
The Spectrum of Germline Nucleotide Variants in Gastric Cancer Patients in the Kyrgyz Republic

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Extraction
2.2. Library Preparation and Sequencing
2.3. Variant Classification and Bioinformatics Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smyth, E.C.; Nilsson, M.; Grabsch, H.I.; van Grieken, N.C.; Lordick, F. Gastric cancer. Lancet 2020, 396, 635–648. [Google Scholar] [CrossRef]
- Rahman, R.; Asombang, A.W.; Ibdah, J.A. Characteristics of gastric cancer in Asia. World J. Gastroenterol. 2014, 20, 4483–4490. [Google Scholar] [CrossRef]
- Axel, E.M. Gastrointestinal cancer statistics. Sib. J. Oncol. 2017, 16, 5–11. [Google Scholar] [CrossRef][Green Version]
- Toygonbekov, A.K.; Soodonbekov, E.T.; Makimbetov, E.K. Epidemiology of Stomach Cancer: Monograph; KRSU: Bishkek, Kyrgyzstan, 2020; Volume 178. [Google Scholar]
- Toigonbekov, A.; Akhunbaev, S.; Umetov, M.; Tumanbaev, A. Some Intense and Standardized Stomach Cancer Disease Indicators in the Kyrgyz Republic. Bull. Sci. Pract. 2020, 6, 169–175. [Google Scholar] [CrossRef]
- WHO Globocan Statistics. 2020. Available online: https://gco.iarc.fr/today/data/factsheets/populations/417-kyrgyzstan-fact-sheets.pdf (accessed on 12 May 2023).
- Sitarz, R.; Skierucha, M.; Mielko, J.; Offerhaus, G.J.A.; Maciejewski, R.; Polkowski, W.P. Gastric cancer: Epidemiology, prevention, classification, and treatment. Cancer Manag. Res. 2018, 10, 239–248. [Google Scholar] [CrossRef]
- Machlowska, J.; Baj, J.; Sitarz, M.; Maciejewski, R.; Sitarz, R. Gastric Cancer: Epidemiology, Risk Factors, Classification, Genomic Characteristics and Treatment Strategies. Int. J. Mol. Sci. 2020, 21, 4012. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Pinheiro, H.; Figueiredo, J.; Seruca, R.; Carneiro, F. Familial gastric cancer: Genetic susceptibility, pathology, and implications for management. Lancet Oncol. 2015, 16, e60–e70. [Google Scholar] [CrossRef] [PubMed]
- Assumpção, P.; Araújo, T.; Khayat, A.; Ishak, G.; Santos, S.; Barra, W.; Acioli, J.F.; Rossi, B.; Assumpção, P. Hereditary gastric cancer: Three rules to reduce missed diagnoses. World J. Gastroenterol. 2020, 26, 1382–1393. [Google Scholar] [CrossRef]
- Sehgal, R.; Sheahan, K.; O’Connell, P.R.; Hanly, A.M.; Martin, S.T.; Winter, D.C. Lynch syndrome: An updated review. Genes 2014, 5, 497–507. [Google Scholar] [CrossRef]
