
Exploring the Functions of Mutant p53 through TP53 Knockout in HaCaT Keratinocytes

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Transcriptomic Analysis
2.3. Proteomic Analysis
2.4. Bioinformatic Analysis
2.5. RT-qPCR
2.6. Migration Assay
2.7. Confocal Fluorescent Microscopy
2.8. Proliferation Assay
2.9. Flow Cytometry
2.10. Statistical Analysis
3. Results
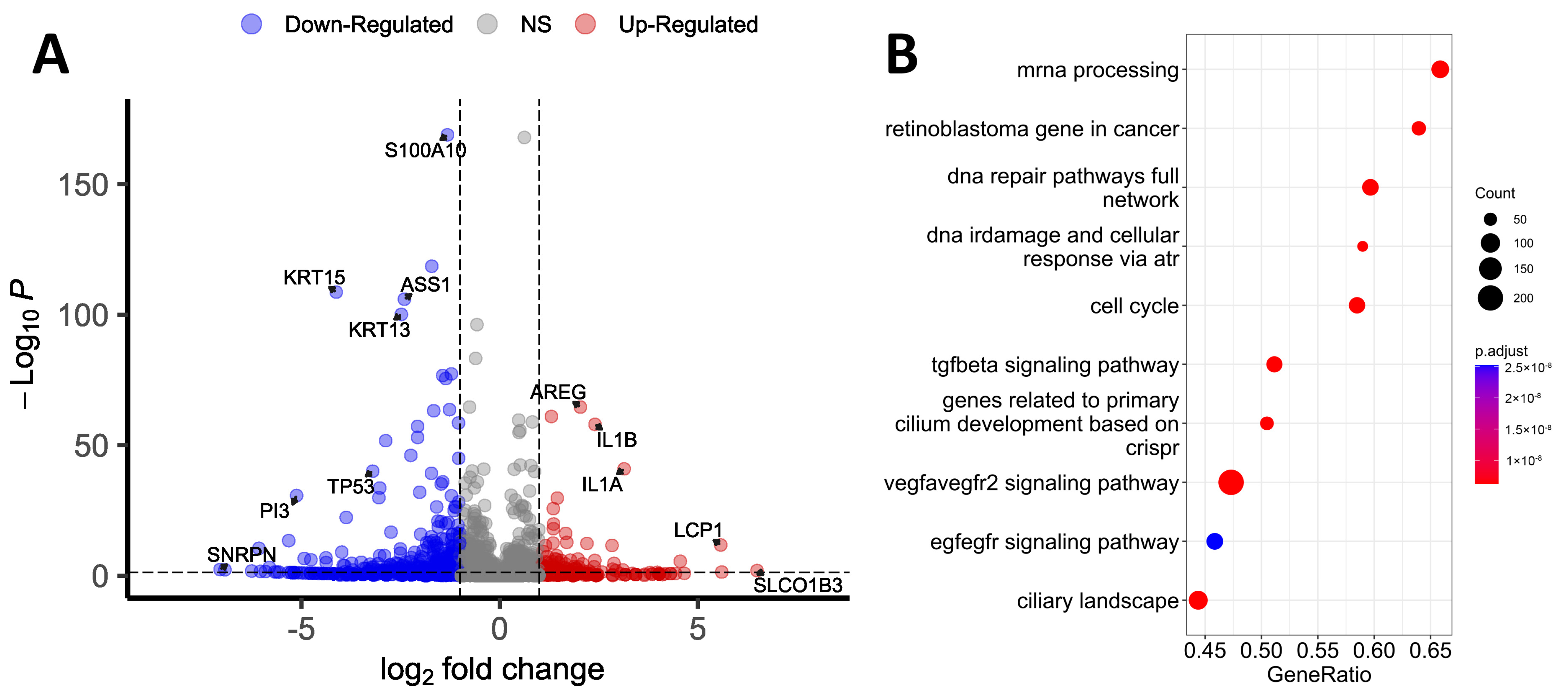
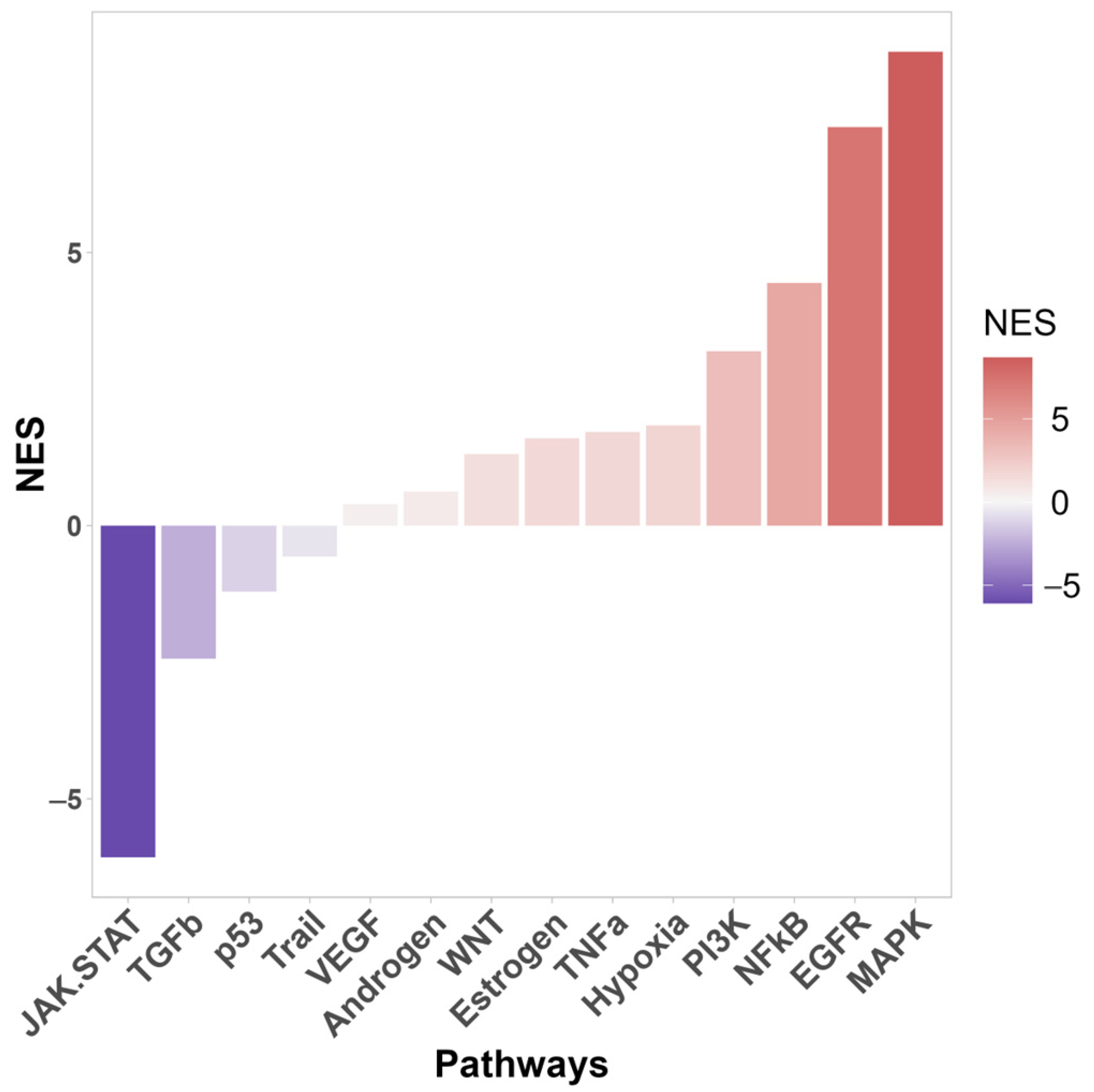
3.1. Transcriptomic Differences
3.2. Proteomic Differences
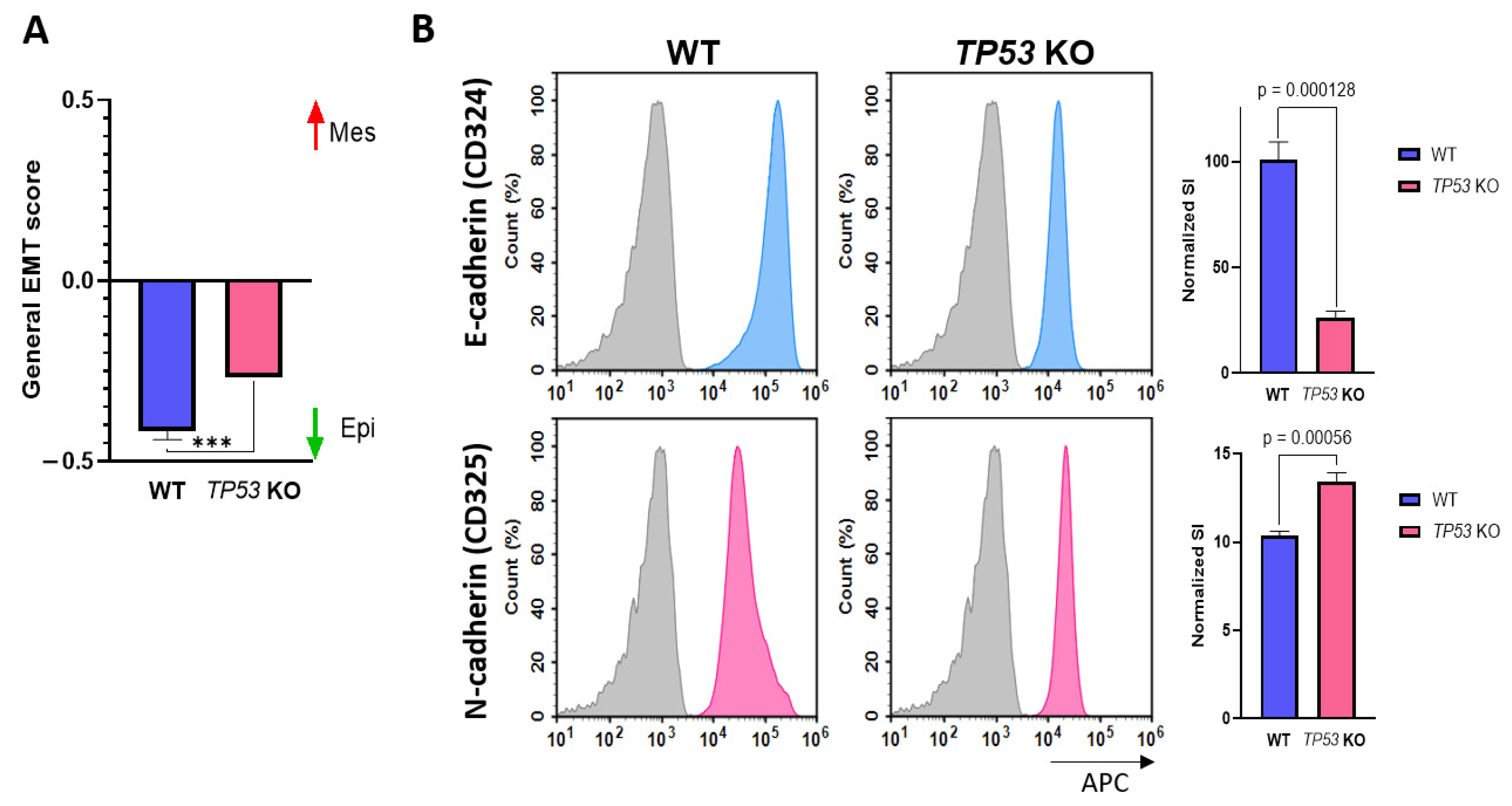
3.3. p53-Defficient HaCaT Cells Display the Features of EMT
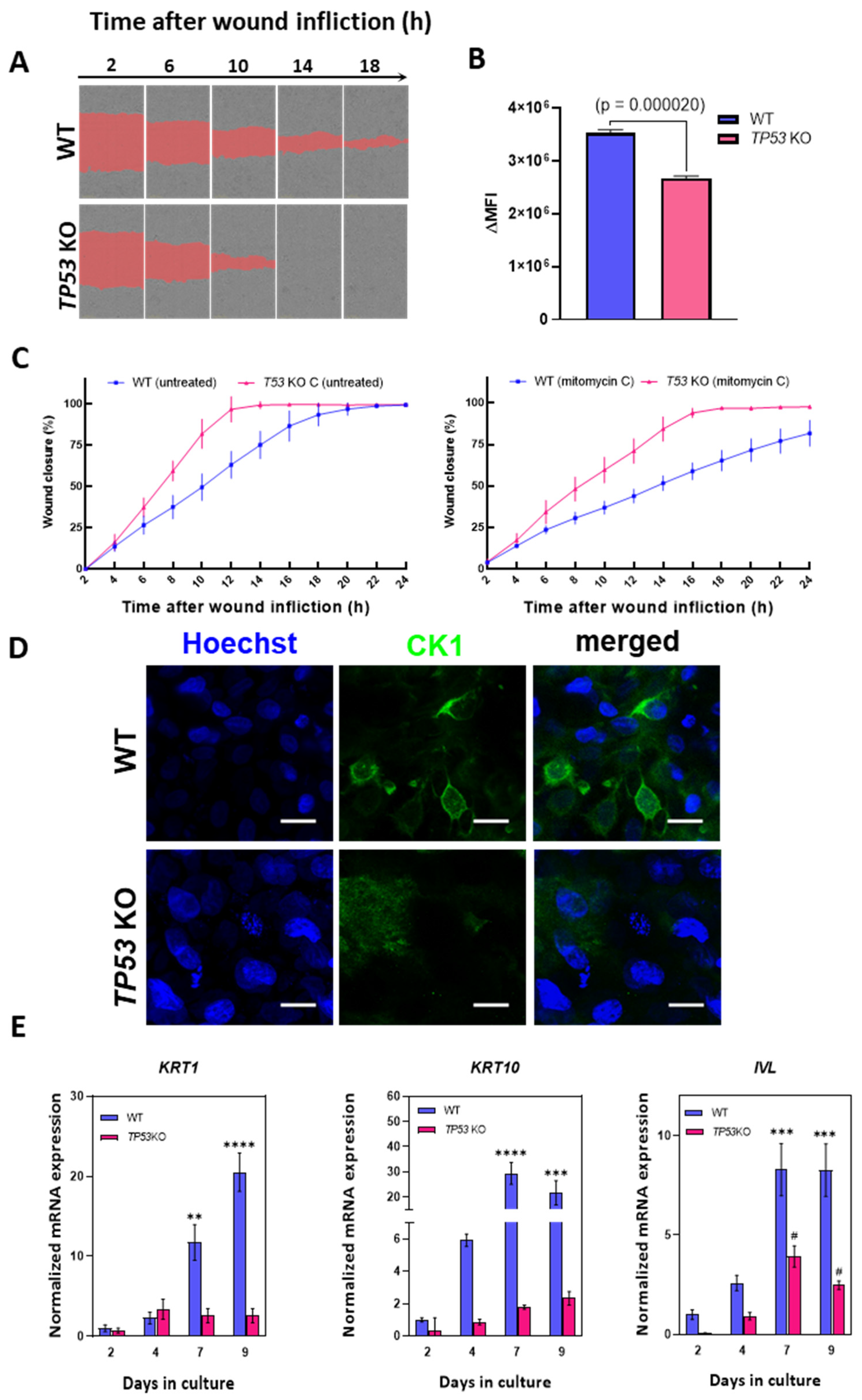
3.4. Knockout of TP53 Enhances Migration and Suppresses Epidermal Differentiation in HaCaT Cells
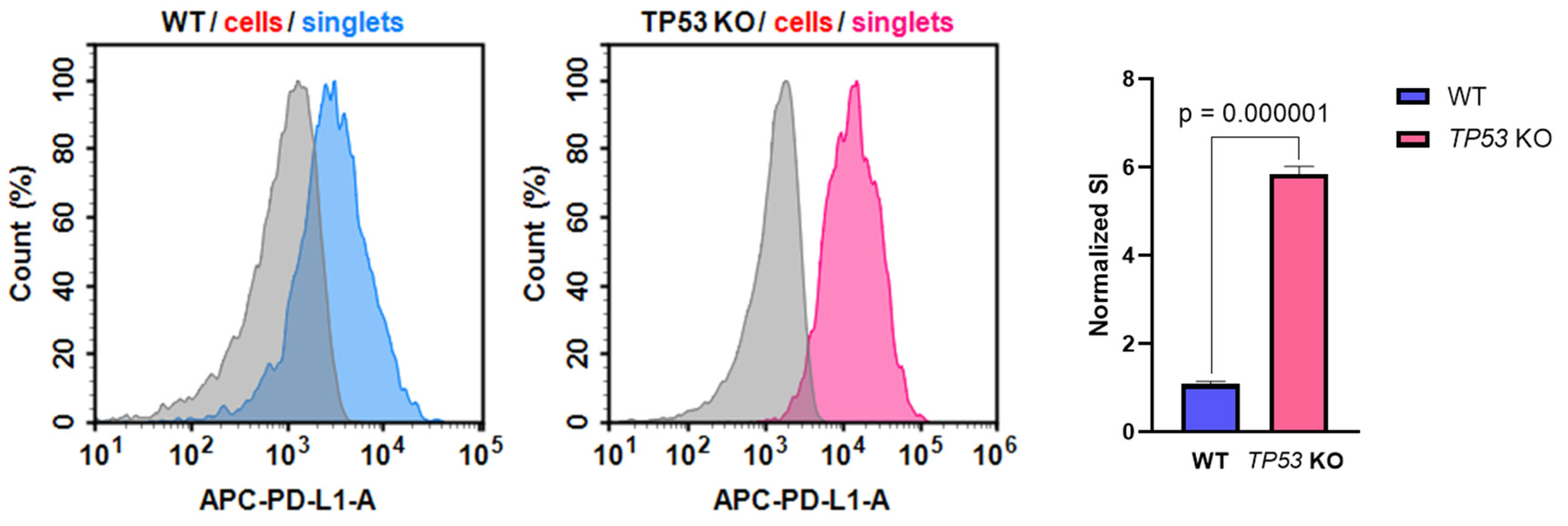
3.5. p53-Defficient HaCaT keratinocytes Possess Altered PD-L1 Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Nehal, K.S.; Bichakjian, C.K. Update on Keratinocyte Carcinomas. N. Engl. J. Med. 2018, 379, 363–374. [Google Scholar] [CrossRef]
- Piipponen, M.; Riihilä, P.; Nissinen, L.; Kähäri, V.-M. The Role of p53 in Progression of Cutaneous Squamous Cell Carcinoma. Cancers 2021, 13, 4507. [Google Scholar] [CrossRef] [PubMed]
- Giglia-Mari, G.; Sarasin, A. TP53 mutations in human skin cancers. Hum. Mutat. 2003, 21, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Colombo, I.; Sangiovanni, E.; Maggio, R.; Mattozzi, C.; Zava, S.; Corbett, Y.; Fumagalli, M.; Carlino, C.; Corsetto, P.A.; Scaccabarozzi, D.; et al. HaCaT Cells as a Reliable In Vitro Differentiation Model to Dissect the Inflammatory/Repair Response of Human Keratinocytes. Mediators Inflamm. 2017, 2017, 7435621. [Google Scholar] [CrossRef]
- Shkrigunov, T.; Kisrieva, Y.; Samenkova, N.; Larina, O.; Zgoda, V.; Rusanov, A.; Romashin, D.; Luzgina, N.; Karuzina, I.; Lisitsa, A.; et al. Comparative proteoinformatics revealed the essentials of SDS impact on HaCaT keratinocytes. Sci. Rep. 2022, 12, 21437. [Google Scholar] [CrossRef]
- Li, J.-Y.; Cui, D.-L.; Xie, Y.-M.; Su, J.-Z.; Zhang, M.-Y.; Niu, Y.-Y.; Xiang, P. Mechanisms of Cd-Induced Cytotoxicity in Normal Human Skin Keratinocytes: Implication for Human Health. Int. J. Mol. Sci. 2022, 23, 11767. [Google Scholar] [CrossRef]
- Lehman, T.A.; Modali, R.; Boukamp, P.; Stanek, J.; Bennett, W.P.; Welsh, J.A.; Metcalf, R.A.; Stampfer, M.R.; Fusenig, N.; Rogan, E.M. p53 mutations in human immortalized epithelial cell lines. Carcinogenesis 1993, 14, 833–839. [Google Scholar] [CrossRef]
