The Role of Short Chain Fatty Acids in Inflammation and Body Health. Int. J. Mol. Sci. 2024, 25, 7379. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 222–232. [Google Scholar] [CrossRef]
- Gandhi, C.R. Pro- and Anti-fibrogenic Functions of Gram-Negative Bacterial Lipopolysaccharide in the Liver. Front. Med. 2020, 7, 130. [Google Scholar] [CrossRef] [PubMed]
- Darfeuille-Michaud, A.; Boudeau, J.; Bulois, P.; Neut, C.; Glasser, A.L.; Barnich, N.; Bringer, M.A.; Swidsinski, A.; Beaugerie, L.; Colombel, J.F. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 2004, 127, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef]
- Schaubeck, M.; Clavel, T.; Calasan, J.; Lagkouvardos, I.; Haange, S.B.; Jehmlich, N.; Basic, M.; Dupont, A.; Hornef, M.; von Bergen, M.; et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut 2016, 65, 225–237. [Google Scholar] [CrossRef]
- Forbes, J.D.; Chen, C.Y.; Knox, N.C.; Marrie, R.A.; El-Gabalawy, H.; de Kievit, T.; Alfa, M.; Bernstein, C.N.; Van Domselaar, G. A comparative study of the gut microbiota in immune-mediated inflammatory diseases-does a common dysbiosis exist? Microbiome 2018, 6, 221. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Armour, C.R.; Nayfach, S.; Pollard, K.S.; Sharpton, T.J. A Metagenomic Meta-analysis Reveals Functional Signatures of Health and Disease in the Human Gut Microbiome. mSystems 2019, 4, e00332-18. [Google Scholar] [CrossRef]
- Alam, M.T.; Amos, G.C.A.; Murphy, A.R.J.; Murch, S.; Wellington, E.M.H.; Arasaradnam, R.P. Microbial imbalance in inflammatory bowel disease patients at different taxonomic levels. Gut Pathog. 2020, 12, 1. [Google Scholar] [CrossRef]
- Pisani, A.; Rausch, P.; Bang, C.; Ellul, S.; Tabone, T.; Marantidis Cordina, C.; Zahra, G.; Franke, A.; Ellul, P. Dysbiosis in the Gut Microbiota in Patients with Inflammatory Bowel Disease during Remission. Microbiol. Spectr. 2022, 10, e0061622. [Google Scholar] [CrossRef]
- Ma, J.; Wang, K.; Wang, J.; Zeng, Q.; Liu, K.; Zheng, S.; Chen, Y.; Yao, J. Microbial Disruptions in Inflammatory Bowel Disease: A Comparative Analysis. Int. J. Gen. Med. 2024, 17, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- Hertz, S.; Anderson, J.M.; Nielsen, H.L.; Schachtschneider, C.; McCauley, K.E.; Özçam, M.; Larsen, L.; Lynch, S.V.; Nielsen, H. Fecal microbiota is associated with extraintestinal manifestations in inflammatory bowel disease. Ann. Med. 2024, 56, 2338244. [Google Scholar] [CrossRef] [PubMed]
- De Caro, C.; Spagnuolo, R.; Quirino, A.; Mazza, E.; Carrabetta, F.; Maurotti, S.; Cosco, C.; Bennardo, F.; Roberti, R.; Russo, E.; et al. Gut Microbiota Profile Changes in Patients with Inflammatory Bowel Disease and Non-Alcoholic Fatty Liver Disease: A Metagenomic Study. Int. J. Mol. Sci. 2024, 25, 5453. [Google Scholar] [CrossRef] [PubMed]
- Scarlata, G.G.M.; Abenavoli, L. Should Routine Diagnostics Implement Gut Microbiota Analysis? Int. J. Gastroenterol. Hepatol. Dis. 2024, 3, e26662906338438. [Google Scholar] [CrossRef]
- Yan, D.; Ye, S.; He, Y.; Wang, S.; Xiao, Y.; Xiang, X.; Deng, M.; Luo, W.; Chen, X.; Wang, X. Fatty acids and lipid mediators in inflammatory bowel disease: From mechanism to treatment. Front. Immunol. 2023, 14, 1286667. [Google Scholar] [CrossRef]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef]
- Hodgkinson, K.; El Abbar, F.; Dobranowski, P.; Manoogian, J.; Butcher, J.; Figeys, D.; Mack, D.; Stintzi, A. Butyrate’s role in human health and the current progress towards its clinical application to treat gastrointestinal disease. Clin. Nutr. 2023, 42, 61–75. [Google Scholar] [CrossRef]
