
Clinical Characteristics of Molecularly Defined Renal Cell Carcinomas


Abstract
1. Introduction
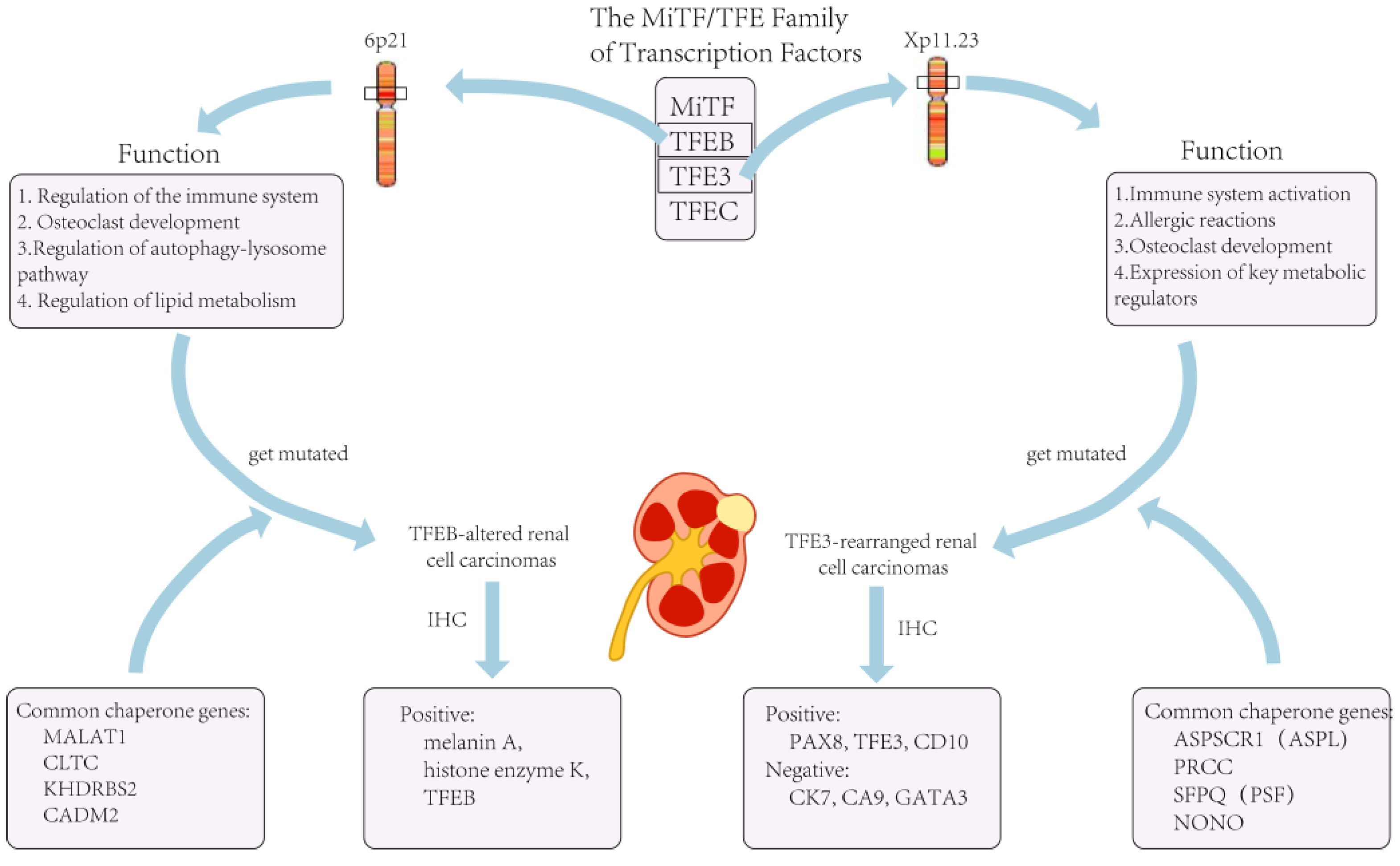
2. TFE3-Rearranged Renal Cell Carcinomas
3. TFEB-Altered Renal Cell Carcinomas
4. Elongin C (ELOC, Formerly TCEB1)-Mutated Renal Cell Carcinoma
5. Fumarate Hydratase-Deficient Renal Cell Carcinoma
6. Succinate Dehydrogenase-Deficient Renal Cell Carcinoma
7. ALK-Rearranged Renal Cell Carcinomas
8. SMARCB1-Deficient Renal Medullary Carcinoma
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Capitanio, U.; Montorsi, F. Renal cancer. Lancet 2016, 10021, 894–906. [Google Scholar] [CrossRef]
- Dibajnia, P.; Cardenas, L.M.; Lalani, A.A. The emerging landscape of neo/adjuvant immunotherapy in renal cell carcinoma. Hum. Vaccin. Immunother. 2023, 1, 2178217. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 6, 394–424. [Google Scholar] [CrossRef]
- Motzer, R.J.; Jonasch, E.; Agarwal, N.; Alva, A.; Baine, M.; Beckermann, K.; Carlo, M.I.; Choueiri, T.K.; Costello, B.A.; Derweesh, I.H.; et al. Kidney Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 1, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Moch, H.; Amin, M.B.; Berney, D.M.; Compérat, E.M.; Gill, A.J.; Hartmann, A.; Menon, S.; Raspollini, M.R.; Rubin, M.A.; Srigley, J.R.; et al. The 2022 World Health Organization Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2022, 5, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.E.; Harris, G.T. Renal Cell Carcinoma: Diagnosis and Management. Am. Fam. Physician 2019, 3, 179–184. [Google Scholar]
- Padala, S.A.; Barsouk, A.; Thandra, K.C.; Saginala, K.; Mohammed, A.; Vakiti, A.; Rawla, P.; Barsouk, A. Epidemiology of Renal Cell Carcinoma. World J. Oncol. 2020, 3, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Yong, C.; Stewart, G.D.; Frezza, C. Oncometabolites in renal cancer. Nat. Rev. Nephrol. 2020, 3, 156–172. [Google Scholar] [CrossRef]
- Navani, V.; Heng., D.Y.C. Treatment Selection in First-line Metastatic Renal Cell Carcinoma-The Contemporary Treatment Paradigm in the Age of Combination Therapy: A Review. JAMA Oncol. 2022, 8, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, J.; Esteller, M. Cancer epigenomics: Beyond genomics. Curr. Opin. Genet. Dev. 2012, 1, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.J.; Turajlic, S.; Rowan, A.; Nicol, D.; Farmery, J.H.R.; O’Brien, T.; Martincorena, I.; Tarpey, P.; Angelopoulos, N.; Yates, L.R.; et al. TRACERx Renal Consortium. Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell 2018, 3, 611–623.e17. [Google Scholar] [CrossRef]
- Xing, T.; He, H. Epigenomics of clear cell renal cell carcinoma: Mechanisms and potential use in molecular pathology. Chin. J. Cancer Res. 2016, 1, 80–91. [Google Scholar]