- Martynova, E.; Pozzi, S.; Basile, V.; Dolfini, D.; Zambelli, F.; Imbriano, C.; Pavesi, G.; Mantovani, R. Gain-of-function p53 mutants have widespread genomic locations partially overlapping with p63. Oncotarget 2012, 3, 132–143. [Google Scholar] [CrossRef]
- Müller, I.; Beissert, S.; Kulms, D. Anti-Apoptotic NF-κB and “Gain of Function” mutp53 in Concert Act Pro-Apoptotic in Response to UVB+IL-1 via Enhanced TNF Production. J. Investig. Dermatol. 2015, 135, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Vikhanskaya, F.; Lee, M.K.; Mazzoletti, M.; Broggini, M.; Sabapathy, K. Cancer-derived p53 mutants suppress p53-target gene expression—Potential mechanism for gain of function of mutant p53. Nucleic Acids Res. 2007, 35, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- El-Mahdy, M.A.; Zhu, Q.; Wang, Q.-E.; Wani, G.; Patnaik, S.; Zhao, Q.; Arafa, E.-S.; Barakat, B.; Mir, S.N.; Wani, A.A. Naringenin Protects HaCaT Human Keratinocytes Against UVB-induced Apoptosis and Enhances the Removal of Cyclobutane Pyrimidine Dimers from the Genome. Photochem. Photobiol. 2008, 84, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Reagan-Shaw, S.; Breur, J.; Ahmad, N. Enhancement of UVB radiation-mediated apoptosis by sanguinarine in HaCaT human immortalized keratinocytes. Mol. Cancer Ther. 2006, 5, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Hengeltraub, S.F.; Gao, F.C.; Leivant, M.A.; Spandau, D.F. Aberrant NF-κB Activity in HaCaT Cells Alters their Response to UVB Signaling. J. Investig. Dermatol. 2006, 126, 1885–1892. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Bhardwaj, A.; Srivastava, S.K.; Arora, S.; Marimuthu, S.; Deshmukh, S.K.; Singh, A.P.; Carter, J.E.; Singh, S. Development and Characterization of a Novel in vitro Progression Model for UVB-Induced Skin Carcinogenesis. Sci. Rep. 2015, 5, 13894. [Google Scholar] [CrossRef] [PubMed]
- Rusanov, A.L.; Romashin, D.D.; Kozhin, P.M.; Karagyaur, M.N.; Loginov, D.S.; Tikhonova, O.V.; Zgoda, V.G.; Luzgina, N.G. Impact of p53 Knockout on Protein Data Set of HaCaT Cells in Confluent and Subconfluent Conditions. Data 2022, 7, 27. [Google Scholar] [CrossRef]
