Abstract
Stalk lodging constitutes a primary constraint on achieving consistently high yields in maize. Genetic improvement of lodging resistance requires the identification of stable quantitative trait loci (QTL) to facilitate the application of genomics-assisted breeding for improving selection efficiency in breeding programs. In this study, we performed a meta-analysis to identify consensus loci and functionally characterized candidate genes associated with stalk lodging-related traits. Through meta-analysis integrating 889 reported lodging-related QTLs using the IBM2 2008 Neighbors high-density genetic map, we identified 67 meta-QTLs (MQTLs), of which 32 were determined as core MQTLs. Among them, 67% were validated by co-localized marker–trait associations from genome-wide association studies (GWAS). Comparative genomics further revealed 40 evolutionarily conserved orthologs via protein alignment with rice lodging genes, while screening of core MQTL regions detected 802 candidate genes with KEGG enrichment implicating galactose degradation II in cell wall reinforcement, supported by transcriptomic evidence of their roles in lignin biosynthesis pathways modulating mechanical strength. In conclusion, the MQTL identified and validated in our study have significant scope in marker-assisted selection (MAS) breeding and map-based cloning programs for improving maize stalk lodging.
1. Introduction
In recent years, escalating natural disasters, pest outbreaks, and altered planting regimes have exacerbated maize lodging risks globally. Global estimates indicate that stalk lodging causes annual yield losses of 5–20% [1], critically compromising production security. For sustainable maize production systems, developing lodging-resistant cultivars through breeding is increasingly recognized as a pivotal strategy to ensure high and stable yields [2,3]. Consequently, elucidating the genetic architecture underlying maize lodging resistance and mining functionally validated candidate genes are essential for precision breeding of lodging-resilient crops [4,5].
Maize lodging manifests as two distinct phenotypes: stalk lodging and root lodging [6], with the former typically causing more severe yield damage during equivalent growth stages [7]. As a polygenic quantitative trait, stalk lodging exhibits complex genetic architecture. QTL mapping enables the identification of genomic intervals controlling lodging resistance through molecular markers, facilitating marker-assisted selection (MAS) in breeding programs. Extensive QTL mapping studies have characterized lodging-related traits across multiple dimensions, including the following: (i) morphological parameters such as plant height, ear height, internode length, and stem diameter [8]; (ii) biochemical constituents encompassing cellulose, hemicellulose, lignin, and soluble sugar content [9,10]; (iii) biomechanical properties measured through rind penetrometer resistance and stalk bending strength [11]; and (iv) anatomical features including vascular bundle density and cross-sectional area [12]. However, substantial variations in genetic backgrounds, population structures, statistical methodologies, and testing environments, combined with limitations in marker density, frequently yield QTL confidence intervals exceeding 10 cM. This compromises the utility of QTL mapping for precision breeding of lodging-resistant maize.
Meta-analysis, employing mathematical models to integrate results from multiple studies, effectively narrows QTL confidence intervals (CI) while enhancing mapping precision and reliability [13]. This methodology has been successfully implemented in major crops including rice [14], maize [15,16], and soybean [17]. Current maize meta-analyses primarily address yield components [18], abiotic stress tolerance [19], and biotic resistance [20,21], with lodging-related traits remaining critically underrepresented. To address this gap, we conducted the first comprehensive meta-analysis of maize stalk lodging QTLs to establish a robust framework for candidate gene discovery and marker-assisted breeding.
In this study, we systematically curated documented QTLs associated with 12 key stalk lodging-related traits: plant height (PH), ear height (EH), rind penetrometer resistance (RPR), internode length (IL), stem diameter (SD), detergent fiber (DF), cellulose (Cel), hemicellulose (Hem), lignin (Lig), soluble sugar content (SSC), stalk bending strength (SBS), and vascular bundle characteristics (Vb). These QTLs were integrated via meta-analysis using a high-density consensus linkage map, generating a refined MQTL landscape for stalk lodging. Subsequent integration of MQTL coordinates with maize reference genome sequences (B73 RefGen_v4) and computational biology approaches enabled systematic candidate gene identification, collectively providing foundational resources for precision breeding initiatives.
2. Materials and Methods
2.1. Collection of Mapping and Quantitative Trait Locus (QTL) Data
QTL information for 12 stalk lodging-related traits including PH, EH, RPR, IL, SD, DF, Cel, Hem, Lig, SSC, SBS, and Vb was collected from 44 publications (2003–2024) [8,9,11,12,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61] retrieved through databases including China National Knowledge Infrastructure (CNKI, https://www.cnki.net/, accessed on 1 December 2024) and the National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/, accessed on 1 December 2024) (Table 1). The compiled data encompassed the following: mapping population size, population type, mapping function, chromosomal position, LOD score, R2 value, genetic map, molecular markers, and genetic distance. These 12 traits were categorized into the following: stalk morphology (SM), stalk strength (SS), stalk chemical composition (SC), stalk anatomical structure (SA). All QTL data were derived from experiments conducted under normal conditions, excluding studies involving stress treatments. QTL position (maximum likelihood position and its confidence interval) and contribution rate (proportion of phenotypic variance explained) are two key parameters of QTL [62]. Where positional data was missing, the midpoint between flanking markers was substituted. For unknown confidence intervals, estimates were calculated using the following formulae [63,64,65]:
where N represents population size and R2 denotes the QTL contribution rate. When LOD scores were unavailable, R2 was estimated using the following formula:
C.I. = 530/(N × R2) (backcross and F2 populations)
C.I. = 163/(N × R2) (recombinant inbred line populations)

Table 1.
Collection of maize stalk lodging-related QTL information.
2.2. QTL Projection and Meta-QTL Analysis
The original QTL information collected in this study was derived from different mapping populations using varied genetic maps and molecular marker types, resulting in limited common markers available for constructing a consensus map. To address this, we utilized the high-density IBM2 2008 Neighbors genetic map (https://www.maizegdb.org/data_center/map, accessed on 20 December 2024) as a reference map. This comprehensive map incorporates diverse marker types and provides unambiguous locus order information for first-generation molecular markers (e.g., RFLP, RAPD) and second-generation markers (e.g., SSR). Meta-QTL analysis was performed using Biomercator v4.2 software [66]. The QTLs and common marker’ position on the reference map are computed by application of the appropriate distance ratio (homothetic projection), which is used to establish a linear, proportionally scaled positional mapping relationship between the common markers shared by two genetic maps. The absolute position of the QTL on the reference map is calculated by computing the confidence interval genetic distance ratio between the reference map and the original map, as well as the relative positional proportion of the QTL relative to the start of the confidence interval on the original map. This allowed the positions of QTLs from different sources, along with the coordinates defining the bounds of their confidence intervals, to be projected onto the IBM2 2008 Neighbors reference map. SNP markers were projected onto the reference map using markers with adjacent physical positions on the maize genome [14]. QTLs whose projected positions fell outside the range of the reference map were excluded.
Meta-QTL analysis employed the Veyrieras two-step algorithm [67]. Using a clustering method based on the Gaussian mixture model, this approach determines how many original QTLs underline the observed MQTLs. The parameters of this model were estimated via the EM algorithm. The QTL model estimated the number of MQTLs per chromosome using the five optional criteria: AIC (Akaike information criterion), AICc (AIC correction), AIC3 (AIC 3 candidate model), BIC (Bayesian information criterion), and AWE (average weight of evidence); we chose the results based on the AIC, which demonstrated strong performance in simulations, with the aim of achieving the comprehensive identification of all potential loci based on predictive accuracy [68]. The 95% confidence interval was used to estimate the interval of the MQTLs. The figures in this study were plotted using R.
2.3. Verification of MQTL with GWAS
To validate the identified MQTLs, marker–trait association (MTA) information from nine independent maize GWAS on lodging resistance was collected [58,69,70,71,72,73,74,75,76]. The physical positions of MTAs from these studies were compared with those of the MQTLs. The MTAs obtained from GWAS near MQTL (500 kb upstream and downstream of the MQTL regions) were considered as validation of the MQTL [77].
2.4. Identifying and Functional Annotation of Candidate Genes
To comprehensively identify and characterize candidate gene functions within MQTL regions, we referenced methods from previous studies [77]: (i) Query genes within the MQTL regions in the maize genome database (http://www.maizegdb.org/, accessed on 6 March 2025). The protein sequences of these genes were downloaded from the NCBI website (http://www.ncbi.nlm.nih.gov/, accessed on 6 March 2025) and their physical positions relative to the B73 reference genome were determined. Cloned rice lodging resistance genes were searched for using the China National Rice Data Center (https://www.ricedata.cn/, accessed on 1 April 2025), China National Knowledge Infrastructure (CNKI), and the National Center for Biotechnology Information (NCBI). BLASTP alignment was performed to compare the homology between the protein sequences of the rice genes and those of the genes within the MQTL regions. The alignment criterion was E < 1 × 10−10 and identity > 60%. (ii) Candidate genes within the breeders’ MQTLs [78] (comprising ≥2 original QTLs, <1 Mb physical size, <4 cM genetic size) were explored. (iii) We identified candidate genes located within the GWAS-MTA-validated MQTL. For MQTLs harboring >3 MTAs and a physical confidence interval longer than 1 Mb, we used a 1 Mb genomic region (500 kb flanking each side of the peak); peak physical positions of the MQTLs were calculated by Saini et al. [79]. To investigate the functions of candidate genes, we performed GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) analyses on candidate genes obtained through three methods, while GO enrichment analysis was conducted using the AgriGO v2 platform (http://systemsbiology.cau.edu.cn/agriGOv2/manual.php, accessed on 3 May 2025) to explore the biological functions of candidate genes. KEGG pathway analysis was performed using the Plant Reactome database (https://plantreactome.gramene.org/index.php?lang=en, accessed on 3 May 2025).