- MacFarland, S.P.; Ebrahimzadeh, J.E.; Zelley, K.; Begum, L.; Bass, L.M.; Brand, R.E.; Dudley, B.; Fishman, D.S.; Ganzak, A.; Karloski, E.; et al. Phenotypic Differences in Juvenile Polyposis Syndrome With or Without a Disease-causing SMAD4/BMPR1A Variant. Cancer Prev. Res. 2021, 14, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.D.; Jeon, C.H. Peutz-Jeghers syndrome with germline mutation of STK11. Ann. Surg. Treat Res. 2014, 86, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Gullo, I.; van der Post, R.S.; Carneiro, F. Recent advances in the pathology of heritable gastric cancer syndromes. Histopathology 2021, 78, 125–147. [Google Scholar] [CrossRef] [PubMed]
- Ajani, J.A.; D’Amico, T.A.; Bentrem, D.J.; Chao, J.; Cooke, D.; Corvera, C.; Das, P.; Enzinger, P.C.; Enzler, T.; Fanta, P.; et al. Gastric Cancer, Version 2.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 167–192. [Google Scholar] [CrossRef] [PubMed]
- Rechsteiner, M.; von Teichman, A.; Rüschoff, J.H.; Fankhauser, N.; Pestalozzi, B.; Schraml, P.; Weber, A.; Wild, P.; Zimmermann, D.; Moch, H. KRAS, BRAF, and TP53 deep sequencing for colorectal carcinoma patient diagnostics. J. Mol. Diagn. 2013, 15, 299–311. [Google Scholar] [CrossRef]
- Chang, Y.S.; Hsu, H.T.; Ko, Y.C.; Yeh, K.T.; Chang, S.J.; Lin, C.Y.; Chang, J.G. Combined mutational analysis of RAS, BRAF, PIK3CA, and TP53 genes in Taiwanese patients with oral squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2014, 118, 110–116.e1. [Google Scholar] [CrossRef]
- Loveday, C.; Josephs, K.; Chubb, D.; Gunning, A.; Izatt, L.; Tischkowitz, M.; Ellard, S.; Turnbull, C. p.Val804Met, the Most Frequent Pathogenic Mutation in RET, Confers a Very Low Lifetime Risk of Medullary Thyroid Cancer. J. Clin. Endocrinol. Metab. 2018, 103, 4275–4282. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F.; et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef]
- VarSome: The Human Genomic Variant Search Engine. Available online: https://academic.oup.com/bioinformatics/article/35/11/1978/5146783 (accessed on 12 May 2023).
- Borg, A.; Haile, R.W.; Malone, K.E.; Capanu, M.; Diep, A.; Törngren, T.; Teraoka, S.; Begg, C.B.; Thomas, D.C.; Concannon, P.; et al. Characterization of BRCA1 and BRCA2 deleterious mutations and variants of unknown clinical significance in unilateral and bilateral breast cancer: The WECARE study. Hum. Mutat. 2010, 31, E1200–E1240. [Google Scholar] [CrossRef]
- Janavičius, R.; Rudaitis, V.; Mickys, U.; Elsakov, P.; Griškevičius, L. Comprehensive BRCA1 and BRCA2 mutational profile in Lithuania. Cancer Genet. 2014, 207, 195–205. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Hart, S.N.; Vijai, J.; Schrader, K.A.; Slavin, T.P.; Thomas, T.; Wubbenhorst, B.; Ravichandran, V.; Moore, R.M.; Hu, C.; et al. Evaluation of ACMG-Guideline-Based Variant Classification of Cancer Susceptibility and Non-Cancer-Associated Genes in Families Affected by Breast Cancer. Am. J. Hum. Genet. 