
Comparative Analysis of Digital Transcriptomics Between Pre- and Post-Treatment Samples of Patients with Locally Advanced Cervical Cancer: A Preliminary Study

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. Macrodissection and RNA Isolation
2.3. Digital Gene Expression Analysis
2.4. Statistical Analysis
3. Results
3.1. Patient Characteristics
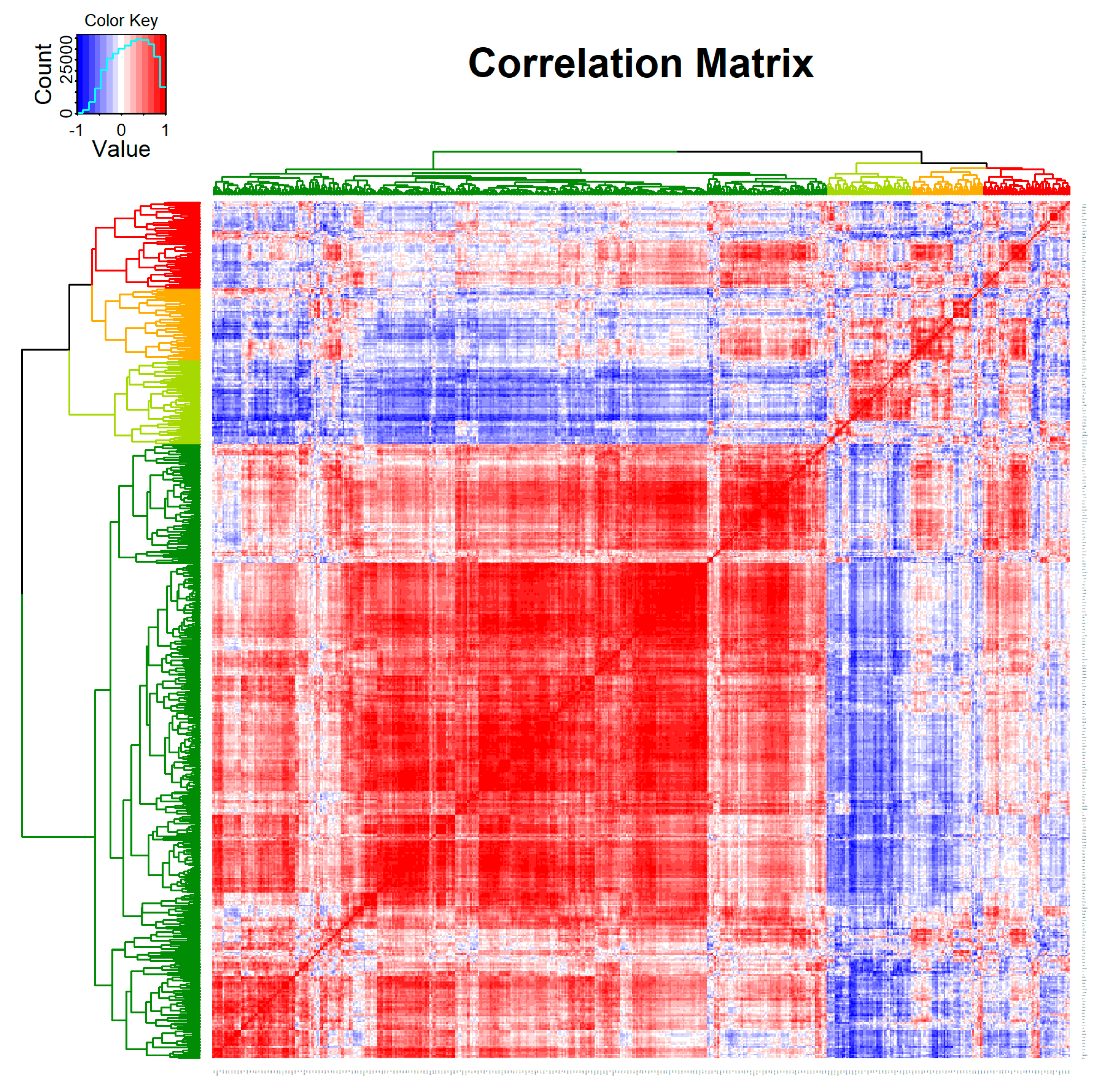