- Trivedi, U.H.; Cézard, T.; Bridgett, S.; Montazam, A.; Nichols, J.; Blaxter, M.; Gharbi, K. Quality control of next-generation sequencing data without a reference. Front. Genet. 2014, 5, 111. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Carlsson, H.; Petersson, S.; Enerbäck, C. Cluster analysis of S100 gene expression and genes correlating to psoriasin (S100A7) expression at different stages of breast cancer development. Int. J. Oncol. 2005, 27, 1473–1481. [Google Scholar] [PubMed]
- Poverennaya, E.V.; Pyatnitskiy, M.A.; Dolgalev, G.V.; Arzumanian, V.A.; Kiseleva, O.I.; Kurbatov, I.Y.; Kurbatov, L.K.; Vakhrushev, I.V.; Romashin, D.D.; Kim, Y.S.; et al. Exploiting Multi-Omics Profiling and Systems Biology to Investigate Functions of TOMM34. Biology 2023, 12, 198. [Google Scholar] [CrossRef]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; Miller, R.A.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. WikiPathways: Connecting communities. Nucleic Acids Res. 2020, 49, D613–D621. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Klinger, B.; Klünemann, M.; Sieber, A.; Uhlitz, F.; Sauer, S.; Garnett, M.J.; Blüthgen, N.; Saez-Rodriguez, J. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat. Commun. 2018, 9, 20. [Google Scholar] [CrossRef]
- Gene Ontology Consortium; Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L.; et al. The Gene Ontology knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar] [CrossRef]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, S.; Huang, R.Y.-J.; Thiery, J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods San Diego Calif. 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Blanchard, G.; Pich, C.; Hohl, D. HaCaT cells as a model system to study primary cilia in keratinocytes. Exp. Dermatol. 2022, 31, 1276–1280. [Google Scholar] [CrossRef]
- Mahagita, C.; Grassl, S.M.; Piyachaturawat, P.; Ballatori, N. Human organic anion transporter 1B1 and 1B3 function as bidirectional carriers and do not mediate GSH-bile acid cotransport. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G271–G278. [Google Scholar] [CrossRef]
- König, J.; Cui, Y.; Nies, A.T.; Keppler, D. Localization and genomic organization of a new hepatocellular organic anion transporting polypeptide. J. Biol. Chem. 2000, 275, 23161–23168. [Google Scholar] [CrossRef]
- Shapanis, A.; Lai, C.; Smith, S.; Coltart, G.; Sommerlad, M.; Schofield, J.; Parkinson, E.; Skipp, P.; Healy, E. Identification of proteins associated with development of metastasis from cutaneous squamous cell carcinomas (cSCCs) via proteomic analysis of primary cSCCs. Br. J. Dermatol. 2021, 184, 709–721. [Google Scholar] [CrossRef]