2016, 98, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Sivapiragasam, A.; Ashok Kumar, P.; Sokol, E.S.; Albacker, L.A.; Killian, J.K.; Ramkissoon, S.H.; Huang, R.S.P.; Severson, E.A.; Brown, C.A.; Danziger, N.; et al. Predictive Biomarkers for Immune Checkpoint Inhibitors in Metastatic Breast Cancer. Cancer Med. 2021, 10, 53–61. [Google Scholar] [CrossRef]
- Chung, J.H.; Dewal, N.; Sokol, E.; Mathew, P.; Whitehead, R.; Millis, S.Z.; Frampton, G.M.; Bratslavsky, G.; Pal, S.K.; Lee, R.J.; et al. Prospective Comprehensive Genomic Profiling of Primary and Metastatic Prostate Tumors. JCO Precis. Oncol. 2019, 3, PO.18.00283. [Google Scholar] [CrossRef]
- Mezina, A.; Philips, N.; Bogus, Z.; Erez, N.; Xiao, R.; Fan, R.; Olthoff, K.M.; Reddy, K.R.; Samadder, N.J.; Nielsen, S.M.; et al. Multigene Panel Testing in Individuals With Hepatocellular Carcinoma Identifies Pathogenic Germline Variants. JCO Precis. Oncol. 2021, 5, PO.21.00079. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted With Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253. [Google Scholar] [CrossRef]
- van der Post, R.S.; Oliveira, C.; Guilford, P.; Carneiro, F. Hereditary gastric cancer: What’s new? Update 2013–2018. Fam. Cancer 2019, 18, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Blair, V.R.; McLeod, M.; Carneiro, F.; Coit, D.G.; D’Addario, J.L.; van Dieren, J.M.; Harris, K.L.; Hoogerbrugge, N.; Oliveira, C.; van der Post, R.S.; et al. Hereditary diffuse gastric cancer: Updated clinical practice guidelines. Lancet Oncol. 2020, 21, e386–e397. [Google Scholar] [CrossRef]
- Slavin, T.P.; Weitzel, J.N.; Neuhausen, S.L.; Schrader, K.A.; Oliveira, C.; Karam, R. Genetics of gastric cancer: What do we know about the genetic risks. Transl. Gastroenterol. Hepatol. 2019, 4, 55. [Google Scholar] [CrossRef]
- Krampitz, G.W.; Norton, J.A. RET gene mutations (genotype and phenotype) of multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma. Cancer 2014, 120, 1920–1931. [Google Scholar] [CrossRef]
- Roskoski, R.; Sadeghi-Nejad, A. Role of RET protein-tyrosine kinase inhibitors in the treatment RET-driven thyroid and lung cancers. Pharmacol. Res. 2018, 128, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Welcsh, P.L.; King, M.C. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum. Mol. Genet. 2001, 10, 705–713. [Google Scholar] [CrossRef]
- Molinaro, E.; Andrikou, K.; Casadei-Gardini, A.; Rovesti, G. BRCA in Gastrointestinal Cancers: Current Treatments and Future Perspectives. Cancers 2020, 12, 3346. [Google Scholar] [CrossRef]