3.2. Transcriptomic Changes in Pre- and Post-Treatment Cervical Carcinoma Samples
3.3. Pathway Enrichment Analysis
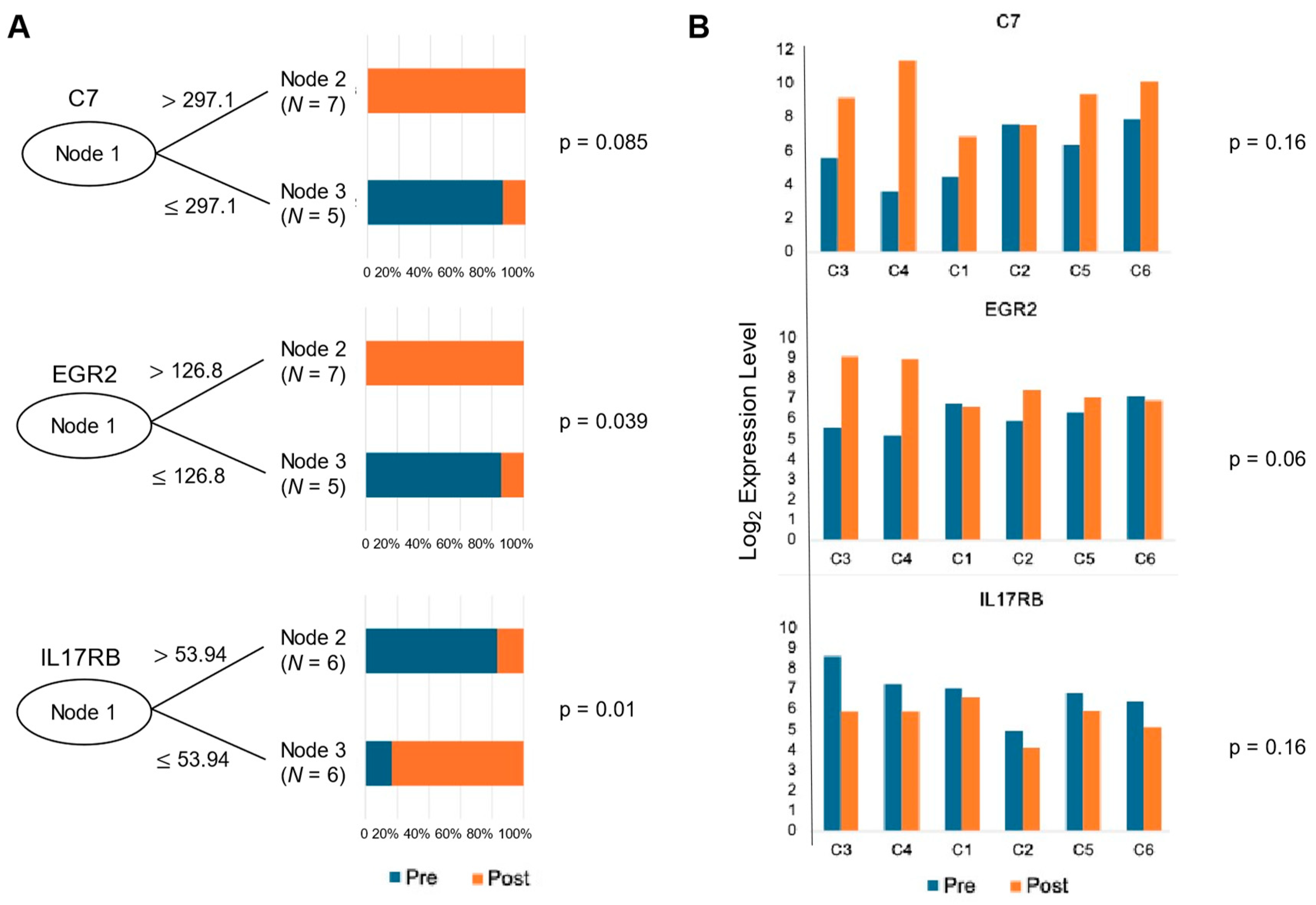
3.4. Predictive Value of Significantly Differentiated Genes for Response
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cervical Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cervical-cancer (accessed on 28 February 2024).