- Valle-Rios, R.; Maravillas-Montero, J.L.; Burkhardt, A.M.; Martinez, C.; Buhren, B.A.; Homey, B.; Gerber, P.A.; Robinson, O.; Hevezi, P.; Zlotnik, A. Isthmin 1 is a secreted protein expressed in skin, mucosal tissues, and NK, NKT, and th17 cells. J. Interferon Cytokine Res. 2014, 34, 795–801. [Google Scholar] [CrossRef]
- Dickinson, S.E.; Khawam, M.; Kirschnerova, V.; Vaishampayan, P.; Centuori, S.M.; Saboda, K.; Calvert, V.S.; Petricoin, E.F.; Curiel-Lewandrowski, C. Increased PD-L1 Expression in Human Skin Acutely and Chronically Exposed to UV Irradiation. Photochem. Photobiol. 2021, 97, 778–784. [Google Scholar] [CrossRef]
- Ock, C.-Y.; Kim, S.; Keam, B.; Kim, M.; Kim, T.M.; Kim, J.-H.; Jeon, Y.K.; Lee, J.-S.; Kwon, S.K.; Hah, J.H.; et al. PD-L1 expression is associated with epithelial-mesenchymal transition in head and neck squamous cell carcinoma. Oncotarget 2016, 7, 15901–15914. [Google Scholar] [CrossRef]
- Yu, Z.; Yu, Q.; Xu, H.; Dai, X.; Yu, Y.; Cui, L.; Chen, Y.; Gu, J.; Zhang, X.; Guo, C.; et al. IL-17A Promotes Psoriasis-Associated Keratinocyte Proliferation through ACT1-Dependent Activation of YAP-AREG Axis. J. Investig. Dermatol. 2022, 142, 2343–2352. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, L.; Zhang, H.; Lu, J.; Zhang, Z.; Wu, H.; Liang, Z. AREG mediates the epithelial-mesenchymal transition in pancreatic cancer cells via the EGFR/ERK/NF-κB signalling pathway. Oncol. Rep. 2020, 43, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef] [PubMed]
- Napoli, S.; Scuderi, C.; Gattuso, G.; Di Bella, V.; Candido, S.; Basile, M.S.; Libra, M.; Falzone, L. Functional Roles of Matrix Metalloproteinases and Their Inhibitors in Melanoma. Cells 2020, 9, 1151. [Google Scholar] [CrossRef] [PubMed]
- Sjoestroem, C.; Khosravi, S.; Cheng, Y.; Safaee Ardekani, G.; Martinka, M.; Li, G. DLC1 expression is reduced in human cutaneous melanoma and correlates with patient survival. Mod. Pathol. 2014, 27, 1203–1211. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, D.; Qian, X.; Rajaram, M.; Durkin, M.E.; Lowy, D.R. DLC1 is the principal biologically-relevant down-regulated DLC family member in several cancers. Oncotarget 2016, 7, 45144–45157. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Kaneko, T.; Li, J.S.; Liu, A.-D.; Voss, C.; Li, S.S.C. A phosphorylation switch controls the spatiotemporal activation of Rho GTPases in directional cell migration. Nat. Commun. 2015, 6, 7721. [Google Scholar] [CrossRef] [PubMed]