- Maccaroni, E.; Giampieri, R.; Lenci, E.; Scortichini, L.; Bianchi, F.; Belvederesi, L.; Brugiati, C.; Pagliaretta, S.; Ambrosini, E.; Berardi, R. BRCA mutations and gastrointestinal cancers: When to expect the unexpected. World J. Clin. Oncol. 2021, 12, 565–580. [Google Scholar] [CrossRef]
- Buckley, K.H.; Niccum, B.A.; Maxwell, K.N.; Katona, B.W. Gastric Cancer Risk and Pathogenesis in BRCA1 and BRCA2 Carriers. Cancers 2022, 14, 5953. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Gong, Y.; Zhao, W.; Han, Z.; Guo, S.; Liu, H.; Peng, X.; Xiao, W.; Li, Y.; Dang, S.; et al. Comprehensive analysis of POLE and POLD1 Gene Variations identifies cancer patients potentially benefit from immunotherapy in Chinese population. Sci. Rep. 2019, 9, 15767. [Google Scholar] [CrossRef] [PubMed]
- Mur, P.; García-Mulero, S.; Del Valle, J.; Magraner-Pardo, L.; Vidal, A.; Pineda, M.; Cinnirella, G.; Martín-Ramos, E.; Pons, T.; López-Doriga, A.; et al. Role of POLE and POLD1 in familial cancer. Genet. Med. 2020, 22, 2089–2100. [Google Scholar] [CrossRef]
- steban-Jurado, C.; Giménez-Zaragoza, D.; Muñoz, J.; Franch-Expósito, S.; Álvarez-Barona, M.; Ocaña, T.; Cuatrecasas, M.; Carballal, S.; López-Cerón, M.; Marti-Solano, M.; et al. POLE and POLD1 screening in 155 patients with multiple polyps and early-onset colorectal cancer. Oncotarget 2017, 8, 26732–26743. [Google Scholar] [CrossRef] [PubMed]
- Siraj, A.K.; Parvathareddy, S.K.; Bu, R.; Iqbal, K.; Siraj, S.; Masoodi, T.; Concepcion, R.M.; Ghazwani, L.O.; AlBadawi, I.; Al-Dayel, F.; et al. Germline POLE and POLD1 proofreading domain mutations in endometrial carcinoma from Middle Eastern region. Cancer Cell Int. 2019, 19, 334. [Google Scholar] [CrossRef]
- Zhu, M.; Cui, H.; Zhang, L.; Zhao, K.; Jia, X.; Jin, H. Assessment of POLE and POLD1 mutations as prognosis and immunotherapy biomarkers for stomach adenocarcinoma. Transl. Cancer Res. 2022, 11, 193–205. [Google Scholar] [CrossRef]
- Mai, P.L.; Best, A.F.; Peters, J.A.; DeCastro, R.M.; Khincha, P.P.; Loud, J.T.; Bremer, R.C.; Rosenberg, P.S.; Savage, S.A. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer 2016, 122, 3673–3681. [Google Scholar] [CrossRef]
- Guimaraes, D.P.; Hainaut, P. TP53: A key gene in human cancer. Biochimie 2002, 84, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef]
- Tan, P.; Yeoh, K.G. Genetics and Molecular Pathogenesis of Gastric Adenocarcinoma. Gastroenterology 2015, 149, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Frebourg, T.; Bajalica Lagercrantz, S.; Oliveira, C.; Magenheim, R.; Evans, D.G.; Hoogerbrugge, N.; Ligtenberg, M.; Kets, M.; Oostenbrink, R.; Sijmons, R.; et al. Guidelines for the Li-Fraumeni and heritable TP53-related cancer syndromes. Eur. J. Hum. Genet. 2020, 28, 1379–1386. [Google Scholar] [CrossRef]