2.5. Analysis of Expression Patterns of Candidate Genes
The expression data (transcript profiling) for candidate genes were retrieved from “qteller” (https://qteller.maizegdb.org/, accessed on 6 March 2025) of maizeGDB using the B73 RefGen_v4 assembly [80]. The whole transcriptomic data encompasses 55 tissues/stages across multiple timepoints and tissues [81], and were available for the following: embryo (16, 18, 20, 22, 24 DAP), endosperm (16, 18, 20, 22, 24 DAP), internode (0, 6, 12, 18, 24, 30 NOPOL), internode (0, 6, 12, 18, 24, 30 POL), whole seed (2, 4, 6, 8, 10, 12, 14, 18, 20, 22, 24 DAP), anthers (R1), brace root (V13), crown root (V7, V13), cob (R1, V18), Silks (R1), Primary root (Z1, Z2, Z3, Z4), Stem (V1, V3), Tassel (V13, V18), Internode elongation zone (V5, V9), Whole primary root (7 d), Whole root system (3, 7 d). The expression level of candidate genes was evaluated by Fragments Per Kilobase of transcript per million mapped reads (FPKM) value, and the genes with FPKM value > 2 in tissue expression were screened for expression analysis [82], and log2 (FPKM + 1) was used for heatmap plotting.
3. Results
3.1. Genomic Distribution of QTLs Associated with Stalk Lodging-Related Traits in Maize Genome
A comprehensive dataset of 889 stalk lodging-related QTLs was compiled from 44 independent studies (2003–2024) [8,9,11,12,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61] for meta-analysis (Tables S1 and S2; Figure 1). The collected QTLs were unevenly distributed across all 10 maize chromosomes, with plant height (PH) having the highest number (187), followed by rind penetrometer resistance (RPR) with 113 and lignin (Lig) with 110. Of the total QTLs, 835 (93.9%) were successfully projected onto the IBM2 2008 Neighbors reference map. The unmapped 54 QTLs (6.1%) were excluded due to their projected positions falling outside the reference map range. Original studies featured population sizes ranging from 118 to 1948 individuals, utilized predominantly SSR, RFLP, and SNP markers, and reported variable QTL densities (1–86 per study). The LOD scores of these QTLs ranged from 0.82 to 36.96, with distribution analysis revealing the following: 127 QTLs (14.3%) had scores < 3, 575 (64.7%) between 3–6, 130 (14.6%) between 6–9, 32 (3.6%) between 9–12, and 25 (2.8%) ≥ 12 (Figure 1a). Individual QTLs explained 1.3–43.0% of phenotypic variation (PVE/R2), with distribution analysis revealing the following: <5% (n = 150, 16.9%), 5–10% (n = 463, 52.1%), 10–15% (n = 173, 19.5%), 15–20% (n = 68, 7.6%), and ≥20% (n = 35, 3.9%) (Figure 1b). The predominance of minor-effect loci (68.95% with R2 < 10%) indicates polygenic control of stalk lodging traits. Twelve traits evaluated under standard conditions were functionally categorized into the following: SM (44.99%), SS (14.96%), SC (37.35%), and SA (2.7%) (Figure 1c).
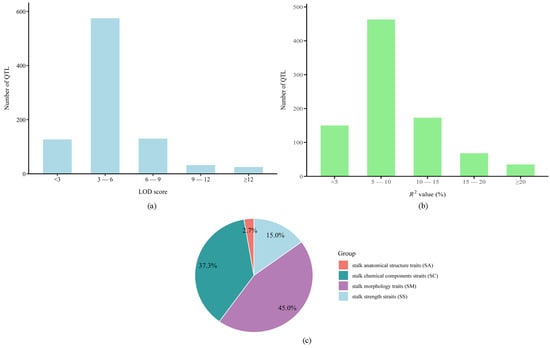
Figure 1.
(a) Distribution of logarithm of odds (LOD) score, (b) distribution of R2 value or phenotypic variance explained (PVE), (c) classification of the traits. SA, stalk anatomical structure; SC, stalk chemical composition; SS, stalk strength.
3.2. Meta-Analysis of QTL for Stalk Lodging-Related Traits in Maize
Meta-analysis of the 835 successfully mapped QTLs identified 67 MQTLs, each integrating 1–54 original QTLs (Table S3; Figure 2 and Figure S1). These 67 MQTLs were distributed across all 10 maize chromosomes, with chromosomal densities ranging from 3 MQTLs (chromosome 3) to 9 MQTLs (chromosomes 9 and 10).
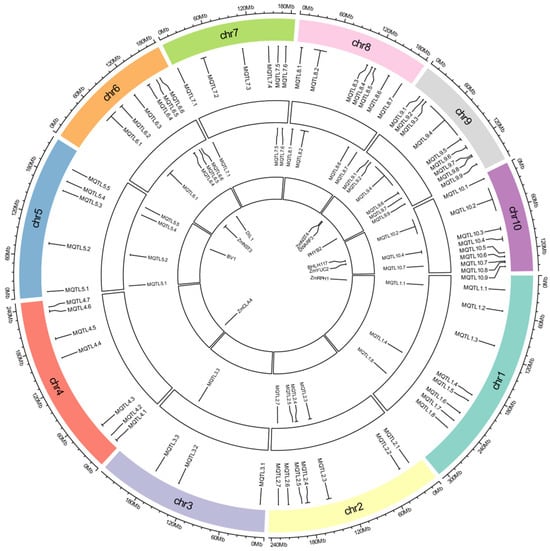
Figure 2.
Circular plot of the stalk lodging-related MQTL in maize. From the inside to the outside: The innermost circle represents the cloned maize lodging resistance genes, the middle circle represents the physical intervals of core MQTL, and the outermost circle represents chromosomes with MQTL intervals.
MQTL confidence intervals spanned 0.06–18.63 cM, with an average of 4.86 cM. MQTL intervals containing a greater number of initial QTLs and traits across diverse genetic backgrounds and environments, are considered more reliable. Among these, MQTL3.3 encompassed 54 initial QTLs and 10 traits, MQTL5.5 encompassed 38 initial QTLs and 7 traits, and MQTL1.1 encompassed 33 initial QTLs and 9 traits. MQTL10.7 and MQTL10.8 both encompassed one initial QTL and one trait. A total of 18 MQTLs had physical intervals less than 1 Mb. Among these, 9 MQTLs had genetic distances less than 4 cM: MQTL1.1, MQTL1.8, MQTL2.5, MQTL2.7, MQTL3.3, MQTL5.5, MQTL6.6, MQTL7.5 and MQTL8.7, respectively.
3.3. Validation of MQTL via GWAS Co-Localization
To validate the meta-QTLs, we cross-referenced their physical positions with marker–trait associations (MTAs) from nine maize lodging resistance GWAS [58,69,70,71,72,73,74,75,76]. A total of 45 MQTL (upstream and downstream 500 kb regions) were found to overlap with 217 MTAs, which were identified in eight GWAS on lodging resistance in maize (Tables S4 and S5). Among them, MQTL9.4 overlapped with 36 MTAs and MQTL6.1 with 17 MTAs, followed by MQTL10.2 overlapping with 12 MTAs, MQTL7.6 and MQTL8.2 overlapping with 11 MTAs, respectively. Furthermore, there were 19 MQTLs overlapping with 3–9 MTAs and 13 MQTL overlapping with 2 MTAs, while 8 MQTLs only overlapped with 1 MTA.
3.4. Functional Characterization of Candidate Genes for Stalk Lodging Within MQTL Regions
Multiple genes regulating maize stalk lodging-related traits were localized within MQTL regions (Figure 2; Table S3): ZmRPH1 (MQTL1.1) encoding a microtubule-associated protein regulating plant and ear height [83]; ZmCLA4 (MQTL4.2) acting as negative regulator of leaf angle through mRNA accumulation modulation, affecting stem gravitropism and cellular development [84]; Bv1 (MQTL5.3) controlling auxin transport-mediated height regulation [85]; ZmNST3 (MQTL6.1) as NAC transcription factor activating cell wall biosynthesis genes to enhance lodging resistance [86]; DIL1 (MQTL6.4) encoding AP2 transcription factor governing internode elongation [87]; ZmNST4 (MQTL9.2) essential for secondary cell wall biosynthesis [88]; DWARF3 (MQTL9.3) cytochrome P450 enzyme in gibberellin-mediated height control [89]; PHYB2 (MQTL9.8) modulating stem elongation [90]; BHLH117 (MQTL10.3) phytochrome-interacting factor (PIF) regulating plant stature [91]; and ZmYUC2 (MQTL10.4) mediating local auxin biosynthesis to modulate brace root angle and lodging resistance [92]. Additionally, cloned lodging resistance genes within ±1 Mb of MQTLs included BRD1 (MQTL1.8), ZmPHYC2 (MQTL5.1), GA20ox5 (MQTL8.7), ZmACS7 (MQTL10.8), ZRP4 (MQTL6.6), and RTH6 (MQTL1.3) [93,94,95,96,97,98].