- Dierks, S.; von Hardenberg, S.; Schmidt, T.; Bremmer, F.; Burfeind, P.; Kaulfuß, S. Leupaxin stimulates adhesion and migration of prostate cancer cells through modulation of the phosphorylation status of the actin-binding protein caldesmon. Oncotarget 2015, 6, 13591–13606. [Google Scholar] [CrossRef][Green Version]
- Wang, Z.; He, T.; Lv, W.; Hu, J. Down-regulation of FBP1 in lung adenocarcinoma cells promotes proliferation and invasion through SLUG mediated epithelial mesenchymal transformation. Transl. Cancer Res. 2023, 12, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Li, Q.-G.; Xue, J.-J.; Wang, Z.; Yuan, X.; Tong, J.-D.; Xu, L.-C. Downregulation of FBP1 Promotes Tumor Metastasis and Indicates Poor Prognosis in Gastric Cancer via Regulating Epithelial-Mesenchymal Transition. PLoS ONE 2016, 11, e0167857. [Google Scholar] [CrossRef] [PubMed]
- Ondruššek, R.; Kvokačková, B.; Kryštofová, K.; Brychtová, S.; Souček, K.; Bouchal, J. Prognostic value and multifaceted roles of tetraspanin CD9 in cancer. Front. Oncol. 2023, 13, 1140738. [Google Scholar] [CrossRef] [PubMed]
- Bloor, B.K.; Seddon, S.V.; Morgan, P.R. Gene expression of differentiation-specific keratins in oral epithelial dysplasia and squamous cell carcinoma. Oral Oncol. 2001, 37, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Hatta, M.; Miyake, Y.; Uchida, K.; Yamazaki, J. Keratin 13 gene is epigenetically suppressed during transforming growth factor-β1-induced epithelial-mesenchymal transition in a human keratinocyte cell line. Biochem. Biophys. Res. Commun. 2018, 496, 381–386. [Google Scholar] [CrossRef]
- Collier, A.E.; Wek, R.C.; Spandau, D.F. Human keratinocyte differentiation requires translational control by the eIF2α kinase GCN2. J. Investig. Dermatol. 2017, 137, 1924–1934. [Google Scholar] [CrossRef]
- Sanz-Gómez, N.; Freije, A.; Gandarillas, A. Keratinocyte Differentiation by Flow Cytometry. Methods Mol. Biol. 2020, 2109, 83–92. [Google Scholar] [CrossRef]
- Teng, Y.; Fan, Y.; Ma, J.; Lu, W.; Liu, N.; Chen, Y.; Pan, W.; Tao, X. The PI3K/Akt Pathway: Emerging Roles in Skin Homeostasis and a Group of Non-Malignant Skin Disorders. Cells 2021, 10, 1219. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [PubMed]