- Herrmann, L.J.; Heinze, B.; Fassnacht, M.; Willenberg, H.S.; Quinkler, M.; Reisch, N.; Zink, M.; Allolio, B.; Hahner, S. TP53 germline mutations in adult patients with adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 2012, 97, E476–E485. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Meng, H.; Yao, L.; Lv, M.; Bai, J.; Zhang, J.; Wang, L.; Ouyang, T.; Li, J.; Wang, T.; et al. Germline Mutations in Cancer Susceptibility Genes in a Large Series of Unselected Breast Cancer Patients. Clin. Cancer Res. 2017, 23, 6113–6119. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.L.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 2018, 173, 355–370. [Google Scholar] [CrossRef]
- Rana, H.Q.; Clifford, J.; Hoang, L.; LaDuca, H.; Black, M.H.; Li, S.; McGoldrick, K.; Speare, V.; Dolinsky, J.S.; Gau, C.L.; et al. Genotype-phenotype associations among panel-based TP53+ subjects. Genet. Med. 2019, 21, 2478–2484. [Google Scholar] [CrossRef]
- Gao, F.; Pan, X.; Dodd-Eaton, E.B.; Recio, C.V.; Montierth, M.D.; Bojadzieva, J.; Mai, P.L.; Zelley, K.; Johnson, V.E.; Braun, D.; et al. A pedigree-based prediction model identifies carriers of deleterious de novo mutations in families with Li-Fraumeni syndrome. Genome Res. 2020, 30, 1170–1180. [Google Scholar] [CrossRef]
- Kato, S.; Han, S.Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429. [Google Scholar] [CrossRef]
- Monti, P.; Campomenosi, P.; Ciribilli, Y.; Iannone, R.; Aprile, A.; Inga, A.; Tada, M.; Menichini, P.; Abbondandolo, A.; Fronza, G. Characterization of the p53 mutants ability to inhibit p73 beta transactivation using a yeast-based functional assay. Oncogene 2003, 22, 5252–5260. [Google Scholar] [CrossRef][Green Version]
- Shiraishi, K.; Kato, S.; Han, S.Y.; Liu, W.; Otsuka, K.; Sakayori, M.; Ishida, T.; Takeda, M.; Kanamaru, R.; Ohuchi, N.; et al. Isolation of temperature-sensitive p53 mutations from a comprehensive missense mutation library. J. Biol. Chem. 2004, 279, 348–355. [Google Scholar] [CrossRef]
- Kotler, E.; Shani, O.; Goldfeld, G.; Lotan-Pompan, M.; Tarcic, O.; Gershoni, A.; Hopf, T.A.; Marks, D.S.; Oren, M.; Segal, E. A Systematic p53 Mutation Library Links Differential Functional Impact to Cancer Mutation Pattern and Evolutionary Conservation. Mol. Cell 2018, 71, 178–190. [Google Scholar] [CrossRef]