- Cervical Cancer Statistics|Key Facts About Cervical Cancer. Available online: https://www.cancer.org/cancer/types/cervical-cancer/about/key-statistics.html (accessed on 1 March 2024).
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Krebs—Cervical Cancer. Available online: https://www.krebsdaten.de/Krebs/EN/Content/Cancer_sites/Cervical_cancer/cervical_cancer_node.html (accessed on 13 June 2024).
- Tewari, K.S.; Sill, M.W.; Long, H.J.; Penson, R.T.; Huang, H.; Ramondetta, L.M.; Landrum, L.M.; Oaknin, A.; Reid, T.J.; Leitao, M.M.; et al. Improved Survival with Bevacizumab in Advanced Cervical Cancer. N. Engl. J. Med. 2014, 370, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Dubot, C.; Lorusso, D.; Caceres, M.V.; Hasegawa, K.; Shapira-Frommer, R.; Tewari, K.S.; Salman, P.; Usta, E.H.; Yañez, E.; et al. Pembrolizumab for Persistent, Recurrent, or Metastatic Cervical Cancer. N. Engl. J. Med. 2021, 385, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Peng, L.; Zhang, Y.; Chen, S.; Lei, Q.; Li, G.; Zhang, C. Identification of Key Genes and Pathways in Cervical Cancer by Bioinformatics Analysis. Int. J. Med. Sci. 2019, 16, 800–812. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Albert Einstein College of Medicine; Analytical Biological Services; Barretos Cancer Hospital; Baylor College of Medicine; Beckman Research Institute of City of Hope; Buck Institute for Research on Aging; Canada’s Michael Smith Genome Sciences Centre; Harvard Medical School; Helen F. Graham Cancer Center &Research Institute at Christiana Care Health Services; et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef]
- Bowden, S.J.; Bodinier, B.; Kalliala, I.; Zuber, V.; Vuckovic, D.; Doulgeraki, T.; Whitaker, M.D.; Wielscher, M.; Cartwright, R.; Tsilidis, K.K.; et al. Genetic variation in cervical preinvasive and invasive disease: A genome-wide association study. Lancet Oncol. 2021, 22, 548–557. [Google Scholar] [CrossRef]
- Qiu, L.; Feng, H.; Yu, H.; Li, M.; You, Y.; Zhu, S.; Yang, W.; Jiang, H.; Wu, X. Characterization of the Genomic Landscape in Cervical Cancer by Next Generation Sequencing. Genes 2022, 13, 287. [Google Scholar] [CrossRef] [PubMed]
- Balacescu, O.; Balacescu, L.; Tudoran, O.; Todor, N.; Rus, M.; Buiga, R.; Susman, S.; Fetica, B.; Pop, L.; Maja, L.; et al. Gene expression profiling reveals activation of the FA/BRCA pathway in advanced squamous cervical cancer with intrinsic resistance and therapy failure. BMC Cancer 2014, 14, 246. [Google Scholar] [CrossRef]
- Mitra, T.; Elangovan, S. Cervical cancer development, chemoresistance, and therapy: A snapshot of involvement of microRNA. Mol. Cell. Biochem. 2021, 476, 4363–4385. [Google Scholar] [CrossRef]
- Bhattacharjee, R.; Dey, T.; Kumar, L.; Kar, S.; Sarkar, R.; Ghorai, M.; Malik, S.; Jha, N.K.; Vellingiri, B.; Kesari, K.K.; et al. Cellular landscaping of cisplatin resistance in cervical cancer. Biomed. Pharmacother. 2022, 153, 113345. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- de Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023, 83, 3861–3867. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Quaas, A.; Noh, K.-W.; Cartolano, M.; Abedpour, N.; Mauch, C.; Quantius, J.; Reinhardt, H.C.; Buettner, R.; Peifer, M.; et al. Integrative Analysis of Pleomorphic Dermal Sarcomas Reveals Fibroblastic Differentiation and Susceptibility to Immunotherapy. Clin. Cancer Res. 2020, 26, 5638–5645. [Google Scholar] [CrossRef]
- R: A Language and Environment for Statistical Computing—ScienceOpen. Available online: https://www.scienceopen.com/book?vid=b164ea90-95d2-43bf-9710-99753c479112 (accessed on 6 July 2024).