- Whyte, J.L.; Smith, A.A.; Helms, J.A. Wnt Signaling and Injury Repair. Cold Spring Harb. Perspect. Biol. 2012, 4, a008078. [Google Scholar] [CrossRef] [PubMed]
- Fullard, N.; Moles, A.; O’Reilly, S.; van Laar, J.M.; Faini, D.; Diboll, J.; Reynolds, N.J.; Mann, D.A.; Reichelt, J.; Oakley, F. The c-Rel Subunit of NF-κB Regulates Epidermal Homeostasis and Promotes Skin Fibrosis in Mice. Am. J. Pathol. 2013, 182, 2109–2120. [Google Scholar] [CrossRef]
- Wullaert, A.; Bonnet, M.C.; Pasparakis, M. NF-κB in the regulation of epithelial homeostasis and inflammation. Cell Res. 2011, 21, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Antonangeli, F.; Natalini, A.; Garassino, M.C.; Sica, A.; Santoni, A.; Di Rosa, F. Regulation of PD-L1 Expression by NF-κB in Cancer. Front. Immunol. 2020, 11, 584626. [Google Scholar] [CrossRef] [PubMed]
- Betzler, A.C.; Theodoraki, M.-N.; Schuler, P.J.; Döscher, J.; Laban, S.; Hoffmann, T.K.; Brunner, C. NF-κB and Its Role in Checkpoint Control. Int. J. Mol. Sci. 2020, 21, 3949. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Takada, K.; Komine-Aizawa, S.; Hirohata, N.; Trinh, Q.D.; Nishina, A.; Kimura, H.; Hayakawa, S. Poly I:C induces collective migration of HaCaT keratinocytes via IL-8. BMC Immunol. 2017, 18, 19. [Google Scholar] [CrossRef]
- Su, L.; Fu, L.; Li, X.; Zhang, Y.; Li, Z.; Wu, X.; Li, Y.; Bai, X.; Hu, D. Loss of CAR promotes migration and proliferation of HaCaT cells, and accelerates wound healing in rats via Src-p38 MAPK pathway. Sci. Rep. 2016, 6, 19735. [Google Scholar] [CrossRef]
- Watt, F.M. Involucrin and other markers of keratinocyte terminal differentiation. J. Investig. Dermatol. 1983, 81, 100s–103s. [Google Scholar] [CrossRef]
- Wilson, V.G. Growth and Differentiation of HaCaT Keratinocytes. In Epidermal Cells: Methods and Protocols; Turksen, K., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2014; pp. 33–41. [Google Scholar] [CrossRef]
- Wang, S.; Drummond, M.L.; Guerrero-Juarez, C.F.; Tarapore, E.; MacLean, A.L.; Stabell, A.R.; Wu, S.C.; Gutierrez, G.; That, B.T.; Benavente, C.A.; et al. Single cell transcriptomics of human epidermis identifies basal stem cell transition states. Nat. Commun. 2020, 11, 4239. [Google Scholar] [CrossRef] [PubMed]