- Pastore, N.; Vainshtein, A.; Klisch, T.J.; Armani, A.; Huynh, T.; Herz, N.J.; Polishchuk, E.V.; Sandri, M.; Ballabio, A. TFE3 Regulates whole-body energy metabolism in cooperation with TFEB. EMBO Mol. Med. 2017, 5, 605–621. [Google Scholar] [CrossRef]
- Argani, P. Translocation carcinomas of the kidney. Genes. Chromosomes Cancer 2022, 5, 219–227. [Google Scholar] [CrossRef]
- Caliò, A.; Segala, D.; Munari, E.; Brunelli, M.; Martignoni, G. MiT Family Translocation Renal Cell Carcinoma: From the Early Descriptions to the Current Knowledge. Cancers 2019, 8, 1110. [Google Scholar] [CrossRef]
- Sukov, W.R.; Hodge, J.C.; Lohse, C.M.; Leibovich, B.C.; Thompson, R.H.; Pearce, K.E.; Wiktor, A.E.; Cheville, J.C. TFE3 rearrangements in adult renal cell carcinoma: Clinical and pathologic features with outcome in a large series of consecutively treated patients. Am. J. Surg. Pathol. 2012, 5, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Akgul, M.; Williamson, S.R.; Ertoy, D.; Argani, P.; Gupta, S.; Caliò, A.; Reuter, V.; Tickoo, S.; Al-Ahmadie, H.A.; Netto, G.J.; et al. Diagnostic approach in TFE3-rearranged renal cell carcinoma: A multi-institutional international survey. J. Clin. Pathol. 2021, 5, 291–299. [Google Scholar] [CrossRef]
- Kmeid, M.; Akgul, M. TFE3 Rearrangement and Expression in Renal Cell Carcinoma. Int. J. Surg. Pathol. 2022, 1, 10668969221108517. [Google Scholar] [CrossRef]
- Caliò, A.; Marletta, S.; Brunelli, M.; Pedron, S.; Portillo, S.C.; Segala, D.; Bariani, E.; Gobbo, S.; Netto, G.; Martignoni, G. TFE3 and TFEB-rearranged renal cell carcinomas: An immunohistochemical panel to differentiate from common renal cell neoplasms. Virchows Arch. 2022, 6, 877–891. [Google Scholar] [CrossRef]
- Argani, P.; Zhong, M.; Reuter, V.E.; Fallon, J.T.; Epstein, J.I.; Netto, G.J.; Antonescu, C.R. TFE3-Fusion Variant Analysis Defines Specific Clinicopathologic Associations Among Xp11 Translocation Cancers. Am. J. Surg. Pathol. 2016, 6, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Argani, P. MiT family translocation renal cell carcinoma. Semin. Diagn. Pathol. 2015, 2, 103–113. [Google Scholar] [CrossRef]
- Tretiakova, M.S. Chameleon TFE3-translocation RCC and How Gene Partners Can Change Morphology: Accurate Diagnosis Using Contemporary Modalities. Adv. Anat. Pathol. 2022, 3, 131–140. [Google Scholar] [CrossRef]
- Green, W.M.; Yonescu, R.; Morsberger, L.; Morris, K.; Netto, G.J.; Epstein, J.I.; Illei, P.B.; Allaf, M.; Ladanyi, M.; Griffin, C.A.; et al. Utilization of a TFE3 break-apart FISH assay in a renal tumor consultation service. Am. J. Surg. Pathol. 2013, 8, 1150–1163. [Google Scholar] [CrossRef]