- Winter, S.E.; Winter, M.G.; Xavier, M.N.; Thiennimitr, P.; Poon, V.; Keestra, A.M.; Laughlin, R.C.; Gomez, G.; Wu, J.; Lawhon, S.D.; et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 2013, 339, 708–711. [Google Scholar] [CrossRef]
- Sinha, S.R.; Haileselassie, Y.; Nguyen, L.P.; Tropini, C.; Wang, M.; Becker, L.S.; Sim, D.; Jarr, K.; Spear, E.T.; Singh, G.; et al. Dysbiosis-Induced Secondary Bile Acid Deficiency Promotes Intestinal Inflammation. Cell Host Microbe 2020, 27, 659–670.e5. [Google Scholar] [CrossRef]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.B.; Yassour, M.; Sauk, J.; Garner, A.; Jiang, X.; Arthur, T.; Lagoudas, G.K.; Vatanen, T.; Fornelos, N.; Wilson, R.; et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 2017, 9, 103. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Artis, D.; Becker, C. The intestinal barrier: A pivotal role in health, inflammation, and cancer. Lancet Gastroenterol. Hepatol. 2025, 10, 573–592. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, Y.; Zhao, X.; Shang, C.; Xiang, M.; Li, L.; Cui, X. Microbiota-derived short-chain fatty acids: Implications for cardiovascular and metabolic disease. Front. Cardiovasc. Med. 2022, 9, 900381. [Google Scholar] [CrossRef]
- Yan, W.; Zhang, K.; Guo, J.; Xu, L. Bile acid-mediated gut-liver axis crosstalk: The role of nuclear receptor signaling in dynamic regulation of inflammatory networks. Front. Immunol. 2025, 16, 1595486. [Google Scholar] [CrossRef]
- He, Y.; Shaoyong, W.; Chen, Y.; Li, M.; Gan, Y.; Sun, L.; Liu, Y.; Wang, Y.; Jin, M. The functions of gut microbiota-mediated bile acid metabolism in intestinal immunity. J. Adv. Res. 2025; in press. [Google Scholar]
- Cebi, M.; Yilmaz, Y. Epithelial barrier hypothesis in the context of nutrition, microbial dysbiosis, and immune dysregulation in metabolic dysfunction-associated steatotic liver. Front. Immunol. 2025, 16, 1575770. [Google Scholar] [CrossRef]
- Wang, B.; Zhou, Z.; Li, L. Gut Microbiota Regulation of AHR Signaling in Liver Disease. Biomolecules 2022, 12, 1244. [Google Scholar] [CrossRef]
- Hou, J.J.; Ma, A.H.; Qin, Y.H. Activation of the aryl hydrocarbon receptor in inflammatory bowel disease: Insights from gut microbiota. Front. Cell Infect. Microbiol. 2023, 13, 1279172. [Google Scholar] [CrossRef]
- Abenavoli, L.; Giubilei, L.; Procopio, A.C.; Spagnuolo, R.; Luzza, F.; Boccuto, L.; Scarpellini, E. Gut Microbiota in Non-Alcoholic Fatty Liver Disease Patients with Inflammatory Bowel Diseases: A Complex Interplay. Nutrients 2022, 14, 5323. [Google Scholar] [CrossRef]
- Schnabl, B.; Damman, C.J.; Carr, R.M. Metabolic dysfunction-associated steatotic liver disease and the gut microbiome: Pathogenic insights and therapeutic innovations. J. Clin. Investig. 2025, 135, e186423. [Google Scholar] [CrossRef]
- Cui, C.; Gao, S.; Shi, J.; Wang, K. Gut-Liver Axis: The Role of Intestinal Microbiota and Their Metabolites in the Progression of Metabolic Dysfunction-Associated Steatotic Liver Disease. Gut Liver 2025, 19, 479–507. [Google Scholar]
- Yu, J.X.; Wu, J.; Chen, X.; Zang, S.G.; Li, X.B.; Wu, L.P.; Xuan, S.H. Gut microbiota in liver diseases: Initiation, development and therapy. Front. Med. 2025, 12, 1615839. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yang, Y.; Lin, S.; Geller, D.A.; Yan, Y. The microenvironment in the development of MASLD-MASH-HCC and associated therapeutic in MASH-HCC. Front. Immunol. 2025, 16, 1569915. [Google Scholar] [CrossRef] [PubMed]