- Masciari, S.; Dewanwala, A.; Stoffel, E.M.; Lauwers, G.Y.; Zheng, H.; Achatz, M.I.; Riegert-Johnson, D.; Foretova, L.; Silva, E.M.; Digianni, L.; et al. Gastric cancer in individuals with Li-Fraumeni syndrome. Genet. Med. 2011, 13, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Nepomuceno, T.C.; De Gregoriis, G.; de Oliveira, F.M.B.; Suarez-Kurtz, G.; Monteiro, A.N.; Carvalho, M.A. The Role of PALB2 in the DNA Damage Response and Cancer Predisposition. Int. J. Mol. Sci. 2017, 18, 1886. [Google Scholar] [CrossRef]
- Fewings, E.; Larionov, A.; Redman, J.; Goldgraben, M.A.; Scarth, J.; Richardson, S.; Brewer, C.; Davidson, R.; Ellis, I.; Evans, D.G.; et al. Germline pathogenic variants in PALB2 and other cancer-predisposing genes in families with hereditary diffuse gastric cancer without CDH1 mutation: A whole-exome sequencing study. Lancet Gastroenterol. Hepatol. 2018, 3, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Carmona, L.G. PALB2 as a familial gastric cancer gene: Is the wait over. Lancet Gastroenterol. Hepatol. 2018, 3, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Levran, O.; Diotti, R.; Pujara, K.; Batish, S.D.; Hanenberg, H.; Auerbach, A.D. Spectrum of sequence variations in the FANCA gene: An International Fanconi Anemia Registry (IFAR) study. Hum. Mutat. 2005, 25, 142–149. [Google Scholar] [CrossRef]
- Benitez, A.; Liu, W.; Palovcak, A.; Wang, G.; Moon, J.; An, K.; Kim, A.; Zheng, K.; Zhang, Y.; Bai, F.; et al. FANCA Promotes DNA Double-Strand Break Repair by Catalyzing Single-Strand Annealing and Strand Exchange. Mol. Cell 2018, 71, 621–628. [Google Scholar] [CrossRef]
- Nepal, M.; Che, R.; Ma, C.; Zhang, J.; Fei, P. FANCD2 and DNA Damage. Int. J. Mol. Sci. 2017, 18, 1804. [Google Scholar] [CrossRef]
- Alcón, P.; Shakeel, S.; Chen, Z.A.; Rappsilber, J.; Patel, K.J.; Passmore, L.A. FANCD2-FANCI is a clamp stabilized on DNA by monoubiquitination of FANCD2 during DNA repair. Nat. Struct. Mol. Biol. 2020, 27, 240–248. [Google Scholar] [CrossRef]
- Yao, C.; Du, W.; Chen, H.; Xiao, S.; Huang, L.; Chen, F.P. Involvement of Fanconi anemia genes FANCD2 and FANCF in the molecular basis of drug resistance in leukemia. Mol. Med. Rep. 2015, 11, 4605–4610. [Google Scholar] [CrossRef][Green Version]
- Deng, W.; Zhao, M.; Liu, Y.; Cao, L.; Yang, M. Fanconi anemia in twins with neutropenia: A case report. Oncol. Lett. 2018, 16, 5325–5330. [Google Scholar] [CrossRef]
- Andreassen, P.R.; D’Andrea, A.D.; Taniguchi, T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004, 18, 1958–1963. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; D’Andrea, A.D. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012, 26, 1393–1408. [Google Scholar] [CrossRef]