- Rosdy, M.; Clauss, L.C. Terminal epidermal differentiation of human keratinocytes grown in chemically defined medium on inert filter substrates at the air-liquid interface. J. Investig. Dermatol. 1990, 95, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Paramio, J.M.; Segrelles, C.; Laín, S.; Gómez-Casero, E.; Lane, D.P.; Lane, E.B.; Jorcano, J.L. p53 is phosphorylated at the carboxyl terminus and promotes the differentiation of human HaCaT keratinocytes. Mol. Carcinog. 2000, 29, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Freije, A.; Molinuevo, R.; Ceballos, L.; Cagigas, M.; Alonso-Lecue, P.; Rodriguez, R.; Menendez, P.; Aberdam, D.; De Diego, E.; Gandarillas, A. Inactivation of p53 in Human Keratinocytes Leads to Squamous Differentiation and Shedding via Replication Stress and Mitotic Slippage. Cell Rep. 2014, 9, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Saxena, K.; Jolly, M.K.; Balamurugan, K. Hypoxia, partial EMT and collective migration: Emerging culprits in metastasis. Transl. Oncol. 2020, 13, 100845. [Google Scholar] [CrossRef]
- Lu, P.; Lu, Y. Born to Run? Diverse Modes of Epithelial Migration. Front. Cell Dev. Biol. 2021, 9, 704939. Available online: https://www.frontiersin.org/articles/10.3389/fcell.2021.704939 (accessed on 20 December 2023). [CrossRef] [PubMed]
- Slater, N.A.; Googe, P.B. PD-L1 expression in cutaneous squamous cell carcinoma correlates with risk of metastasis. J. Cutan. Pathol. 2016, 43, 663–670. [Google Scholar] [CrossRef]
- Schaper, K.; Köther, B.; Hesse, K.; Satzger, I.; Gutzmer, R. The pattern and clinicopathological correlates of programmed death-ligand 1 expression in cutaneous squamous cell carcinoma. Br. J. Dermatol. 2017, 176, 1354–1356. [Google Scholar] [CrossRef]
- Sunshine, J.C.; Nguyen, P.L.; Kaunitz, G.J.; Cottrell, T.R.; Berry, S.; Esandrio, J.; Xu, H.; Ogurtsova, A.; Bleich, K.B.; Cornish, T.C.; et al. PD-L1 Expression in Melanoma: A Quantitative Immunohistochemical Antibody Comparison. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 4938–4944. [Google Scholar] [CrossRef]
- Roger, L.; Gadea, G.; Roux, P. Control of cell migration: A tumour suppressor function for p53? Biol. Cell 2006, 98, 141–152. [Google Scholar] [CrossRef]