- Royston, J.P. Algorithm AS 181: The W Test for Normality. J. R. Stat. Soc. Ser. C (Appl. Stat.) 1982, 31, 176–180. [Google Scholar] [CrossRef]
- Bauer, D.F. Constructing Confidence Sets Using Rank Statistics. J. Am. Stat. Assoc. 1972, 67, 687. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene Set Analysis Toolkit with Revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2011, 40, D109–D114. [Google Scholar] [CrossRef]
- Hothorn, T.; Hornik, K.; Zeileis, A. Unbiased Recursive Partitioning: A Conditional Inference Framework. J. Comput. Graph. Stat. 2006, 15, 651–674. [Google Scholar] [CrossRef]
- Würzner, R. Modulation of complement membrane attack by local C7 synthesis. Clin. Exp. Immunol. 2000, 121, 8–10. [Google Scholar] [CrossRef]
- Chen, Z.; Yan, X.; Du, G.-W.; Tuoheti, K.; Bai, X.-J.; Wu, H.-H.; Zhang, R.-J.; Xiao, G.-F.; Liu, T.-Z. Complement C7 (C7), a Potential Tumor Suppressor, Is an Immune-Related Prognostic Biomarker in Prostate Cancer (PC). Front. Oncol. 2020, 10, 1532. [Google Scholar] [CrossRef] [PubMed]
- Ying, L.; Zhang, F.; Pan, X.; Chen, K.; Zhang, N.; Jin, J.; Wu, J.; Feng, J.; Yu, H.; Jin, H.; et al. Complement component 7 (C7), a potential tumor suppressor, is correlated with tumor progression and prognosis. Oncotarget 2016, 7, 86536–86546. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Hu, W.; Xie, Y.; Wu, H.; Jia, Z.; Zhang, Z.; Zhang, X. Functional genetic variants in complement component 7 confer susceptibility to gastric cancer. PeerJ 2022, 10, e12816. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, K.J.; Tourtellotte, W.G.; Millbrandt, J.; Baraban, J.M. The EGR family of transcription-regulatory factors: Progress at the interface of molecular and systems neuroscience. Trends Neurosci. 1999, 22, 167–173. [Google Scholar] [CrossRef]
- Harris, J.E.; Bishop, K.D.; Phillips, N.E.; Mordes, J.P.; Greiner, D.L.; Rossini, A.A.; Czech, M.P. Early Growth Response Gene-2, a Zinc-Finger Transcription Factor, Is Required for Full Induction of Clonal Anergy in CD4+ T Cells. J. Immunol. 2004, 173, 7331–7338. [Google Scholar] [CrossRef]
- Lazarevic, V.; Zullo, A.J.; Schweitzer, M.N.; Staton, T.L.; Gallo, E.M.; Crabtree, G.R.; Glimcher, L.H. The gene encoding early growth response 2, a target of the transcription factor NFAT, is required for the development and maturation of natural killer T cells. Nat. Immunol. 2009, 10, 306–313. [Google Scholar] [CrossRef]
- Unoki, M.; Nakamura, Y. EGR2 induces apoptosis in various cancer cell lines by direct transactivation of BNIP3L and BAK. Oncogene 2003, 22, 2172–2185. [Google Scholar] [CrossRef]
- Urieli-Shoval, S.; Linke, R.P.; Matzner, Y. Expression and function of serum amyloid A, a major acute-phase protein, in normal and disease states. Curr. Opin. Hematol. 2000, 7, 64–69. [Google Scholar] [CrossRef]