To identify candidate genes regulating stalk lodging traits within MQTL regions, we implemented three complementary approaches. First, systematic screening of maize homologs for 280 rice lodging resistance genes identified 40 maize orthologs physically located within MQTL intervals through rigorous protein sequence alignment (Table S6). Most of these orthologous genes in maize exhibit similar functions affecting stalk lodging-related traits. For example, Zm00001d005775 (MQTL2.4) affects lodging-related traits similar to rice orthologous gene OsCesA4, with both being associated with plant height and stalk strength. Zm00001d045463 (MQTL9.2) and its rice orthologous gene OsSWN1 exhibit conserved functions, affecting internode length and lignin biosynthesis. These results indicate that the functions of these candidate genes are likely conserved in maize and rice. As a second approach, employing a breeder-oriented selection threshold (physical interval < 1 Mb, genetic distance < 4 cM, ≥2 initial QTLs) [99], we identified nine high-confidence MQTLs (MQTL1.1, 1.8, 2.5, 2.7, 3.3, 5.5, 6.6, 7.5, 8.7) yielding 149 candidate genes. For the third approach, prioritizing MQTLs co-localizing with >3 marker–trait associations (MTAs) revealed 24 MTA-MQTLs (MQTL1.4, 2.3, 2.4, 5.1, 5.2, 5.4, 6.1, 6.4, 6.5, 7.1, 7.5, 7.6, 8.1, 8.2, 8.6, 9.1, 9.2, 9.4, 9.6, 9.7, 9.9, 10.2, 10.4, 10.7), with the exclusion of overlapping MQTL7.5 and analysis of 19 large-interval (>1 Mb) within ±500 kb of peaks and four compact-interval (<1 Mb: MQTL5.1, 6.5, 7.1, 10.7) loci identifying 614 candidate genes; of the non-redundant integration of these nine breeder MQTLs and 24 MTA-MQTLs, 32 core MQTLs were identified (Figure 2). Core MQTLs are defined as meta-QTLs with physical intervals smaller than 1 Mb, containing at least two initial QTLs, and validated by independent GWAS signals, whose synthesis with maize–rice orthologs revealed 802 candidate genes regulating lodging resistance; we also established a comprehensive scoring system for these genes based on the four types: breeder’s MQTL localization (1 point), GWAS co-localization validation (1 point), rice ortholog with functional support (1 point), and expression in relevant tissues (1 point). Based on the accumulated evidence points, all 802 candidate genes were divided into three confidence tiers: high-confidence genes (3 points): 44 genes (5.49%); medium-confidence genes (2 points): 566 genes (70.57%); low-confidence genes (1 point): 192 genes (23.94%) (Table S7).
3.5. Functional Annotation of Candidate Genes
We next performed GO and KEGG pathway enrichment analysis of the identified 802 candidate genes to determine their functional classification. Among them, 601 genes had GO annotation (Figure 3). The most abundant GO terms related to biological processes were cellular processes (GO:000998, 600/601, 99.83%), metabolic processes (GO:0008152, 564/601, 93.84%) and cellular metabolic processes (GO:0044237, 524/601, 87.19%). GO terms related to molecular function—binding (GO:0005488, 412/601, 68.55%), catalytic activity (GO:0003824, 377/601, 62.73%) and heterocyclic compound binding (GO:1901363, 247/601, 41.10%)—were also highly enriched. Cell part (GO:0044464, 456/601, 75.87%), cell (GO:0005623, 456/601, 75.87%) and intracellular (GO:0005622, 412/601, 68.55%) were enriched in the cellular component’s annotation. The KEGG metabolic pathway is significantly enriched with metabolism and regulation, mainly involving carbohydrate metabolism: galactose degradation II, cellulose biosynthesis, UDP-L-arabinose biosynthesis and transport, and UDP-D-xylose biosynthesis; hormone signaling, transport, and metabolism: Strigolactone signaling; and secondary metabolism: galactosylcyclitol biosynthesis, Myo-inositol biosynthesis, and Polyisoprenoid biosynthesis (Figure 4).
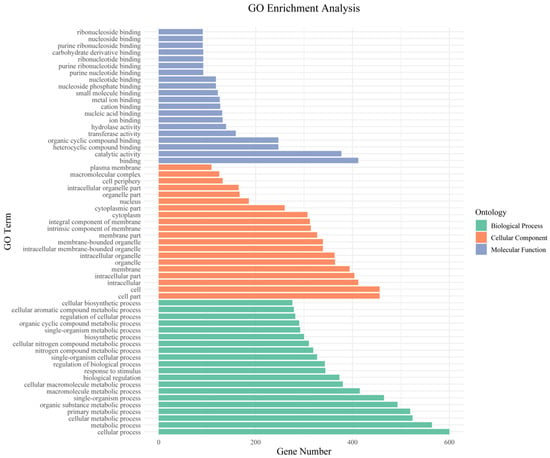
Figure 3.
Enrichment of gene ontology (GO) terms for stalk lodging-related candidate genes identified in MQTL regions.
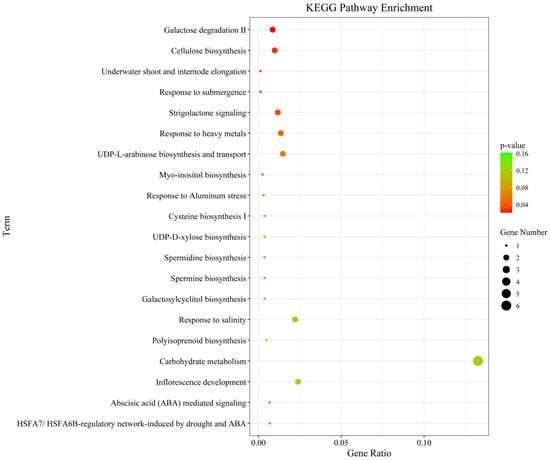
Figure 4.
Top 20 KEGG enrichment pathways for the candidate genes identified for the MQTL regions.
3.6. Expression Analysis of Identified Candidate Genes for Stalk Lodging in Maize
Expression patterns of candidate genes across developmental stages and tissues were analyzed using the qTeller database. Of 802 candidate genes, 601 exhibited detectable expression, with 406 showing high expression levels (Table S7). Here, we focused on 189 candidate genes (40 maize–rice orthologous genes and 149 candidate genes within the breeders’ MQTL regions), of which 114 candidate genes with highly specific expression in various tissues were visualized (Table S8; Figure 5). According to their different expression patterns, these 114 candidate genes were divided into three categories. In the first category, the expression levels were high in almost all tissues and stages. Among them, Zm00001d017526 had the highest expression in all tissues and is involved in multiple stress responses; Zm00001d038653, Zm00001d032346, Zm00001d021810, Zm00001d033186, Zm00001d021815, Zm00001d052494, and Zm00001d038658 were also highly expressed in all tissues. The second type was highly expressed only in some tissues: Zm00001d038648 and Zm00001d046112 are highly expressed in internode and root stages. The third type of gene was expressed in all tissues but the expression level was rather low.
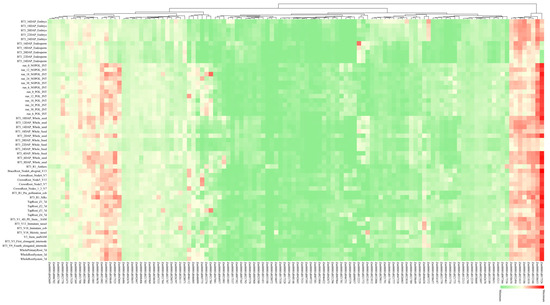
Figure 5.
Heatmap of co-expression patterns for high-confidence lodging resistance candidate genes (≥2 FPKM) across diverse stem tissues in maize. Red represents high expression while green represents low expression.
4. Discussion
In recent decades, extensive QTL mapping studies have been conducted. However, the use of diverse genetic materials, population types, and statistical methods make it difficult to comprehensively detect all QTLs controlling target traits in individual mapping experiments. Due to limitations in marker density, the identified QTLs often exhibit large genetic distances in their confidence intervals, resulting in lower effectiveness. Consequently, these QTLs struggle to be effectively applied in breeding practices. Meta-QTL analysis can overcome these limitations by integrating QTL results obtained across different environments and genetic backgrounds to identify highly reliable consensus genomic regions. Khahani et al. [14] collected 1052 QTLs for yield-related traits to perform meta-analysis in rice and obtained 114 MQTLs with an average of 4.85 cM CI in the resulting MQTLs. Chen et al. [17] conducted a meta-analysis for quality-related traits with 1034 initial QTL in soybean, and 212 MQTLs were found. Karnatam et al. [15] also performed meta-analysis for root-related traits in maize with 917 original QTL and identified 68 MQTLs.