- Strati, F.; Lattanzi, G.; Amoroso, C.; Facciotti, F. Microbiota-targeted therapies in inflammation resolution. Semin. Immunol. 2022, 59, 101599. [Google Scholar] [CrossRef] [PubMed]
- Zikou, E.; Koliaki, C.; Makrilakis, K. The Role of Fecal Microbiota Transplantation (FMT) in the Management of Metabolic Diseases in Humans: A Narrative Review. Biomedicines 2024, 12, 1871. [Google Scholar] [CrossRef]
- Abenavoli, L.; Spagnuolo, R.; Scarlata, G.G.M.; Gambardella, M.L.; Boccuto, L.; Méndez-Sánchez, N.; Luzza, F. Metabolic Dysfunction-Associated Steatotic Liver Disease in Patients with Inflammatory Bowel Diseases: A Pilot Study. Life 2024, 14, 1226. [Google Scholar] [CrossRef]
- Chen, A.T.; Wu, X.; Ye, G.; Li, W. Editorial: Machine learning and deep learning applications in pathogenic microbiome research. Front. Cell Infect. Microbiol. 2024, 14, 1429197. [Google Scholar]
- Popa, S.L.; Ismaiel, A.; Cristina, P.; Cristina, M.; Chiarioni, G.; David, L.; Dumitrascu, D.L. Non-Alcoholic Fatty Liver Disease: Implementing Complete Automated Diagnosis and Staging. A Systematic Review. Diagnostics 2021, 11, 1078. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Model | Study Groups | Main Findings | Reference |
|---|---|---|---|
| Human | MASH vs. obese vs. healthy controls | Distinct enterotypes based on disease status; MASH enriched in Proteobacteria, Enterobacteriaceae, E. coli; elevated blood ethanol only in MASH group, suggesting endogenous ethanol production as a pathogenic mechanism; overlap in microbial signatures supports a continuum from obesity to MASH. | [26] |
| Human | Obese vs. MASLD vs. MASH vs. healthy controls | Reduced microbial diversity in obesity and MASLD; MASH enriched in Lachnospiraceae, Ruminococcus, Dorea; decreased Oscillospira in steatosis; volatile organic compounds (2-butanone, 4-methyl-2-pentanone) differed between MASH and controls; microbiota/metabolite signatures distinguished healthy vs. diseased, but prediction model couldn’t distinguish MASLD from MASH. | [27] |
| Human | MASLD patients with/without MASH-fibrosis | Increased Proteobacteria, Enterobacteriaceae; decreased Ruminococcus obeum, Eubacterium rectale in MASH-fibrosis group; machine-learning prediction model achieved AUC = 0.92, outperforming standard clinical scores. | [29] |
| Human | Biopsy-proven MASH patients stratified by BMI | Fibrosis ≥ F2 linked to Lactobacillus increase; lean MASH: ↓ Faecalibacterium, ↓ Ruminococcus, ↓ Lactobacillus; obese MASH: ↑ Lactobacillus; overweight MASH: ↓ Bifidobacterium; lean MASH patients showed alpha diversity comparable to healthy controls, suggesting distinct microbiota-related mechanisms in lean vs. obese phenotypes. | [30] |
| Human | MASLD and MASH cohorts | Advanced fibrosis linked to ↓ SCFA-producing taxa (Oscillospiraceae, Lachnospiraceae, Ruminococcus) and ↑ pro-inflammatory genera (Veillonella, Streptococcus, Klebsiella); microbial taxa consistently associated with histological severity across cohorts. | [31] |
| Human | Obese MASLD and MASH groups | ↑ Roseibacillus, Peptostreptococcus, Bifidobacterium, Streptomyces in MASH; Proteobacteria linked to severe phenotype; obese MASH associated with stronger inflammatory/fibrotic profile compared to lean MASH. | [34] |