- Betts, J.C.; Cheshire, J.K.; Akira, S.; Kishimoto, T.; Woo, P. The role of NF-kappa B and NF-IL6 transactivating factors in the synergistic activation of human serum amyloid A gene expression by interleukin-1 and interleukin-6. J. Biol. Chem. 1993, 268, 25624–25631. [Google Scholar] [CrossRef]
- Edbrooke, M.R.; Foldi, J.; Cheshire, J.K.; Li, F.; Faulkes, D.J.; Woo, P. Constitutive and NF-κB—Like proteins in the regulation of the serum amyloid a gene by interleukin 1. Cytokine 1991, 3, 380–388. [Google Scholar] [CrossRef]
- Lee, H.Y.; Kim, M.-K.; Park, K.S.; Bae, Y.H.; Yun, J.; Park, J.-I.; Kwak, J.-Y.; Bae, Y.-S. Serum amyloid A stimulates matrix-metalloproteinase-9 upregulation via formyl peptide receptor like-1-mediated signaling in human monocytic cells. Biochem. Biophys. Res. Commun. 2005, 330, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Vallon, R.; Freuler, F.; Desta-Tsedu, N.; Robeva, A.; Dawson, J.; Wenner, P.; Engelhardt, P.; Boes, L.; Schnyder, J.; Tschopp, C.; et al. Serum Amyloid A (apoSAA) Expression Is Up-Regulated in Rheumatoid Arthritis and Induces Transcription of Matrix Metalloproteinases. J. Immunol. 2001, 166, 2801–2807. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Wang, H.; Lu, D.; Xie, X.; Chen, X.; Peng, J.; Hu, Q.; Shi, G.; Liu, S. Expression of serum amyloid A in uterine cervical cancer. Diagn. Pathol. 2014, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Emberley, E.D.; Murphy, L.C.; Watson, P.H. S100 proteins and their influence on pro-survival pathways in cancer. Biochem. Cell Biol. 2004, 82, 508–515. [Google Scholar] [CrossRef]
- Tian, T.; Hua, Z.; Kong, Y.; Wang, L.Z.; Liu, X.Y.; Han, Y.; Zhou, X.M.; Cui, Z.M. The mechanism of S100A7 inducing the migration and invasion in cervical cancer cells. Zhonghua Zhong Liu Za Zhi 2023, 45, 375–381. [Google Scholar] [CrossRef]
- Bie, Q.; Song, H.; Chen, X.; Yang, X.; Shi, S.; Zhang, L.; Zhao, R.; Wei, L.; Zhang, B.; Xiong, H.; et al. IL-17B/IL-17RB signaling cascade contributes to self-renewal and tumorigenesis of cancer stem cells by regulating Beclin-1 ubiquitination. Oncogene 2021, 40, 2200–2216. [Google Scholar] [CrossRef]
- Huang, C.-K.; Yang, C.-Y.; Jeng, Y.-M.; Chen, C.-L.; Wu, H.-H.; Chang, Y.-C.; Ma, C.; Kuo, W.-H.; Chang, K.-J.; Shew, J.-Y.; et al. Autocrine/paracrine mechanism of interleukin-17B receptor promotes breast tumorigenesis through NF-κB-mediated antiapoptotic pathway. Oncogene 2013, 33, 2968–2977. [Google Scholar] [CrossRef]
- Laprevotte, E.; Cochaud, S.; du Manoir, S.; Lapierre, M.; Dejou, C.; Philippe, M.; Giustiniani, J.; Frewer, K.A.; Sanders, A.J.; Jiang, W.G.; et al. The IL-17B-IL-17 receptor B pathway promotes resistance to paclitaxel in breast tumors through activation of the ERK1/2 pathway. Oncotarget 2017, 8, 113360–113372. [Google Scholar] [CrossRef]