In this study, we integrated 889 stalk lodging-related QTLs using the IBM2 2008 Neighbors reference genetic linkage map for meta-analysis, identifying 67 consensus MQTL. Comparative analysis revealed 26 MQTLs overlapping with previous studies (Table S9) [15,100], with most exhibiting significantly reduced confidence intervals. Notably, MQTL6.1 showed a highly consistent positional overlap across previous studies. Additionally, MQTL2.5, MQTL2.7, MQTL6.1, MQTL6.6, and MQTL8.6 had two overlapping intervals from previous studies. This indicates that multiple loci with distinct genetic effects collectively contribute to maize stalk lodging resistance. Furthermore, we identified nine breeder’s MQTLs with narrow physical intervals (<1 Mb) and 95% confidence intervals <4 cM. The genomic regions defined by these MQTLs represent high-confidence candidate regions for future gene cloning and serve as robust targets for marker-assisted selection in breeding for improved maize lodging resistance.
The integration of marker–trait associations (MTAs) from GWAS provides robust validation for MQTLs, reinforcing the stability and reliability of candidate genes within these genomic regions. Daryani et al. [101] found that a total of 52 MQTLs co-localized with 171 MTAs, while Li et al. [78] documented at least one MTA overlapping with 31 of 64 MQTLs. In our study, 45 MQTLs (within ±500 kb flanking regions) co-localized with 217 MTAs, validating 67.2% (45/67) of identified MQTLs through independent GWAS evidence. In particular, further characterization of these MQTLs will facilitate the identification of associated genes, playing a critical role in advancing our understanding of the molecular mechanisms underlying stalk lodging resistance in maize.
Through integration of three distinct methodologies, 802 candidate genes were identified within the MQTL intervals. GO enrichment analysis revealed statistically significant associations between enriched biological processes and maize lodging resistance. Notably, single-organism processes encompassing signal transduction and cell wall biosynthesis pathways were implicated [102]. Ions function as enzymatic cofactors in critical processes including cell wall lignification and signal transduction, directly influencing stalk structural integrity. The OsGLR3.4-mediated Ca2+ flux is essential for actin filament organization and vesicle trafficking; knockout of OsGLR3.4 in rice results in brassinosteroid (BR)-regulated growth defects, reduced BR sensitivity, and dwarf phenotypes due to suppressed internode elongation [103]. Integral membrane components synergistically regulate cell wall biosynthesis, thereby modulating lodging resistance. In rice, STRONG1 stabilizes cortical microtubules to guide cellulose synthase complex movement along microtubular trajectories, enabling transverse deposition of cellulose microfibrils that significantly enhance secondary wall thickness and mechanical resistance to lodging [104]. These findings collectively demonstrate that lodging resistance emerges through synergistic interactions of multiple mechanisms.
Further KEGG analysis indicated significant enrichment in metabolic and regulatory pathways. Research confirms that specific metabolic regulatory pathways substantially influence stalk strength. For instance, Strigolactones (SLs)—phytohormones involved in shoot branching suppression—interact with auxin signaling to regulate stem and root thickening [105]. Myo-inositol contributes directly to plant cell wall construction by providing precursors for pectin and hemicellulose synthesis, serving as a key determinant of stalk mechanical strength [106]. Cellulose biosynthesis, mediated by RTH6-encoded cellulose synthase, shapes maize root system architecture to enhance lodging resistance [98].
Galactose degradation II emerged as the most significantly enriched pathway (Figure 6), involving two key genes. Zm00001d032346 (located in MQTL1.6) encodes UDP-glucose 4-epimerase (UGE), which catalyzes the conversion of UDP-galactose to UDP-glucose in galactose degradation II. Previous analysis demonstrated that Zm00001d032346 (UGE4) represents a candidate gene potentially involved in ferulic acid metabolism, while diferulates (DFAs) promote cell wall cross-linking, reinforcing cell wall rigidity in maize [107,108]. As the ortholog of rice Osfc24, it encodes UGE catalyzing UDP-glucose (UDP-Glc) to UDP-galactose (UDP-Gal) conversion. UDP-Gal serves as substrate for carbohydrate, glycolipid, and glycoprotein biosynthesis. Osfc24 mutants exhibit brittle leaves and internodes with reduced sclerenchyma thickness, altered cell wall composition, and disrupted cellulose microfibril orientation, collectively compromising mechanical strength [109]. Zm00001d013245 (in MQTL5.1) encodes UDP-glucose 6-dehydrogenase (UGD), converting UDP-glucose to UDP-glucuronate. By analyzing the transcriptional regulatory network of GA-regulated lignin biosynthesis, Zm00001d013245 is co-expressed with multiple genes related to cell wall synthesis and was significantly correlated with multiple lignin synthesis metabolites in maize [110], mirroring its rice ortholog UGD4 (Os12g0443500) that enhances stress resistance through cell wall modification [111].
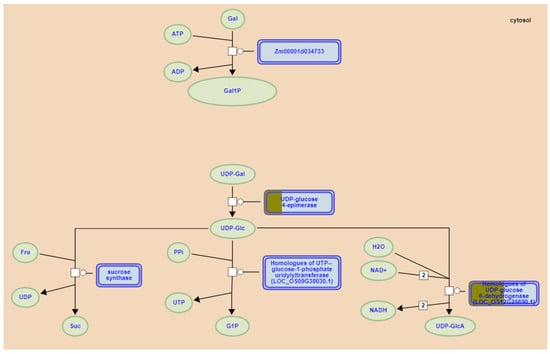
Figure 6.
Regulation of Galactose degradation Il by lodging resistance candidate genes in maize (https://plantreactome.gramene.org/PathwayBrowser/#/R-ZMA-1119452 accessed on 3 May 2025). The brown in the blue box represents candidate genes.
The second most enriched pathway was cellulose biosynthesis. Cellulose, a linear 1,4-linked homopolymer of β-D-glucopyranose, forms stable crystalline microfibrils through intermolecular interactions that provide structural support [112] and serves as the primary component conferring rigidity to plant cell walls [113]. Two key genes drive this pathway: Zm00001d023810 (located in MQTL10.2) encodes cellulose synthase-like D1, which participates in cell wall polysaccharide formation during cell division [114]. This gene is orthologous to rice DNL1 (encoding cellulose synthase-like D4) which regulates plant height [115]. Zm00001d005775 (CesA7) encodes a cellulose synthase A catalytic subunit controlling plant height [116] orthologous to rice OsCesA4 which participates in cellulose synthesis, forms part of the cellulose synthase complex for secondary cell wall formation, and critically influences stem strength [117].
The maize lodging resistance genes ZmRPH1, ZmCLA4, BV1, ZmNST3, DIL1, ZmNST4, DWARF3, PHYB2, BHLH117, and ZmYUC2 have been identified in the MQTL regions [83,84,85,86,87,88,89,90,91,92]. Notably, PHYB2 (located in MQTL9.8) encodes phytochrome B2, which regulates key aspects of seedling development such as mesocotyl elongation and chloroplast gene expression. This gene modulates multiple agronomic traits including plant height, ear height, stalk diameter, leaf sheath length, and internode length, while significantly influencing flowering time plasticity in maize. BHLH117 (MQTL10.3) encodes a phytochrome-interacting factor governing plant height determination and salt stress tolerance, thereby impacting yield stability. DIL1 (MQTL6.4) functions as an AP2 transcription factor that reduces plant height through internode length modulation. Additionally, it modifies leaf architecture to enhance high-density planting adaptability, contributing to yield improvement through optimized canopy structure.
This study leveraged the conserved genomic synteny between maize and rice (Poaceae family) to elucidate gene functions through ortholog analysis. Orthologous sequences revealed physiological function conservation, enhancing our understanding of maize gene networks. For instance, rice genes DNL1 [115], OsCesA4 [117], and OsSWN1 [118]—known regulators of lodging resistance—exhibit parallel functions in maize, demonstrating the utility of cross-species homology for candidate gene identification. We identified 40 maize–rice orthologous lodging resistance genes within MQTL regions, indicating evolutionarily conserved trait regulation across gramineous species.
Tissue-specific expression profiling revealed 406 candidate genes exhibiting high tissue specificity in internodes, roots, and seeds, with 114 visualized (Figure 5; Table S8). These genes showed pronounced expression in tissues functionally linked to stalk lodging resistance. For instance, Zm00001d017526 (plasma membrane intrinsic protein PIP1b) displays ubiquitous strong expression across maize tissues. This gene mediates drought and salt stress responses while regulating plant growth; notably, knockout of its rice ortholog OsPIP reduces plant height [119]. Additionally, Zm00001d046112 (spermine synthase) potentially regulates stalk sugar content, with its rice ortholog OsSPMS1 encoding spermine synthase that negatively regulates cell expansion and significantly impacts stalk development [120]. Collectively, integrating maize–rice ortholog functional data with tissue expression patterns identified multiple high-confidence lodging resistance candidate genes within MQTL regions.