- Li, J.; Sipple, J.; Maynard, S.; Mehta, P.A.; Rose, S.R.; Davies, S.M.; Pang, Q. Fanconi anemia links reactive oxygen species to insulin resistance and obesity. Antioxid. Redox Signal. 2012, 17, 1083–1098. [Google Scholar] [CrossRef]
- Li, J.; Du, W.; Maynard, S.; Andreassen, P.R.; Pang, Q. Oxidative stress-specific interaction between FANCD2 and FOXO3a. Blood 2010, 115, 1545–1548. [Google Scholar] [CrossRef]
- Vasimuddin, M.; Misra, S.; Li, H.; Aluru, S. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. In Proceedings of the 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), Rio de Janeiro, Brazil, 20–24 May 2019; pp. 314–324. [Google Scholar]
- Broadinstitute/Picard: A Set of Command Line Tools (in Java) for Manipulating High-Throughput Sequencing (HTS) Data and Formats Such as SAM/BAM/CRAM and VCF. 2019. Available online: https://github.com/broadinstitute/picard (accessed on 12 May 2023).
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv 2018. [Google Scholar] [CrossRef]
- Kurian, A.W.; Li, Y.; Hamilton, A.S.; Ward, K.C.; Hawley, S.T.; Morrow, M.; McLeod, M.C.; Jagsi, R.; Katz, S.J. Gaps in Incorporating Germline Genetic Testing Into Treatment Decision-Making for Early-Stage Breast Cancer. J. Clin. Oncol. 2017, 35, 2232–2239. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| SDHB, PTCH2, MUTYH, NTRK1, SDHC, CDC73, PARP1, FH, RET, BMPR1A, PTEN, SUFU, HRAS, CDKN1C, ABCC8, WT1, EXT2, SDHAF2, MEN1, MRE11A, ATM, SDHD, CDKN1B, CDK4, POLE, BRCA2, RB1, FANCM, MAX, RAD51B, MLH3, DICER1, GREM1, FANCI, BLM, NTHL1, TSC2, SLX4, PALB2, CDH1, FANCA, RPA1, TP53, FLCN, NF1, RAD51D, CDK12, ERBB2, SMARCE1, BRCA1, HOXB13, RAD51C, PPM1D, BRIP1, AXIN2, RHBDF2, RBBP8, SMAD4, STK11, SMARCA4, ERCC2, POLD1, GEN1, ALK, EPCAM, MSH2, MSH6, FANCL, TMEM127, PMS1, BARD1, SMARCB1, CHEK2, NF2, FANCD2, VHL, MLH1, BAP1, GATA2, ATR, PHOX2B, PDGFRA, KIT, FAM175A, SDHA, TERT, MSH3, APC, RAD50, CTNNA1, SPINK1, FANCE, PMS2, EGFR, RINT1, MET, POT1, PRSS1, XRCC2, PPP2R2A, RPS20, NBN, EXT1, RECQL4, CDKN2A, FANCG, FANCC, PTCH1, GALNT12, TSC1, FANCB. |
| Characteristics | Number of Patients (%) | |
|---|---|---|
| Gender | Female | 35 (31%) |
| Male | 78 (69%) | |
| Histological types (Lauren) | Intestinal type | 77 (68.1%) |
| Diffuse type | 21 (18.6%) | |
| Mixed type | 15 (13.3%) | |
| Helicobacter pylori | H. pylori + | 16 (14.2%) |
| H. pylori − | 97 (85.8%) | |
| Stage of cancer | I | 5 (4.4%) |
| II | 16 (14.2%) | |
| III | 61 (54%) | |
| IV | 31 (27.4%) | |
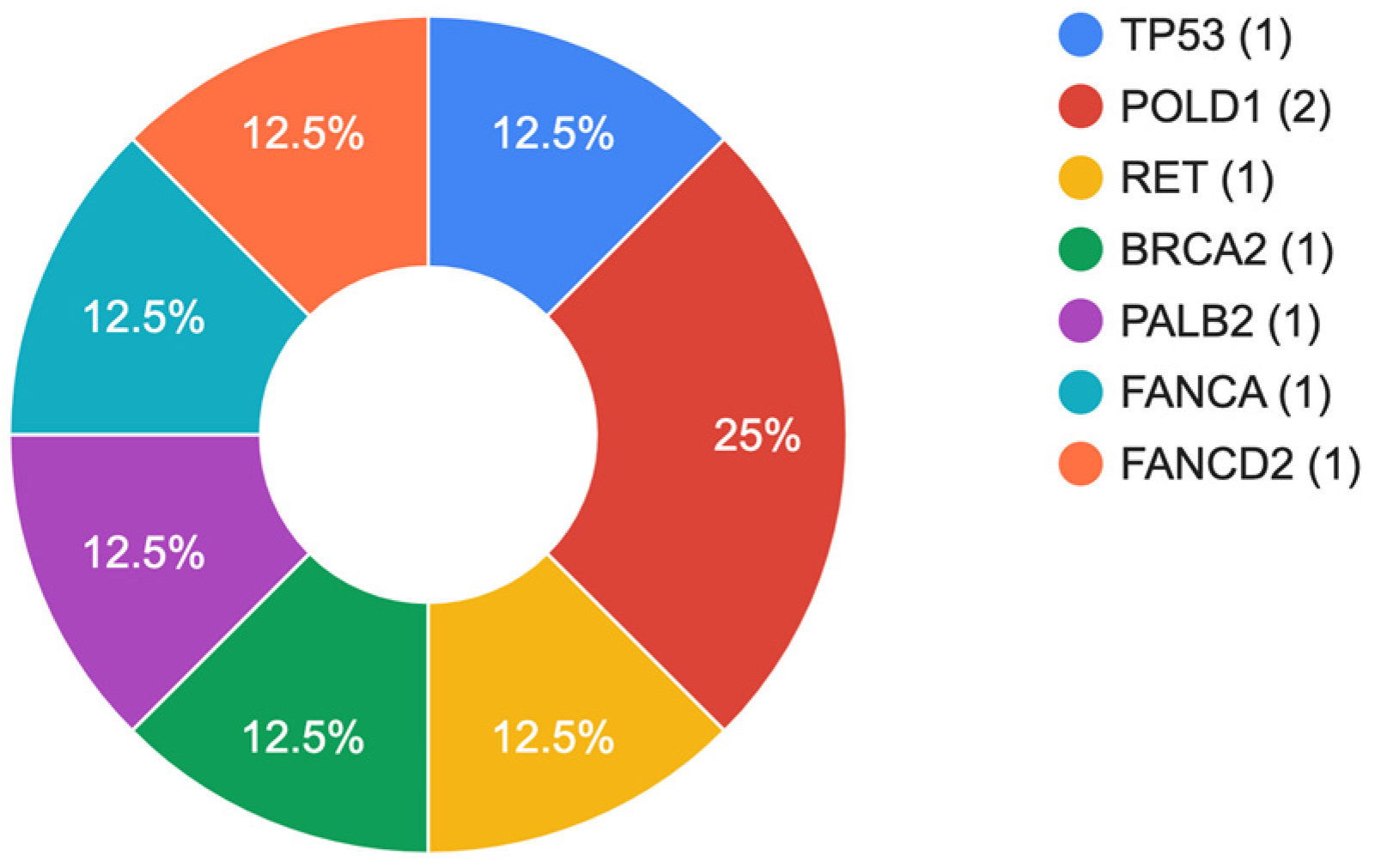
| № | Gender (Age) | Lauren Classification | Stage | Gene | Chromosomal Change | Coding | Protein | ACMG | Cancer Cases in Family History | Pathogenicity Scores * | GnomAD Exomes | Literature |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Male (63) | D a | IIIB | TP53 | chr17:7675140G>A | c.472C>T | p.Arg158Cys | P d | No |