- Wu, H.-H.; Hwang-Verslues, W.W.; Lee, W.-H.; Huang, C.-K.; Wei, P.-C.; Chen, C.-L.; Shew, J.-Y.; Lee, E.Y.-H.; Jeng, Y.-M.; Tien, Y.-W.; et al. Targeting IL-17B–IL-17RB signaling with an anti–IL-17RB antibody blocks pancreatic cancer metastasis by silencing multiple chemokines. J. Exp. Med. 2015, 212, 333–349. [Google Scholar] [CrossRef]
- Hervouet, C.; Luci, C.; Rol, N.; Rousseau, D.; Kissenpfennig, A.; Malissen, B.; Czerkinsky, C.; Anjuère, F. Langerhans Cells Prime IL-17–Producing T Cells and Dampen Genital Cytotoxic Responses following Mucosal Immunization. J. Immunol. 2010, 184, 4842–4851. [Google Scholar] [CrossRef]
- Alves, J.J.P.; Fernandes, T.A.A.d.M.; Cobucci, R.N.O.; Lanza, D.C.F.; Bezerra, F.L.; de Araújo, J.M.G.; Andrade, V.S.; Fernandes, J.V. Th17 response in patients with cervical cancer. Oncol. Lett. 2018, 16, 6215–6227. [Google Scholar] [CrossRef] [PubMed]
- Punt, S.; Fleuren, G.J.; Kritikou, E.; Lubberts, E.; Trimbos, J.B.; Jordanova, E.S.; Gorter, A. Angels and demons: Th17 cells represent a beneficial response, while neutrophil IL-17 is associated with poor prognosis in squamous cervical cancer. OncoImmunology 2015, 4, e984539. [Google Scholar] [CrossRef] [PubMed]
- Stuart, L.M.; Ezekowitz, R.A.B. Phagocytosis: Elegant Complexity. Immunity 2005, 22, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, E.; Oliveira, C.R.; Pereira, C.M. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol. Dis. 2008, 30, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, N.A. The complement system in prion diseases. Curr. Opin. Immunol. 2004, 16, 587–593. [Google Scholar] [CrossRef]
- Rothenberg, E.V.; Taghon, T. Molecular genetics of T cell development. Annu. Rev. Immunol. 2005, 23, 601–649. [Google Scholar] [CrossRef]
- Da Silva, D.M.; Enserro, D.M.; Mayadev, J.S.; Skeate, J.G.; Matsuo, K.; Pham, H.Q.; Lankes, H.A.; Moxley, K.M.; Ghamande, S.A.; Lin, Y.G.; et al. Immune Activation in Patients with Locally Advanced Cervical Cancer Treated with Ipilimumab Following Definitive Chemoradiation (GOG-9929). Clin. Cancer Res. 2020, 26, 5621–5630. [Google Scholar] [CrossRef]
- Onyango, C.G.; Ogonda, L.; Guyah, B.; Shiluli, C.; Ganda, G.; Orang’o, O.E.; Patel, K. Novel biomarkers with promising benefits for diagnosis of cervical neoplasia: A systematic review. Infect. Agents Cancer 2020, 15, 68. [Google Scholar] [CrossRef]
- Gupta, S.; Maheshwari, A.; Parab, P.; Mahantshetty, U.; Hawaldar, R.; Sastri Chopra, S.; Kerkar, R.; Engineer, R.; Tongaonkar, H.; Ghosh, J.; et al. Neoadjuvant chemotherapy followed by radical surgery versus concomitant chemotherapy and radiotherapy in patients with stage IB2, IIA, or IIB squamous cervical cancer: A randomized controlled trial. J. Clin. Oncol. 2018, 36, 1548–1555. [Google Scholar] [CrossRef]
- Huang, H.-J.; Chang, T.-C.; Hong, J.-H.; Tseng, C.-J.; Chou, H.-H.; Huang, K.-G.; Lai, C.-H. Prognostic value of age and histologic type in neoadjuvant chemotherapy plus radical surgery for bulky (>=4 cm) stage IB and IIA cervical carcinoma. Int. J. Gynecol. Cancer 2003, 13, 204–211. [Google Scholar] [CrossRef]