| Human | MASLD vs. MASH | Both MASLD and MASH: ↑ Bacteroidetes, ↑ Fusobacteria, ↓ Firmicutes; MASH: ↑ Megamonas, ↑ Fusobacterium; MASLD: ↓ Prevotella; MASH also showed altered glucose metabolism pathways and impaired flavonoid/flavonol biosynthesis, indicating functional as well as taxonomic dysbiosis. | [35] |
| Human | Pediatric MASLD vs. pediatric MASH | MASH: ↑ Alistipes, ↓ Peptostreptococcaceae; ↑ Bacteroides uniformis, ↑ Lachnospiraceae bacterium 7_1_58FAA, ↑ Eubacterium ventriosum, ↑ Roseburia; findings suggest broader microbial shifts may be crucial in progression and improvement of MASLD/MASH, supporting microbiota-based diagnostic profiling. | [36] |
| Model | Study Groups | Main Findings | Reference |
|---|---|---|---|
| Human | CD patients | Isolation of AIEC from ileal biopsies; AIEC adhere to and invade epithelial cells, survive in macrophages, induce TNF-α and chronic inflammation; AIEC strains implicated in initiating and perpetuating ileal inflammation. | [42] |
| Human | IBD patients vs. healthy controls | Reduced F. prausnitzii abundance in CD group, especially in post-operative recurrence; F. prausnitzii induces IL-10 with anti-inflammatory action; selective reduction suggests loss of key butyrate-producer contributes to recurrence risk. | [43] |
| Human | Pediatric treatment-naïve CD vs. controls | Ileal mucosa enriched in Enterobacteriaceae, Fusobacterium, Peptostreptococcus; depleted Bacteroides, Faecalibacterium in CD group; biopsy-associated microbiota distinguished CD more effectively than fecal samples. | [44] |
| Human | UC patients vs. healthy controls | ↓ Butyrate-producers (Roseburia hominis, F. prausnitzii); ↑ Mucin-degraders (Ruminococcus gnavus); microbial imbalance reflects impaired SCFA production and mucosal barrier degradation in UC. | [45] |
| Animal | Mice models of CD-like ileitis | Germ-free mice protected; antibiotics reduce ileitis; transfer of dysbiotic microbiota induces ileitis; E. coli LF82 on its own is insufficient; inflammation requires complex microbial communities; gut dysbiosis downregulates Paneth cell antimicrobial genes (lysozyme, cryptdin-2). | [46] |
| Human | IBD vs. other immune-mediated inflammatory disorders | ↓ Clostridia, ↓ Bacteroides; ↑ Enterobacteriaceae; core shifts shared with other immune disorders but also IBD-specific alterations, indicating both shared and disease-specific dysbiosis. | [47] |
| Human | IBD patients vs. controls | ↓ Firmicutes (F. prausnitzii, Roseburia, Eubacterium hallii); ↑ Proteobacteria (E. coli, Klebsiella pneumoniae); compositional changes correlated with disease activity, more pronounced during flares. | [48] |
| Human | IBD patients vs. controls | Altered composition across Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria; families enriched in IBD showed poor network connectivity, whereas depleted taxa were highly interconnected, supporting loss of ecosystem stability. | [50] |
| Human | IBD in clinical remission vs. healthy controls | ↓ Alpha diversity, distinct beta diversity; healthy: ↑ Akkermansia, Oscillibacter, Coprococcus; IBD: ↑ Flavonoid-degraders; Enterobacteriaceae formed highly interconnected modules sustaining subclinical inflammation even in remission. | [51] |
| Human | UC vs. CD | UC: ↑ Streptococcus, ↓ Alistipes; CD: ↑ Lachnoclostridium, Fusobacterium, Cloacibacillus, Erysipelatoclostridium; ↓ Faecalibacterium, Roseburia, Haemophilus; activity-dependent: further ↓ anti-inflammatory taxa (Roseburia, Coprococcus, Ruminiclostridium) in active disease vs. remission. | [52] |