Pathogenic-22 Uncertai-9 Benign-1 | ƒ = 0.00000796 | [21,22] |
| 2 | Male (60) | D a | IIIB | RET | chr10:43119548G>A | c.2410G>A | p.Val804Met | P d | No |
Pathogenic-5 Uncertai-12 Benign-3 | ƒ = 0.000125 | [23,24] |
| BRCA2 | chr13:32340763delA | c.6410del | p.Asn2137MetfsTer31 | P d | - | Not found | [25,26] | |||||
| 3 | Female (57) | M b | IIIB | POLD1 | chr19:50414912delT | c.2486del | p.Leu829ArgfsTer59 | LP e | No | - | Not found | - |
| 4 | Male (74) | I c | IIIB | POLD1 | chr19:50416486G>T | c.2911G>T | p.Glu971Ter | LP e | Yes |
Pathogenic-4 Uncertai-3 Benign-2 | Not found | - |
| 5 | Male (62) | I c | IIIB | PALB2 | chr16:23630456insACGACTT | c.1692_1698dup | p.His567LysfsTer13 | LP e | No | - | Not found | - |
| 6 | Male (70) | I c | IIIB | FANCA | chr16:89816550C>T | c.66G>A | p.Trp22Ter | P d | No |
Uncertai-3 Benign-7 | Not found | [27,28,29,30,31] |
| 7 | Male (58) | D a | III | FANCD2 | chr3:10081422delGC | c.3182_3183del | p.Cys1061LeufsTer21 | LP e | No | - | Not found | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilyalov, A.; Nikolaev, S.; Danishevich, A.; Khatkov, I.; Makhmudov, K.; Isakova, Z.; Bakirov, N.; Omurbaev, E.; Osipova, A.; Ramaldanov, R.; et al. The Spectrum of Germline Nucleotide Variants in Gastric Cancer Patients in the Kyrgyz Republic. Curr. Issues Mol. Biol. 2023, 45, 6383-6394. https://doi.org/10.3390/cimb45080403
Bilyalov A, Nikolaev S, Danishevich A, Khatkov I, Makhmudov K, Isakova Z, Bakirov N, Omurbaev E, Osipova A, Ramaldanov R, et al. The Spectrum of Germline Nucleotide Variants in Gastric Cancer Patients in the Kyrgyz Republic. Current Issues in Molecular Biology. 2023; 45(8):6383-6394. https://doi.org/10.3390/cimb45080403
Chicago/Turabian StyleBilyalov, Airat, Sergey Nikolaev, Anastasiia Danishevich, Igor Khatkov, Komron Makhmudov, Zhainagul Isakova, Nurbek Bakirov, Ernis Omurbaev, Alena Osipova, Ramaldan Ramaldanov, and et al. 2023. "The Spectrum of Germline Nucleotide Variants in Gastric Cancer Patients in the Kyrgyz Republic" Current Issues in Molecular Biology 45, no. 8: 6383-6394. https://doi.org/10.3390/cimb45080403
APA StyleBilyalov, A., Nikolaev, S., Danishevich, A., Khatkov, I., Makhmudov, K., Isakova, Z., Bakirov, N., Omurbaev, E., Osipova, A., Ramaldanov, R., Shagimardanova, E., Kiyasov, A., Gusev, O., & Bodunova, N. (2023). The Spectrum of Germline Nucleotide Variants in Gastric Cancer Patients in the Kyrgyz Republic. Current Issues in Molecular Biology, 45(8), 6383-6394. https://doi.org/10.3390/cimb45080403