- Mereu, L.; Pecorino, B.; Ferrara, M.; Tomaselli, V.; Scibilia, G.; Scollo, P. Neoadjuvant Chemotherapy plus Radical Surgery in Locally Advanced Cervical Cancer: Retrospective Single-Center Study. Cancers 2023, 15, 5207. [Google Scholar] [CrossRef] [PubMed]
- Zubizarreta, E.; Fidarova, E.; Healy, B.; Rosenblatt, E. Need for Radiotherapy in Low and Middle Income Countries—The Silent Crisis Continues. Clin. Oncol. 2015, 27, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Buda, A.; Borghese, M.; Puppo, A.; Perotto, S.; Novelli, A.; Borghi, C.; Olearo, E.; Tripodi, E.; Surace, A.; Bar, E.; et al. Neoadjuvant Chemotherapy Prior Fertility-Sparing Surgery in Women with FIGO 2018 Stage IB2 Cervical Cancer: A Systematic Review. Cancers 2022, 14, 797. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | C1 | C2 | C3 | C4 | C5 | C6 |
|---|---|---|---|---|---|---|
| Median Age in yeas | 34 | 55 | 36 | 52 | 46 | 39 |
| Date of first Diagnosis | Jan 2019 | Jan 2018 | Jan 2016 | Feb 2018 | Aug 2021 | May 2018 |
| FIGO stage 2018 | IB3 | IB1 | IIB | IIB | IIIB | IIB |
| Histological type | SCC | AC | AC | SCC | SCC | SCC |
| Grading | G3 | G3 | G1 | G3 | n.d. | G2 |
| Lymphovascular invasion | pos. | neg. | neg. | neg. | pos. | pos. |
| PFS | 17 | 17 | n.d. | n.d. | 1 | 18 |
| OS | 59 | n.d. | n.d. | n.d. | 11 | n.d. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baek, S.; Mairinger, F.D.; Borchert, S.; Zhao, Y.; Ratiu, D.; Mallmann, P.K.; Pilch, H.; Noh, K.-W. Comparative Analysis of Digital Transcriptomics Between Pre- and Post-Treatment Samples of Patients with Locally Advanced Cervical Cancer: A Preliminary Study. Curr. Issues Mol. Biol. 2024, 46, 12075-12087. https://doi.org/10.3390/cimb46110716
Baek S, Mairinger FD, Borchert S, Zhao Y, Ratiu D, Mallmann PK, Pilch H, Noh K-W. Comparative Analysis of Digital Transcriptomics Between Pre- and Post-Treatment Samples of Patients with Locally Advanced Cervical Cancer: A Preliminary Study. Current Issues in Molecular Biology. 2024; 46(11):12075-12087. https://doi.org/10.3390/cimb46110716
Chicago/Turabian StyleBaek, Sunhwa, Fabian Dominik Mairinger, Sabrina Borchert, Yue Zhao, Dominik Ratiu, Peter Konrad Mallmann, Henryk Pilch, and Ka-Won Noh. 2024. "Comparative Analysis of Digital Transcriptomics Between Pre- and Post-Treatment Samples of Patients with Locally Advanced Cervical Cancer: A Preliminary Study" Current Issues in Molecular Biology 46, no. 11: 12075-12087. https://doi.org/10.3390/cimb46110716
APA StyleBaek, S., Mairinger, F. D., Borchert, S., Zhao, Y., Ratiu, D., Mallmann, P. K., Pilch, H., & Noh, K.-W. (2024). Comparative Analysis of Digital Transcriptomics Between Pre- and Post-Treatment Samples of Patients with Locally Advanced Cervical Cancer: A Preliminary Study. Current Issues in Molecular Biology, 46(11), 12075-12087. https://doi.org/10.3390/cimb46110716