- Gadea, G.; de Toledo, M.; Anguille, C.; Roux, P. Loss of p53 promotes RhoA-ROCK-dependent cell migration and invasion in 3D matrices. J. Cell Biol. 2007, 178, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Izumi, H.; Onitsuka, T.; Miyamoto, N.; Kashiwagi, E.; Kidani, A.; Hirano, G.; Takahashi, M.; Naito, S.; Kohno, K. Twist and p53 reciprocally regulate target genes via direct interaction. Oncogene 2008, 27, 5543–5553. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-J.; Chao, C.-H.; Xia, W.; Yang, J.-Y.; Xiong, Y.; Li, C.-W.; Yu, W.-H.; Rehman, S.K.; Hsu, J.L.; Lee, H.-H.; et al. p53 regulates epithelial-mesenchymal transition (EMT) and stem cell properties through modulating miRNAs. Nat. Cell Biol. 2011, 13, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Sasaki, Y.; Kobashi, K.; Takeda, K.; Nakagaki, T.; Idogawa, M.; Tokino, T. CRKL oncogene is downregulated by p53 through miR-200s. Cancer Sci. 2015, 106, 1033–1040. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| GAPDH | 5′- TCGACAGTCAGCCGCATCTTCTTT -3′ | 5′- ACCAAATCCGTTGACTCCGACCTT -3′ |
| ACTB | 5′- TCAGAAGGATTCCTATGTGGGCGA -3′ | 5′- CACGCAGCTCATTGTAGAAGGTGT -3′ |
| KRT10 | 5′- AGCATGGCAACTCACATCA -3′ | 5′- GTCGATCTGAAGCAGGATGTT -3′ |
| KRT1 | 5′- GCGGACAAATGCAGAGAATG -3′ | 5′- TGCTTGGTAGAGTGCTGTAAG -3′ |
| IVL | 5′- 5′-CCAAAGCCTCTGCCTCAG -3′ | 5′- GTATTGACTGGAGGAGGAACAG -3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romashin, D.; Rusanov, A.; Arzumanian, V.; Varshaver, A.; Poverennaya, E.; Vakhrushev, I.; Netrusov, A.; Luzgina, N. Exploring the Functions of Mutant p53 through TP53 Knockout in HaCaT Keratinocytes. Curr. Issues Mol. Biol. 2024, 46, 1451-1466. https://doi.org/10.3390/cimb46020094
Romashin D, Rusanov A, Arzumanian V, Varshaver A, Poverennaya E, Vakhrushev I, Netrusov A, Luzgina N. Exploring the Functions of Mutant p53 through TP53 Knockout in HaCaT Keratinocytes. Current Issues in Molecular Biology. 2024; 46(2):1451-1466. https://doi.org/10.3390/cimb46020094
Chicago/Turabian StyleRomashin, Daniil, Alexander Rusanov, Viktoriia Arzumanian, Alexandra Varshaver, Ekaterina Poverennaya, Igor Vakhrushev, Alexander Netrusov, and Nataliya Luzgina. 2024. "Exploring the Functions of Mutant p53 through TP53 Knockout in HaCaT Keratinocytes" Current Issues in Molecular Biology 46, no. 2: 1451-1466. https://doi.org/10.3390/cimb46020094
APA StyleRomashin, D., Rusanov, A., Arzumanian, V., Varshaver, A., Poverennaya, E., Vakhrushev, I., Netrusov, A., & Luzgina, N. (2024). Exploring the Functions of Mutant p53 through TP53 Knockout in HaCaT Keratinocytes. Current Issues in Molecular Biology, 46(2), 1451-1466. https://doi.org/10.3390/cimb46020094