- Lee, H.J.; Shin, D.H.; Kim, S.Y.; Hwang, C.S.; Lee, J.H.; Park, W.Y.; Choi, K.U.; Kim, J.Y.; Lee, C.H.; Sol, M.Y.; et al. TFE3 translocation and protein expression in renal cell carcinoma are correlated with poor prognosis. Histopathology 2018, 5, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Zhang, Y.; Mannan, R.; Skala, S.L.; Rangaswamy, R.; Chinnaiyan, A.; Su, F.; Cao, X.; Zelenka-Wang, S.; McMurry, L.; et al. TRIM63 is a sensitive and specific biomarker for MiT family aberration-associated renal cell carcinoma. Mod. Pathol. 2021, 8, 1596–1607. [Google Scholar] [CrossRef]
- Aldera, A.P.; Ramburan, A.; John, J. TFE3-rearranged renal cell carcinoma with osseous metaplasia and indolent behaviour. Urol. Case Rep. 2022, 42, 102041. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Argani, P.; Jungbluth, A.A.; Chen, Y.B.; Tickoo, S.K.; Fine, S.W.; Gopalan, A.; Al-Ahmadie, H.A.; Sirintrapun, S.J.; Sanchez, A.; et al. TFEB Expression Profiling in Renal Cell Carcinomas: Clinicopathologic Correlations. Am. J. Surg. Pathol. 2019, 11, 1445–1461. [Google Scholar] [CrossRef] [PubMed]
- Argani, P.; Reuter, V.E.; Zhang, L.; Sung, Y.S.; Ning, Y.; Epstein, J.I.; Netto, G.J.; Antonescu, C.R. TFEB-amplified Renal Cell Carcinomas: An Aggressive Molecular Subset Demonstrating Variable Melanocytic Marker Expression and Morphologic Heterogeneity. Am. J. Surg. Pathol. 2016, 11, 1484–1495. [Google Scholar] [CrossRef]
- Wyvekens, N.; Rechsteiner, M.; Fritz, C.; Wagner, U.; Tchinda, J.; Wenzel, C.; Kuithan, F.; Horn, L.C.; Moch, H. Histological and molecular characterization of TFEB-rearranged renal cell carcinomas. Virchows Arch. 2019, 5, 625–631. [Google Scholar] [CrossRef]
- Williamson, S.R.; Grignon, D.J.; Cheng, L.; Favazza, L.; Gondim, D.D.; Carskadon, S.; Gupta, N.S.; Chitale, D.A.; Kalyana-Sundaram, S.; Palanisamy, N. Renal Cell Carcinoma with Chromosome 6p Amplification Including the TFEB Gene: A Novel Mechanism of Tumor Pathogenesis? Am. J. Surg. Pathol. 2017, 3, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.R.; Eble, J.N.; Palanisamy, N. Sclerosing TFEB-rearrangement renal cell carcinoma: A recurring histologic pattern. Hum. Pathol. 2017, 62, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Johnson, S.H.; Vasmatzis, G.; Porath, B.; Rustin, J.G.; Rao, P.; Costello, B.A.; Leibovich, B.C.; Thompson, R.H.; Cheville, J.C.; et al. TFEB-VEGFA (6p21.1) co-amplified renal cell carcinoma: A distinct entity with potential implications for clinical management. Mod. Pathol. 2017, 7, 998–1012. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, R.G.; Gorelick, A.N.; Makarov, V.; Blum, K.A.; Silagy, A.W.; Freeman, B.; Chowell, D.; Marcon, J.; Mano, R.; Sanchez, A.; et al. Putative Drivers of Aggressiveness in TCEB1-mutant Renal Cell Carcinoma: An Emerging Entity with Variable Clinical Course. Eur. Urol. Focus. 