Meta-QTL analysis allows QTL detected from independent experiments to be grouped into classes, and a consensus estimation of QTL position to be made [13]. When collecting relevant QTL information, it is necessary to ensure accuracy, authenticity, and QTL numbers, because (i) differences in mapping population and meta-QTL analysis method can reduce precision in identifying MQTLs position; (ii) the ordering of the markers on some genetic maps can differ locally from their order on physical maps, leading to map errors, and (iii) MQTL analysis is less reliable with fewer QTLs per chromosome [68]. Although candidate genes were identified and functionally analyzed in this study, several limitations persist. The use of a ±500 kb window for GWAS co-localization, though commonly adopted, may not account for local variation in linkage disequilibrium patterns. Furthermore, while conserved orthologs provide functional clues, direct evidence of gene functions in maize stem strength is still limited for many candidates. Future work should focus on integrating multi-omics data to build regulatory networks for screening high-confidence candidate genes, followed by functional validation using genome editing techniques.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cimb47100792/s1.
Author Contributions
Conceptualization, F.J. and X.L. (Xiaohui Li); data collection, H.F., C.Z., W.Q., X.Z., J.D. and X.L. (Xueyan Liu); meta-analysis, H.F., C.Z., F.J. and X.L. (Xiaohui Li); visualization, H.F. and C.Z.; writing and editing, H.F., C.Z., W.Q., F.J. and X.L. (Xiaohui Li); supervision, F.J. and X.L. (Xiaohui Li); project administration, F.J. All authors have read and agreed to the published version of the manuscript.
Funding
The research was supported by the Agricultural Science and Technology Innovation Program of Jilin Province (Grant No. CXGC2025SJ013).
Data Availability Statement
All relevant data are within the paper and its Supplementary Materials.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Zuber, M.S.; Kang, M.S. Corn lodging slowed by sturdier stalk. Crops Soils 1978, 3, 18–19. [Google Scholar]
- Ibitoye, D.O.; Akin-Idowu, P.E. Marker-assisted-selection (MAS): A fast track to increase genetic gain in horticultural crop breeding. Afr. J. Biotechnol. 2010, 9, 8889–8895. [Google Scholar]
- Stendal, C.; Casler, M.D.; Jung, G. Marker-assisted selection for neutral detergent fiber in smooth bromegrass. Crop Sci. 2006, 46, 303–311. [Google Scholar] [CrossRef]
- Xue, J.; Zhao, Y.; Gou, L.; Shi, Z.; Yao, M.; Zhang, W. How high plant density of maize affects basal internode development and strength formation. Crop Sci. 2016, 56, 3295–3306. [Google Scholar] [CrossRef]
- Xie, L.; Wen, D.; Wu, C.; Zhang, C. Transcriptome analysis reveals the mechanism of internode development affecting maize stalk strength. BMC Plant Biol. 2022, 22, 49. [Google Scholar] [CrossRef]
- Xue, J.; Ming, B.; Xie, R.; Wang, K.; Hou, P.; Li, S. Evaluation of maize lodging resistance based on the critical wind speed of stalk breaking during the late growth stage. Plant Methods 2020, 16, 148. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ma, W.; Peng, J.; Chen, Z. Study on yield loss of summer maize due to lodging at the big fare stage and grain filling stage. Sci. Agric. Sin. 2015, 48, 3952–3964. [Google Scholar]
- Zhu, L.; Chen, J.; Li, D.; Zhang, J.; Huang, Y.; Zhao, Y.; Song, Z.; Liu, Z. QTL mapping for stalk related traits in maize (Zea mays L.) under different densities. J. Integr. Agric. 2013, 12, 218–228. [Google Scholar] [CrossRef]
- Barrière, Y.; Méchin, V.; Lefevre, B.; Maltese, S. QTLs for agronomic and cell wall traits in a maize RIL progeny derived from a cross between an old minnesota13 line and a modern Iodent line. Theor. Appl. Genet. 2012, 125, 531–549. [Google Scholar] [CrossRef] [PubMed]
- Meents, M.J.; Watanabe, Y.; Samuels, A.L. The cell biology of secondary cell wall biosynthesis. Ann. Bot. 2018, 121, 1107–1125. [Google Scholar] [CrossRef]
- Zhang, Y.; Liang, T.; Chen, M.; Zhang, Y.; Wang, T.; Lin, H.; Rong, T.; Zou, C.; Liu, P.; Lee, M.; et al. Genetic dissection of stalk lodging related traits using an IBM Syn10 DH population in maize across three environments (Zea mays L.). Mol. Genet. Genomics 2019, 294, 1277–1288. [Google Scholar] [CrossRef]
- Huang, C.; Chen, Q.; Xu, G.; Xu, D.; Tian, J.; Tian, F. Identification and fine mapping of quantitative trait loci for the number of vascular bundle in maize stem. J. Integr. Plant Biol. 2015, 58, 81–90. [Google Scholar] [CrossRef]
- Goffinet, B.; Gerber, S. Quantitative trait loci: A meta-analysis. Genetics 2000, 155, 463–473. [Google Scholar] [CrossRef]
- Khahani, B.; Tavakol, E.; Shariati, V.; Fornara, F. Genome wide screening and comparative genome analysis for Meta-QTLs, ortho-MQTLs and candidate genes controlling yield and yield-related traits in rice. BMC Genom. 2020, 21, 294. [Google Scholar] [CrossRef] [PubMed]
- Karnatam, K.S.; Chhabra, G.; Saini, D.K.; Singh, R.; Kaur, G.; Praba, U.P.; Kumar, P.; Goyal, S.; Sharma, P.; Ranjan, R.; et al. Genome-wide meta-analysis of QTLs associated with root traits and implications for maize breeding. Int. J. Mol. Sci. 2023, 24, 6135. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zhang, J.; Qi, J.; Yue, R.; Han, X.; Yan, S.; Lu, C.; Fu, X.; Chen, N.; Ku, L.; et al. Analysis of meta-quantitative trait loci and their candidate genes related to leaf shape in maize. Chin. Bull. Bot. 2018, 53, 487–501. [Google Scholar]
- Chen, H.; Pan, X.; Wang, F.; Liu, C.; Wang, X.; Li, Y.; Zhang, Q. Novel QTL and meta-QTL mapping for major quality traits in soybean. Front. Plant Sci. 2021, 12, 774270. [Google Scholar] [CrossRef]
- Li, J.Z.; Zhang, Z.W.; Li, Y.L.; Wang, Q.L.; Zhou, Y.G. QTL consistency and meta-analysis for grain yield components in three generations in maize. Theor. Appl. Genet. 2010, 122, 771–782. [Google Scholar] [CrossRef]
- Sheoran, S.; Gupta, M.; Kumari, S.; Kumar, S.; Rakshit, S. Meta-QTL analysis and candidate genes identification for various abiotic stresses in maize (Zea mays L.) and their implications in breeding programs. Mol. Breed. 2022, 42, 26. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, H.J.; Zhang, C.X.; Wang, Q.Y.; Li, X.H. Meta-analysis of constitutive QTLs for disease resistance in maize and its synteny conservation in the rice genome. Genet. Mol. Res. 2015, 14, 961–970. [Google Scholar] [CrossRef]
- Badji, A.; Otim, M.; Machida, L.; Odong, T.; Kwemoi, D.B.; Okii, D.; Agbahoungba, S.; Mwila, N.; Kumi, F.; Ibanda, A.; et al. Maize combined insect resistance genomic regions and their co-localization with cell wall constituents revealed by tissue-specific QTL meta-analyses. Front. Plant Sci. 2018, 9, 895. [Google Scholar] [CrossRef]
- Flint-Garcia, S.A.; McMullen, M.D.; Darrah, L.L. Genetic relationship of stalk strength and ear height in maize. Crop Sci. 2003, 43, 23–31. [Google Scholar] [CrossRef][Green Version]
- Zheng, Z.P.; Liu, X.H. Genetic analysis of agronomic traits associated with plant architecture by QTL mapping in maize. Genet. Mol. Res. 2013, 12, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Ordás, B.; Malvar, R.; Santiago, R.; Butrón, A. QTL mapping for mediterranean corn borer resistance in European flint germplasm using recombinant inbred lines. BMC Genom. 2010, 11, 174. [Google Scholar] [CrossRef]
- Yang, C.; Liu, J.; Rong, T.Z. Detection of quantitative trait loci for ear row number in F2 populations of maize. Genet. Mol. Res. 2015, 14, 14229–14238. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Zheng, Y.; Zhang, Z.; Xu, S. Mapping of QTL associated with waterlogging tolerance during the seedling stage in maize. Ann. Bot. 2007, 99, 1067–1081. [Google Scholar] [CrossRef]