| Human | IBD with extraintestinal manifestations | ↓ Agathobacter, ↓ Blautia; ↑ Eggerthella; specific dysbiosis pattern associated with extraintestinal manifestations. | [53] |
| Human | IBD vs. healthy controls | ↑ F/B ratio; F/B imbalance suggested as potential biomarker for IBD. | [54] |
| Disease Stage | Gut Microbiota Changes | Reference |
|---|---|---|
| MASLD | Increased abundance of Proteobacteria and Firmicutes; decreased Bacteroidetes; enrichment of Escherichia and Klebsiella spp.; reduced microbial diversity; decline of SCFA-producing genera (Roseburia, Faecalibacterium); functional signatures include impaired bile acid metabolism and enhanced LPS-mediated endotoxemia. | [71] |
| MASH | Increase of Proteobacteria; reduction of Bifidobacterium and F. prausnitzii; enrichment of pro-inflammatory taxa (Dorea, Ruminococcus, Megamonas); decreased abundance of Oscillospiraceae; functional alterations include reduced butyrate and propionate biosynthesis, impaired flavonoid metabolism, and increased ethanol production. | [72] |
| Liver fibrosis | Enrichment of Streptococcus and Veillonella spp.; reduction in butyrate-producing species (Roseburia, Eubacterium rectale); increased ethanol-producing bacteria (Klebsiella pneumoniae); reduced microbial diversity and expansion of lactic acid-producing bacteria. | [71] |
| Liver cirrhosis | Dominance of Enterobacteriaceae and Streptococcaceae; depletion of autochthonous taxa (Lachnospiraceae, Ruminococcaceae); increased oral-origin taxa (Veillonella, Streptococcus, Prevotella); reduced Akkermansia muciniphila; decreased SCFA production and increased endotoxin release. | [73] |
| HCC | Increased abundance of E. coli, Enterococcus faecalis, and Klebsiella spp.; lower levels of beneficial SCFA-producing bacteria (Faecalibacterium, Roseburia, Blautia); enrichment of pro-inflammatory and carcinogenesis-associated taxa (Bacteroides, Fusobacterium, Clostridium cluster XI); microbiota signatures linked to secondary bile acid overproduction, DNA-damaging metabolites, and impaired immune surveillance. | [74] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarlata, G.G.M.; Morano, D.; Ismaiel, A.; Spagnuolo, R.; Luzza, F.; Dumitrascu, D.L.; Abenavoli, L. Gut Microbiota Changes in Metabolic Dysfunction-Associated Steatohepatitis and Inflammatory Bowel Disease: Common Pathogenic Features. Curr. Issues Mol. Biol. 2025, 47, 847. https://doi.org/10.3390/cimb47100847
Scarlata GGM, Morano D, Ismaiel A, Spagnuolo R, Luzza F, Dumitrascu DL, Abenavoli L. Gut Microbiota Changes in Metabolic Dysfunction-Associated Steatohepatitis and Inflammatory Bowel Disease: Common Pathogenic Features. Current Issues in Molecular Biology. 2025; 47(10):847. https://doi.org/10.3390/cimb47100847
Chicago/Turabian StyleScarlata, Giuseppe Guido Maria, Domenico Morano, Abdulrahman Ismaiel, Rocco Spagnuolo, Francesco Luzza, Dan Lucian Dumitrascu, and Ludovico Abenavoli. 2025. "Gut Microbiota Changes in Metabolic Dysfunction-Associated Steatohepatitis and Inflammatory Bowel Disease: Common Pathogenic Features" Current Issues in Molecular Biology 47, no. 10: 847. https://doi.org/10.3390/cimb47100847
APA StyleScarlata, G. G. M., Morano, D., Ismaiel, A., Spagnuolo, R., Luzza, F., Dumitrascu, D. L., & Abenavoli, L. (2025). Gut Microbiota Changes in Metabolic Dysfunction-Associated Steatohepatitis and Inflammatory Bowel Disease: Common Pathogenic Features. Current Issues in Molecular Biology, 47(10), 847. https://doi.org/10.3390/cimb47100847