2021, 2, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, P.; Wang, L.; Wang, J.; Ji, X.; Li, Y.; Shi, H.; Li, Y.; Zhang, W.; Jiang, Y. Analysis of clinicopathological and molecular features of ELOC(TCEB1)-mutant renal cell carcinoma. Pathol. Res. Pract. 2022, 235, 153960. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J. Molecular genetics of clear-cell renal cell carcinoma. J. Clin. Oncol. 2014, 18, 1968–1976. [Google Scholar] [CrossRef]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 8, 860–867. [Google Scholar] [CrossRef]
- Shah, R.B. Renal Cell Carcinoma with Fibromyomatous Stroma-The Whole Story. Adv. Anat. Pathol. 2022, 3, 168–177. [Google Scholar] [CrossRef]
- Hakimi, A.A.; Tickoo, S.K.; Jacobsen, A.; Sarungbam, J.; Sfakianos, J.P.; Sato, Y.; Morikawa, T.; Kume, H.; Fukayama, M.; Homma, Y.; et al. TCEB1-mutated renal cell carcinoma: A distinct genomic and morphological subtype. Mod. Pathol. 2015, 6, 845–853. [Google Scholar] [CrossRef]
- Shah, R.B.; Stohr, B.A.; Tu, Z.J.; Gao, Y.; Przybycin, C.G.; Nguyen, J.; Cox, R.M.; Rashid-Kolvear, F.; Weindel, M.D.; Farkas, D.H.; et al. “Renal Cell Carcinoma with Leiomyomatous Stroma” Harbor Somatic Mutations of TSC1, TSC2, MTOR, and/or ELOC (TCEB1): Clinicopathologic and Molecular Characterization of 18 Sporadic Tumors Supports a Distinct Entity. Am. J. Surg. Pathol. 2020, 5, 571–581. [Google Scholar] [CrossRef]
- Lindner, A.K.; Tulchiner, G.; Seeber, A.; Siska, P.J.; Thurnher, M.; Pichler, R. Targeting strategies in the treatment of fumarate hydratase deficient renal cell carcinoma. Front. Oncol. 2022, 12, 906014. [Google Scholar] [CrossRef]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 12, 1326–1338. [Google Scholar] [CrossRef]
- Singh, N.P.; Vinod, P.K. Integrative analysis of DNA methylation and gene expression in papillary renal cell carcinoma. Mol. Genet. Genom. 2020, 3, 807–824. [Google Scholar] [CrossRef]
- Arts, R.J.; Novakovic, B.; Ter Horst, R.; Carvalho, A.; Bekkering, S.; Lachmandas, E.; Rodrigues, F.; Silvestre, R.; Cheng, S.C.; Wang, S.Y.; et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab. 2016, 6, 807–819. [Google Scholar] [CrossRef]
- Ge, X.; Li, M.; Yin, J.; Shi, Z.; Fu, Y.; Zhao, N.; Chen, H.; Meng, L.; Li, X.; Hu, Z.; et al. Fumarate inhibits PTEN to promote tumorigenesis and therapeutic resistance of type2 papillary renal cell carcinoma. Mol. Cell 2022, 7, 1249–1260.e7. [Google Scholar] [CrossRef]
- Sun, G.; Zhang, X.; Liang, J.; Pan, X.; Zhu, S.; Liu, Z.; Armstrong, C.M.; Chen, J.; Lin, W.; Liao, B.; et al. Integrated Molecular Characterization of Fumarate Hydratase-deficient Renal Cell Carcinoma. Clin. Cancer Res. 2021, 6, 1734–1743. [Google Scholar] [CrossRef]