- Fei, J.; Lu, J.; Jiang, Q.; Liu, Z.; Yao, D.; Qu, J.; Liu, S.; Guan, S.; Ma, Y. Maize plant architecture trait QTL mapping and candidate gene identification based on multiple environments and double populations. BMC Plant Biol. 2022, 22, 110. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Su, G.; Zhang, J.; Wang, G. Genetic analysis and QTL mapping of maize yield and associate agronomic traits under semi-arid land condition. Afr. J. Biotechnol. 2008, 7, 1829–1838. [Google Scholar] [CrossRef]
- Jiménez-Galindo, J.C.; Ordás, B.; Butrón, A.; Samayoa, L.F.; Malvar, R.A. QTL mapping for yield and resistance against mediterranean corn borer in maize. Front. Plant Sci. 2017, 8, 698. [Google Scholar] [CrossRef]
- Osman, K.A.; Tang, B.; Wang, Y.; Chen, J.; Yu, F.; Li, L.; Han, X.; Zhang, Z.; Yan, J.; Zheng, Y.; et al. Dynamic QTL analysis and candidate gene mapping for waterlogging tolerance at maize seedling stage. PLoS ONE 2013, 8, e79305. [Google Scholar] [CrossRef]
- Li, K.; Yan, J.; Li, J.; Yang, X. Genetic architecture of rind penetrometer resistance in two maize recombinant inbred line populations. BMC Plant Biol. 2014, 14, 152. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhang, H.; Xin, W.; Ma, K.; Du, D.; Yu, C.; Liu, Y. Dissecting the genetic basis of flowering time and height related-traits using two doubled haploid populations in maize. Plants 2021, 10, 1585. [Google Scholar] [CrossRef]
- Ku, L.; Cao, L.; Wei, X.; Su, H.; Tian, Z.; Guo, S.; Zhang, L.; Ren, Z.; Wang, X.; Zhu, Y.; et al. Genetic dissection of internode length above the uppermost ear in four RIL populations of maize (Zea mays L.). G3 Genes Genomes Genet. 2015, 5, 281–289. [Google Scholar] [CrossRef]
- Luo, M.; Zhao, Y.; Zhang, R.; Xing, J.; Duan, M.; Li, J.; Wang, N.; Wang, W.; Zhang, S.; Chen, Z.; et al. Mapping of a major QTL for salt tolerance of mature field-grown maize plants based on SNP markers. BMC Plant Biol. 2017, 17, 140. [Google Scholar] [CrossRef]
- Lima, M.D.L.A.; de Souza, C.L., Jr.; Bento, D.A.V.; De Souza, A.P.; Carlini-Garcia, L.A. Mapping QTL for grain yield and plant traits in a tropical maize population. Mol. Breed. 2006, 17, 227–239. [Google Scholar] [CrossRef]
- Tang, J.; Ma, X.; Teng, W.; Yan, J.; Wu, W.; Dai, J.; Li, J. Detection of quantitative trait loci and heterotic loci for plant height using an immortalized F2 population in maize. Chin. Sci. Bull. 2007, 52, 477–483. [Google Scholar] [CrossRef]
- Wang, H.; He, Y.; Wang, S. QTL mapping of general combining abilities of four traits in maize using a high-density genetic map. J. Integr. Agric. 2017, 16, 1700–1707. [Google Scholar] [CrossRef]
- Zhang, W.; Li, Z.; Fang, H.; Zhang, M.; Duan, L. Analysis of the genetic basis of plant height-related traits in response to ethylene by QTL mapping in maize (Zea mays L.). PLoS ONE 2018, 13, e0193072. [Google Scholar] [CrossRef]
- Li, X.; Zhou, Z.; Ding, J.; Wu, Y.; Zhou, B.; Wang, R.; Ma, J.; Wang, S.; Zhang, X.; Xia, Z.; et al. Combined linkage and association mapping reveals QTL and candidate genes for plant and ear height in maize. Front. Plant Sci. 2016, 7, 833. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Cheng, S.; Wang, S.; Yu, T.; Wang, Y.; Xu, P.; Xu, X.; Zhou, Q.; Hou, X.; Zhang, G.; et al. Characterization and fine mapping of qRPR1-3 and qRPR3-1, two major QTLs for rind penetrometer resistance in maize. Front. Plant Sci. 2022, 13, 944539. [Google Scholar] [CrossRef]
- Yan, J.; Tang, H.; Huang, Y.; Shi, Y.; Li, J.; Zheng, Y. Dynamic analysis of QTL for plant height at different developmental stages in maize (Zea mays L.). Chin. Sci. Bull. 2003, 48, 2601–2607. [Google Scholar] [CrossRef]
- Meng, Y.; Li, J.; Liu, J.; Hu, H.; Li, W.; Liu, W.; Chen, S. Ploidy effect and genetic architecture exploration of stalk traits using DH and its corresponding haploid populations in maize. BMC Plant Biol. 2016, 16, 50. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lemmon, Z.H.; Doebley, J.F. Genetic dissection of a genomic region with pleiotropic effects on domestication traits in maize reveals multiple linked QTL. Genetics 2014, 198, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhang, Q.; Zhang, Y.; Li, X.; Huang, X. Dissection of the genetic architecture of plant height and ear height in maize (Zea mays L.). J. Fudan Univ. 2016, 55, 605–613. [Google Scholar]
- Cardinal, A.; Lee, M.; Moore, K. Genetic mapping and analysis of quantitative trait loci affecting fiber and lignin content in maize. Theor. Appl. Genet. 2003, 106, 866–874. [Google Scholar] [CrossRef]
- Barrière, Y.; Thomas, J.; Denoue, D. QTL mapping for lignin content, lignin monomeric composition, p-hydroxycinnamate content, and cell wall digestibility in the maize recombinant inbred line progeny F838 × F286. Plant Sci. 2008, 175, 585–595. [Google Scholar] [CrossRef]
- Penning, B.W.; Sykes, R.W.; Babcock, N.C.; Dugard, C.K.; Held, M.A.; Klimek, J.F.; Shreve, J.T.; Fowler, M.; Ziebell, A.; Davis, M.F.; et al. Genetic determinants for enzymatic digestion of lignocellulosic biomass are independent of those for lignin abundance in a maize recombinant inbred population. Plant Physiol. 2014, 165, 1475–1487. [Google Scholar] [CrossRef]
- Courtial, A.; Thomas, J.; Reymond, M.; Méchin, V.; Grima-Pettenati, J.; Barrière, Y. Targeted linkage map densification to improve cell wall related QTL detection and interpretation in maize. Theor. Appl. Genet. 2013, 126, 1151–1165. [Google Scholar] [CrossRef]
- Wang, H.W.; Han, J.; Sun, W.T.; Chen, S.J. Genetic analysis and QTL mapping of stalk digestibility and kernel composition in a high-oil maize mutant (Zea mays L.). Plant Breed. 2010, 129, 318–326. [Google Scholar] [CrossRef]
- Hu, H.; Liu, W.; Fu, Z.; Homann, L.; Technow, F.; Wang, H.; Song, C.; Li, S.; Melchinger, A.E.; Chen, S. QTL mapping of stalk bending strength in a recombinant inbred line maize population. Theor. Appl. Genet. 2013, 126, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Krakowsky, M.D.; Lee, M.; Coors, J.G. Quantitative trait loci for cell-wall components in recombinant inbred lines of maize (Zea mays L.) I: Stalk tissue. Theor. Appl. Genet. 2005, 111, 337–346. [Google Scholar] [CrossRef]
- Virlouvet, L.; Hage, F.E.; Griveau, Y.; Jacquemot, M.-P.; Gineau, E.; Baldy, A.; Legay, S.; Horlow, C.; Combes, V.; Bauland, C.; et al. Water deficit-responsive QTLs for cell wall degradability and composition in maize at silage stage. Front. Plant Sci. 2019, 10, 488. [Google Scholar] [CrossRef]
- Lorenzana, R.E.; Lewis, M.F.; Jung, H.J.G.; Bernardo, R. Quantitative trait loci and trait correlations for maize stover cell wall composition and glucose release for cellulosic ethanol. Crop Sci. 2010, 50, 541–555. [Google Scholar] [CrossRef]
- Wei, M.; Li, X.; Li, J.; Fu, J.; Wang, Y.; Li, Y. QTL detection for stover yield and quality traits using two connected populations in high-oil maize. Plant Physiol. Biochem. 2009, 47, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, H.; Hu, X.; Ma, F.; Wu, Y.; Wang, Q.; Liu, Z.; Huang, C. Genetic and quantitative trait locus analysis of cell wall components and forage digestibility in the Zheng58×HD568 maize RIL population at anthesis stage. Front. Plant Sci. 2017, 8, 1472. [Google Scholar] [CrossRef]
- He, K.; Chang, L.; Cui, T.; Qu, J.; Guo, D.; Xu, S.; Zhang, X.; Zhang, R.; Xue, J.; Liu, J. Mapping QTL plant height and ear height in maize under multi-environments. Sci. Agric. Sin. 2016, 49, 1443–1452. [Google Scholar]
- Yu, R.; Tian, X.; Liu, B.; Duan, Y.; Li, T.; Zhang, X.; Zhang, X.; Hao, Y.; Li, Q.; Xue, J.; et al. Dissecting the genetic architecture of lodging related traits by genome-wide association study and linkage analysis in maize. Acta Agron. Sin. 2022, 48, 138–150. [Google Scholar] [CrossRef]