- Gleeson, J.P.; Nikolovski, I.; Dinatale, R.; Zucker, M.; Knezevic, A.; Patil, S.; Ged, Y.; Kotecha, R.R.; Shapnik, N.; Murray, S.; et al. Comprehensive Molecular Characterization and Response to Therapy in Fumarate Hydratase-Deficient Renal Cell Carcinoma. Clin. Cancer Res. 2021, 10, 2910–2919. [Google Scholar] [CrossRef]
- Yu, Y.F.; He, S.M.; Wu, Y.C.; Xiong, S.W.; Shen, Q.; Li, Y.Y.; Yang, F.; He, Q.; Li, X.S. Clinicopathological features and prognosis of fumarate hydratase deficient renal cell carcinoma. Beijing Da Xue Xue Bao Yi Xue Ban 2021, 4, 640–646. [Google Scholar]
- Lau, H.D.; Chan, E.; Fan, A.C.; Kunder, C.A.; Williamson, S.R.; Zhou, M.; Idrees, M.T.; Maclean, F.M.; Gill, A.J.; Kao, C.S. A Clinicopathologic and Molecular Analysis of Fumarate Hydratase-deficient Renal Cell Carcinoma in 32 Patients. Am. J. Surg. Pathol. 2020, 1, 98–110. [Google Scholar] [CrossRef]
- Muller, M.; Guillaud-Bataille, M.; Salleron, J.; Genestie, C.; Deveaux, S.; Slama, A.; de Paillerets, B.B.; Richard, S.; Benusiglio, P.R.; Ferlicot, S. Pattern multiplicity and fumarate hydratase (FH)/S-(2-succino)-cysteine (2SC) staining but not eosinophilic nucleoli with perinucleolar halos differentiate hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinomas from kidney tumors without FH gene alteration. Mod. Pathol. 2018, 6, 974–983. [Google Scholar]
- Chen, Y.B.; Brannon, A.R.; Toubaji, A.; Dudas, M.E.; Won, H.H.; Al-Ahmadie, H.A.; Fine, S.W.; Gopalan, A.; Frizzell, N.; Voss, M.H.; et al. Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cancer: Recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am. J. Surg. Pathol. 2014, 5, 627–637. [Google Scholar] [CrossRef]
- Nikolovski, I.; Carlo, M.I.; Chen, Y.B.; Vargas, H.A. Imaging features of fumarate hydratase-deficient renal cell carcinomas: A retrospective study. Cancer Imaging 2021, 1, 24. [Google Scholar] [CrossRef]
- Wu, G.; Liu, G.; Wang, J.; Pan, S.; Luo, Y.; Xu, Y.; Kong, W.; Sun, P.; Xu, J.; Xue, W.; et al. MR Spectroscopy for Detecting Fumarate Hydratase Deficiency in Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome. Radiology 2022, 3, 631–639. [Google Scholar] [CrossRef]
- Liu, Y.; Dong, Y.; Gu, Y.; Xu, H.; Fan, Y.; Li, X.; Dong, L.; Zhou, L.; Yang, X.; Wang, C. GATA3 aids in distinguishing fumarate hydratase-deficient renal cell carcinoma from papillary renal cell carcinoma. Ann. Diagn. Pathol. 2022, 60, 152007. [Google Scholar] [CrossRef]
- Grubb, R.L.; Franks, M.E.; Toro, J.; Middelton, L.; Choyke, L.; Fowler, S.; Torres-Cabala, C.; Glenn, G.M.; Choyke, P.; Merino, M.J.; et al. Hereditary leiomyomatosis and renal cell cancer: A syndrome associated with an aggressive form of inherited renal cancer. J. Urol. 2007, 6, 2074–2079. [Google Scholar] [CrossRef]