- Zhao, B.; Li, K.; Wang, M.; Liu, Z.; Yin, P.; Wang, W.; Li, Z.; Li, X.; Zhang, L.; Han, Y.; et al. Genetic basis of maize stalk strength decoded via linkage and association mapping. Plant J. 2024, 117, 1558–1573. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Shao, C.; Liu, H.; He, Y.; Yin, Z. QTL mapping of maize’s stalk diameter and rind penetrometer resistance. Mol. Plant Breed. 2024, 1–11. [Google Scholar]
- Zhang, M.; Zhang, S.; Song, X.; Zhang, Z.; Lu, H.; Chen, G.; Hao, D.; Mao, Y.; Shi, M.; Xue, L.; et al. QTL analysis and genomic selection of rind penetrometer resistance in waxy maize. J. Jiangsu Agric. Sci. 2024, 40, 1191–1198. [Google Scholar]
- Du, Y.X. QTL Mapping of Vascular Bundle Related Traits in Maize Stem; Hebei Agriculture University: Baoding, China, 2018. [Google Scholar]
- Lu, X.; Li, X.; Xie, C.; Hao, Z.; Ji, H.; Shi, L.; Zhang, S. Comparative QTL mapping of resistance to sugarcane mosaic viruses maize based on bioinformatics. Hereditas 2008, 30, 101–108. [Google Scholar]
- Darvasi, A.; Soller, M. A simple method to calculate resolving power and confidence interval of QTL map location. Behav. Genet. 1997, 27, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Weller, J.; Soller, M. An analytical formula to estimate confidence interval of QTL location with a saturated genetic map as a function of experimental design. Theor. Appl. Genet. 2004, 109, 1224–1229. [Google Scholar] [CrossRef]
- Nagelkerke, N.J.D. A note on a general definition of the coefficient of determination. Biometrika 1991, 78, 691–692. [Google Scholar] [CrossRef]
- Arcade, A.; Labourdette, A.; Falque, M.; Mangin, B.; Chardon, F.; Charcosset, A.; Joets, J. BioMercator: Integrating genetic maps and QTL towards discovery of candidate genes. Bioinformatics 2004, 20, 2324–2326. [Google Scholar] [CrossRef]
- Veyrieras, J.B.; Goffinet, B.; Charcosset, A. MetaQTL: A package of new computational methods for the meta-analysis of QTL mapping experiments. BMC Bioinform. 2007, 8, 49. [Google Scholar] [CrossRef]
- Courtois, B.; Ahmadi, N.; Khowaja, F.; Price, A.H.; Rami, J.F.; Frouin, J.; Hamelin, C.; Ruiz, M. Rice root genetic architecture: Meta-analysis from a drought QTL database. Rice 2009, 2, 115–128. [Google Scholar] [CrossRef]
- Wang, H.; Li, K.; Hu, X.; Liu, Z.; Wu, Y.; Huang, C. Genome-wide association analysis of forage quality in maize mature stalk. BMC Plant Biol. 2016, 16, 227. [Google Scholar] [CrossRef]
- Peiffer, J.A.; Flint-Garcia, S.A.; Leon, N.D.; McMullen, M.D.; Kaeppler, S.M.; Buckler, E.S. The genetic architecture of maize stalk strength. PLoS ONE 2013, 8, e67066. [Google Scholar] [CrossRef]
- Li, K.; Wang, H.; Hu, X.; Liu, Z.; Wu, Y.; Huang, C. Genome-wide association study reveals the genetic basis of stalk cell wall components in maize. PLoS ONE 2016, 11, e0158906. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, C.; Mora, F.; Scapim, C.A.; Coan, M. Genome-wide haplotype-based association analysis of key traits of plant lodging and architecture of maize identifies major determinants for leaf angle: hapLA4. PLoS ONE 2019, 14, e0212925. [Google Scholar] [CrossRef]
- Mazaheri, M.; Heckwolf, M.; Vaillancourt, B.; Gage, J.L.; Burdo, B.; Heckwolf, S.; Barry, K.; Lipzen, A.; Ribeiro, C.B.; Kono, T.J.Y.; et al. Genome-wide association analysis of stalk biomass and anatomical traits in maize. BMC Plant Biol. 2019, 19, 45. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, P.; Zhang, X.; Zheng, Q.; Chen, M.; Ge, F.; Li, Z.; Sun, W.; Guan, Z.; Liang, T.; et al. Multi-locus genome-wide association study reveals the genetic architecture of stalk lodging resistance-related traits in maize. Front. Plant Sci. 2018, 9, 611. [Google Scholar] [CrossRef]
- Guan, H.H. Quantitative Trait Loci (QTL) Mapping for Correlation Characters of Stem Lodging-Resistance and Their Combining Ability in Maize; Hebei Agriculture University: Baoding, China, 2018. [Google Scholar]
- Xie, L.Y. Genome-Wide Association Studies and Important Gene Mining of Maize Stalk Strength Related Traits; Hebei Agriculture University: Baoding, China, 2021. [Google Scholar]
- Li, R.; Wang, Y.; Li, D.; Guo, Y.; Zhou, Z.; Zhang, M.; Zhang, Y.; Würschum, T.; Liu, W. Meta-quantitative trait loci analysis and candidate gene mining for drought tolerance-associated traits in maize (Zea mays L.). Mol. sci. 2024, 25, 4295. [Google Scholar] [CrossRef]
- Li, N.; Miao, Y.; Ma, J.; Zhang, P.; Chen, T.; Liu, Y.; Che, Z.; Shahinnia, F.; Yang, D. Consensus genomic regions for grain quality traits in wheat revealed by Meta-QTL analysis and in silico transcriptome integration. Plant Genome 2023, 16, e20336. [Google Scholar] [CrossRef]
- Saini, D.K.; Srivast, P.; Pal, N.; Gupta, P.K. Meta-QTLs, ortho-meta-QTLs and candidate genes for grain yield and associated traits in wheat (Triticum aestivum L.). Theor. Appl. Genet. 2022, 135, 1049–1081. [Google Scholar] [CrossRef] [PubMed]
- Woodhouse, M.R.; Sen, S.; Schott, D.; Portwood, J.L.; Freeling, M.; Walley, J.W.; Andorf, C.M.; Schnable, J.C.; Kelso, J. qTeller: A tool for comparative multi-genomic gene expression analysis. Bioinformatics 2021, 38, 236–242. [Google Scholar] [CrossRef]
- Stelpflug, S.C.; Sekhon, R.S.; Vaillancourt, B.; Hirsch, C.N.; Buell, C.R.; de Leon, N.; Kaeppler, S.M. An Expanded Maize Gene Expression Atlas based on RNA Sequencing and its Use to Explore Root Development. Plant Genome 2016, 9, 25. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and abundance estimation from RNA-Seq reveals thousands of new transcripts and switching among isoforms. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ge, F.; Qiang, Z.; Zhu, L.; Zhang, S.; Chen, L.; Wang, X.; Li, J.; Fu, Y. Maize ZmRPH1 encodes a microtubule-associated protein that controls plant and ear height. Plant Biotechnol. J. 2020, 18, 1345–1347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ku, L.; Han, Z.; Guo, S.; Liu, H.; Zhang, Z.; Cao, L.; Cui, X.; Chen, Y. The ZmCLA4 gene in the qLA4-1 QTL controls leaf angle in maize (Zea mays L.). J. Exp. Bot. 2014, 65, 5063–5076. [Google Scholar] [CrossRef]
- Avila, L.M.; Cerrudo, D.; Swanton, C.; Lukens, L. Brevis plant1, a putative inositol polyphosphate 5-phosphatase, is required for internode elongation in maize. J. Exp. Bot. 2016, 67, 1577–1588. [Google Scholar] [CrossRef]
- Ren, Z.; Zhang, D.; Cao, L.; Zhang, W.; Zheng, H.; Liu, Z.; Han, S.; Dong, Y.; Zhu, F.; Liu, H.; et al. Functions and regulatory framework of ZmNST3 in maize under lodging and drought stress. Plant Cell Environ. 2020, 43, 2272–2286. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Guo, M.; Yang, F.; Duncan, K.; Jackson, D.; Rafalski, A.; Wang, S.; Li, B. Mutations in an AP2 transcription factor-like gene affect internode length and leaf shape in maize. PLoS ONE 2012, 7, e37040. [Google Scholar] [CrossRef]
- Xiao, W.; Yang, Y.; Yu, J. ZmNST3 and ZmNST4 are master switches for secondary wall deposition in maize (Zea mays L.). Plant Sci. 2018, 266, 83–94. [Google Scholar] [CrossRef]
- Winkler, R.G.; Helentjaris, T. The maize Dwarf3 gene encodes a cytochrome P450- mediated early step in Gibberellin biosynthesis. Plant Cell 1995, 7, 1307–1317. [Google Scholar]