- Choi, Y.; Keam, B.; Kim, M.; Yoon, S.; Kim, D.; Choi, J.G.; Seo, J.Y.; Park, I.; Lee, J.L. Bevacizumab Plus Erlotinib Combination Therapy for Advanced Hereditary Leiomyomatosis and Renal Cell Carcinoma-Associated Renal Cell Carcinoma: A Multicenter Retrospective Analysis in Korean Patients. Cancer Res. Treat. 2019, 4, 1549–1556. [Google Scholar] [CrossRef]
- Xu, Y.; Kong, W.; Cao, M.; Wang, J.; Wang, Z.; Zheng, L.; Wu, X.; Cheng, R.; He, W.; Yang, B.; et al. Genomic Profiling and Response to Immune Checkpoint Inhibition plus Tyrosine Kinase Inhibition in FH-Deficient Renal Cell Carcinoma. Eur. Urol. 2023, 2, 163–172. [Google Scholar] [CrossRef]
- Williamson, S.R.; Eble, J.N.; Amin, M.B.; Gupta, N.S.; Smith, S.C.; Sholl, L.M.; Montironi, R.; Hirsch, M.S.; Hornick, J.L. Succinate dehydrogenase-deficient renal cell carcinoma: Detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod. Pathol. 2015, 1, 80–94. [Google Scholar] [CrossRef]
- Wang, G.; Rao, P. Succinate Dehydrogenase-Deficient Renal Cell Carcinoma: A Short Review. Arch. Pathol. Lab. Med. 2018, 10, 1284–1288. [Google Scholar] [CrossRef]
- Tsai, T.H.; Lee, W.Y. Succinate Dehydrogenase-Deficient Renal Cell Carcinoma. Arch. Pathol. Lab. Med. 2019, 5, 643–647. [Google Scholar] [CrossRef]
- Gill, A.J. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology 2018, 1, 106–116. [Google Scholar] [CrossRef]
- Gill, A.J.; Hes, O.; Papathomas, T.; Šedivcová, M.; Tan, P.H.; Agaimy, A.; Andresen, P.A.; Kedziora, A.; Clarkson, A.; Toon, C.W.; et al. Succinate dehydrogenase (SDH)-deficient renal carcinoma: A morphologically distinct entity: A clinicopathologic series of 36 tumors from 27 patients. Am. J. Surg. Pathol. 2014, 12, 1588–1602. [Google Scholar] [CrossRef]
- Aggarwal, R.K.; Luchtel, R.A.; Machha, V.; Tischer, A.; Zou, Y.; Pradhan, K.; Ashai, N.; Ramachandra, N.; Albanese, J.M.; Yang, J.I.; et al. Functional succinate dehydrogenase deficiency is a common adverse feature of clear cell renal cancer. Proc. Natl. Acad. Sci. USA 2021, 39, e2106947118. [Google Scholar] [CrossRef]
- Sun, A.; Liu, Z.; Wang, T.; Xing, J. Succinate dehydrogenase-deficient renal cell carcinoma: A case report and review of the literature. Asian J. Surg. 2021, 4, 692–693. [Google Scholar] [CrossRef]
- Gill, A.J.; Pachter, N.S.; Chou, A.; Young, B.; Clarkson, A.; Tucker, K.M.; Winship, I.M.; Earls, P.; Benn, D.E.; Robinson, B.G.; et al. Renal tumors associated with germline SDHB mutation show distinctive morphology. Am. J. Surg. Pathol. 2011, 10, 1578–1585. [Google Scholar] [CrossRef]