- Sheehan, M.J.; Kennedy, L.M.; Costich, D.E.; Brutnel, T.P. Subfunctionalization of PhyB1 and PhyB2 in the control of seedling and mature plant traits in maize. Plant J. 2007, 49, 338–353. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Zhao, Y.; Shen, R.; Wang, B.; Xie, Y.; Ma, X.; Zheng, Z.; Wang, H. Characterization of maize phytochrome-interacting factors in light signaling and photomorphogenesis. Plant Physiol. 2019, 181, 789–803. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, B.; Zhuo, C.; Xie, Y.; Zhang, X.; Liu, Y.; Zhang, G.; Ding, H.; Zhao, B.; Tian, M.; et al. Local auxin biosynthesis regulates brace root angle and lodging resistance in maize. New Phytol. 2023, 238, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Castorina, G.; Persico, M.; Zilio, M.; Sangiorgio, S.; Carabelli, L.; Consonni, G. The maize lilliputian1 (lil1) gene, encoding a brassinosteroid cytochrome P450 C-6 oxidase, is involved in plant growth and drought response. Ann. Bot. 2018, 122, 227–238. [Google Scholar] [CrossRef]
- Li, Q.; Wu, G.; Zhao, Y.; Wang, B.; Zhao, B.; Kong, D.; Wei, H.; Chen, C.; Wang, H. CRISPR/Cas9-mediated knockout and overexpression studies reveal a role of maize phytochrome C in regulating flowering time and plant height. Plant Biotechnol. J. 2020, 18, 2520–2532. [Google Scholar] [CrossRef]
- Paciorek, T.; Chiapelli, B.J.; Wang, J.Y.; Paciorek, M.; Yang, H.; Sant, A.; Val, D.L.; Boddu, J.; Liu, K.; Gu, C.Y.; et al. Targeted suppression of gibberellin biosynthetic genes ZmGA20ox3 and ZmGA20ox5 produces a short stature maize ideotype. Plant Biotechnol. J. 2022, 20, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, L.; Liu, M.; Dong, Z.; Li, Q.; Fei, S.; Xiang, H.; Liu, B.; Jin, W. Maize plant architecture Is regulated by the ethylene biosynthetic gene ZmACS7. Plant Physiol. 2020, 183, 1184–1199. [Google Scholar] [CrossRef] [PubMed]
- Held, B.M.; Wang, H.; John, I.; Wurtele, E.S.; Colbert, J.T. An mRNA putatively coding for an O-Methyltransferase accumulates preferentially in maize roots and is located predominantly in the region of the endodermis. Plant Physiol. 1993, 102, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Sommer, M.L.; Meyer, A.; Wang, D.; Klaus, A.; Stöcker, T.; Marcon, C.; Schoof, H.; Haberer, G.; Schön, C.-C.; et al. Cold mediates maize root hair developmental plasticity via epidermis-specific transcriptomic responses. Plant Physiol. 2024, 196, 2105–2120. [Google Scholar] [CrossRef]
- Guo, K.; Chen, T.; Zhang, P.; Liu, Y.; Che, Z.; Shahinnia, F.; Yang, D. Meta-QTL analysis and in-silico transcriptome assessment for controlling chlorophyll traits in common wheat. Plant Genome 2023, 16, e20294. [Google Scholar] [CrossRef]
- Wang, S.; Li, H.; Dong, Z.; Wang, C.; Wei, X.; Long, Y.; Wan, X. Genetic structure and molecular mechanism underlying the stalk lodging traits in maize (Zea may L.). Comput. Struct. Biotechnol. J. 2023, 21, 485–494. [Google Scholar]
- Daryani, P.; Darzi Ramandi, H.; Dezhsetan, S.; Mirdar Mansuri, R.; Hosseini Salekdeh, G.; Shobbar, Z.-S. Pinpointing genomic regions associated with root system architecture in rice through an integrative meta-analysis approach. Theor. Appl. Genet. 2022, 135, 81–106. [Google Scholar] [CrossRef]
- Bai, Y.; Zhao, X.; Yao, X.; Li, X.; Wu, K. Research progress on crop lodging resistance related traits and mechanism of signal transduction. Plant Sci. J. 2021, 39, 102–109. [Google Scholar]
- Yu, B.; Wu, Q.; Li, X.; Zeng, R.; Min, Q.; Huang, J. GLUTAMATERECEPTOR-like gene OsGLR3.4 is required for plant growth and systemic wound signaling in rice (Oryza sativa). New Phytol. 2022, 233, 1238–1256. [Google Scholar] [CrossRef]
- Zhao, Y.; Gao, J.; Wang, X.; Rashid, M.A.R.; Wu, Z.; Ma, Z.; Wu, H.; Xu, B.; Wu, Z.; Gu, Y.; et al. Elite haplotype of STRONG1 enhances rice yield by improving lodging resistance, panicle and plant architecture. Nat. Commun. 2025, 16, 5894. [Google Scholar] [CrossRef]
- Agustia, J.; Herolda, S.; Schwarza, M.; Sancheza, P.; Ljungb, K.; Dunc, E.A.; Brewerc, P.B.; Beveridgec, C.A.; Siebererd, T.; Sehra, E.M.; et al. Strigolactone signaling is required for auxin-dependent stimulation of secondary growth in plants. Proc. Natl. Acad. Sci. USA 2011, 108, 20242–20247. [Google Scholar] [CrossRef]
- Loewus, F.; Kelly, S.; Neufeld, E. Metabolism of myo inositol in plants: Conversion to pectin, hemicellulose, D-xylose, and sugar acids. Proc. Natl. Acad. Sci. USA 1962, 48, 421–425. [Google Scholar] [CrossRef]
- Barros-Rios, J.; Malvar, R.A.; Jung, H.J.G.; Bunzel, M.; Santiago, R. Divergent selection for ester-linked diferulates in maize pith stalk tissues. Effects on cell wall composition and degradability. Phytochemistry 2012, 83, 43–50. [Google Scholar] [CrossRef]
- López-Malvar, A.; Butrón, A.; Samayoa, L.F.; Figueroa-Garrido, D.J.; Malvar, R.A.; Santiago, R. Genome-wide association analysis for maize stem Cell Wall-bound Hydroxycinnamates. BMC Plant Biol. 2019, 19, 519. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Hu, H.; Wang, Y.; Hu, Z.; Ren, S.; Li, J.; He, B.; Wang, Y.; Xia, T.; Chen, P.; et al. A novel rice fragile culm 24 mutant encodes a UDP-glucose epimerase that affects cell wall properties and photosynthesis. J. Exp. Bot. 2020, 71, 2956–2969. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Feng, Y.; Wang, J.; Zhang, Y.; Yi, F.; Li, Z.; Zhang, M. Integrated metabolome and transcriptome analysis of gibberellins mediated the circadian rhythm of leaf elongation by regulating lignin synthesis in maize. Int. J. Mol. Sci. 2024, 25, 2705. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cheah, B.H.; Fang, Y.F.; Kuang, Y.-H.; Lin, S.-C.; Liao, C.-T.; Huang, S.-H.; Lin, Y.-F.; Chuang, W.-P. Transcriptomics identifies key defense mechanisms in rice resistant to both leaf feeding and phloem feeding herbivores. BMC Plant Biol. 2021, 21, 306. [Google Scholar] [CrossRef]
- Kim, S.H.; Lee, C.M.; Kafle, K. Characterization of crystalline cellulose in biomass: Basic principles, applications, and limitations of XRD, NMR, IR, Raman, and SFG. Korean J. Chem. Eng. 2013, 30, 2127–2141. [Google Scholar] [CrossRef]
- Brulé, V.; Rafsanjani, A.; Pasini, D.; Western, T.L. Hierarchies of plant stiffness. Plant Sci. 2016, 250, 79–96. [Google Scholar] [CrossRef]
- Yang, X.; Ren, J.; Lin, X.; Yang, Z.; Deng, X.; Ke, Q. Melatonin alleviates chromium toxicity in maize by modulation of cell wall polysaccharides biosynthesis, glutathione metabolism, and antioxidant capacity. Int. J. Mol. Sci. 2023, 24, 3816. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Lin, Z.; Li, Q.; Wu, H.; Xiang, C.; Wang, J. DNL1, encodes cellulose synthase-like D4, is a major QTL for plant height and leaf width in rice (Oryza sativa L.). Biochem. Biophy Res. Commun. 2015, 457, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Jiang, C.; Zhang, W.; Wang, H.; Li, K.; Liu, X.; Liu, Z.; Wu, Y.; Huang, C.; Hu, X. Morphological characterization and transcriptome analysis of new dwarf and narrow-Leaf (dnl2) mutant in maize. Int. J. Mol. Sci. 2022, 23, 795. [Google Scholar] [CrossRef]
- Tanaka, K.; Murata, K.; Yamazaki, M.; Onosato, K.; Miyao, A.; Hirochika, H. Three distinct rice cellulose synthase catalytic subunit genes required for cellulose synthesis in the secondary wall. Plant Physiol. 2003, 133, 73–83. [Google Scholar] [CrossRef]
- Chai, M.; Bellizzi, M.; Wan, C.; Cui, Z.; Li, Y.; Wang, G. The NAC transcription factor OsSWN1 regulates secondary cell wall development in Oryza sativa. J. Plant Biol. 2015, 58, 44–51. [Google Scholar] [CrossRef]
- Huang, X.; Wang, Z.; Huang, J.; Peng, S.; Xiong, D. Mesophyll conductance variability of rice aquaporin knockout lines at different growth stages and growing environments. Plant J. 2021, 107, 1503–1512. [Google Scholar] [CrossRef]
- Chen, J.; Cao, J.; Bian, Y.; Zhang, H.; Li, X.; Wu, Z.; Guo, G.; Lv, G. Identification of genetic variations and candidate genes responsible for stalk sugar content and agronomic traits in fresh corn via GWAS across multiple environments. Int. J. Mol. Sci. 2022, 23, 13490. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).