- Paik, J.Y.; Toon, C.W.; Benn, D.E.; High, H.; Hasovitz, C.; Pavlakis, N.; Clifton-Bligh, R.J.; Gill, A.J. Renal carcinoma associated with succinate dehydrogenase B mutation: A new and unique subtype of renal carcinoma. J. Clin. Oncol. 2014, 6, e10–e13. [Google Scholar] [CrossRef]
- Linehan, W.M.; Ricketts, C.J. The metabolic basis of kidney cancer. Semin. Cancer Biol. 2013, 1, 46–55. [Google Scholar] [CrossRef]
- Yu, W.; Wang, Y.; Jiang, Y.; Zhang, W.; Li, Y. Genetic analysis and clinicopathological features of ALK-rearranged renal cell carcinoma in a large series of resected Chinese renal cell carcinoma patients and literature review. Histopathology 2017, 1, 53–62. [Google Scholar] [CrossRef]
- Jeanneau, M.; Gregoire, V.; Desplechain, C.; Escande, F.; Tica, D.P.; Aubert, S.; Leroy, X. ALK rearrangements-associated renal cell carcinoma (RCC) with unique pathological features in an adult. Pathol. Res. Pract. 2016, 11, 1064–1066. [Google Scholar] [CrossRef]
- Gorczynski, A.; Czapiewski, P.; Korwat, A.; Budynko, L.; Prelowska, M.; Okon, K.; Biernat, W. ALK-rearranged renal cell carcinomas in Polish population. Pathol. Res. Pract. 2019, 12, 152669. [Google Scholar] [CrossRef]
- Hang, J.F.; Chung, H.J.; Pan, C.C. ALK-rearranged renal cell carcinoma with a novel PLEKHA7-ALK translocation and metanephric adenoma-like morphology. Virchows Arch. 2020, 6, 921–929. [Google Scholar] [CrossRef]
- Kuroda, N.; Sugawara, E.; Kusano, H.; Yuba, Y.; Yorita, K.; Takeuchi, K. A review of ALK-rearranged renal cell carcinomas with a focus on clinical and pathobiological aspects. Pol. J. Pathol. 2018, 2, 109–113. [Google Scholar] [CrossRef]
- Debelenko, L.V.; Daw, N.; Shivakumar, B.R.; Huang, D.; Nelson, M.; Bridge, J.A. Renal cell carcinoma with novel VCL-ALK fusion: New representative of ALK-associated tumor spectrum. Mod. Pathol. 2011, 3, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Sukov, W.R.; Hodge, J.C.; Lohse, C.M.; Akre, M.K.; Leibovich, B.C.; Thompson, R.H.; Cheville, J.C. ALK alterations in adult renal cell carcinoma: Frequency, clinicopathologic features and outcome in a large series of consecutively treated patients. Mod. Pathol. 2012, 11, 1516–1525. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.; Bazhenova, L.; Camidge, D.R.; Solomon, B.J.; Herman, J.; Kain, T.; Bang, Y.J.; Kwak, E.L.; Shaw, A.T.; Salgia, R.; et al. Rapid and dramatic radiographic and clinical response to an ALK inhibitor (crizotinib, PF02341066) in an ALK translocation-positive patient with non-small cell lung cancer. J. Thorac. Oncol. 2010, 12, 2044–2046. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Nakajima, T.; Takeuchi, K.; Soda, M.; Mano, H.; Iizasa, T.; Matsui, Y.; Yoshino, M.; Shingyoji, M.; Itakura, M.; et al. ALK fusion gene positive lung cancer and 3 cases treated with an inhibitor for ALK kinase activity. Lung Cancer 2012, 1, 66–72. [Google Scholar] [CrossRef]
- Pal, S.K.; Bergerot, P.; Dizman, N.; Bergerot, C.; Adashek, J.; Madison, R.; Chung, J.H.; Ali, S.M.; Jones, J.O.; Salgia, R. Responses to Alectinib in ALK-rearranged Papillary Renal Cell Carcinoma. Eur. Urol. 2018, 1, 124–128. [Google Scholar] [CrossRef]
- Wang, X.; Haswell, J.R.; Roberts, C.W. Molecular pathways: SWI/SNF (BAF) complexes are frequently mutated in cancer--mechanisms and potential therapeutic insights. Clin. Cancer Res. 2014, 1, 21–27. [Google Scholar] [CrossRef]
- Msaouel, P.; Malouf, G.G.; Su, X.; Yao, H.; Tripathi, D.N.; Soeung, M.; Gao, J.; Rao, P.; Coarfa, C.; Creighton, C.J.; et al. Comprehensive Molecular Characterization Identifies Distinct Genomic and Immune Hallmarks of Renal Medullary Carcinoma. Cancer Cell 2020, 5, 720–734.e13. [Google Scholar] [CrossRef]
- Agaimy, A. The expanding family of SMARCB1(INI1)-deficient neoplasia: Implications of phenotypic, biological, and molecular heterogeneity. Adv. Anat. Pathol. 2014, 6, 394–410. [Google Scholar] [CrossRef]
- Hong, A.L.; Tseng, Y.Y.; Wala, J.A.; Kim, W.J.; Kynnap, B.D.; Doshi, M.B.; Kugener, G.; Sandoval, G.J.; Howard, T.P.; Li, J.; et al. Renal medullary carcinomas depend upon SMARCB1 loss and are sensitive to proteasome inhibition. Elife 2019, 8, e44161. [Google Scholar] [CrossRef]
- Al-Daghmin, A.; Gaashan, M.; Haddad, H. Atypical presentation of renal medullary carcinoma: A case report and review of the literature. Urol. Case Rep. 2018, 22, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Holland, P.; Merrimen, J.; Pringle, C.; Wood, L.A. Renal medullary carcinoma and its association with sickle cell trait: A case report and literature review. Curr. Oncol. 2020, 1, e53–e56. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Hong, A.L. Recent Advances in Renal Medullary Carcinoma. Int. J. Mol. Sci. 2022, 13, 7097. [Google Scholar] [CrossRef] [PubMed]
- Beckermann, K.E.; Sharma, D.; Chaturvedi, S.; Msaouel, P.; Abboud, M.R.; Allory, Y.; Bourdeaut, F.; Calderaro, J.; de Cubas, A.A.; Derebail, V.K.; et al. Renal Medullary Carcinoma: Establishing Standards in Practice. J. Oncol. Pract. 2017, 7, 414–421. [Google Scholar] [CrossRef]
- Lopez-Beltran, A.; Cheng, L.; Raspollini, M.R.; Montironi, R. SMARCB1/INI1 Genetic Alterations in Renal Medullary Carcinomas. Eur. Urol. 2016, 6, 1062–1064. [Google Scholar] [CrossRef]
- Scarpelli, M.; Mazzucchelli, R.; Lopez-Beltran, A.; Cheng, L.; De Nictolis, M.; Santoni, M.; Montironi, R. Renal cell carcinoma with rhabdoid features and loss of INI1 expression in an individual without sickle cell trait. Pathology 2014, 7, 653–655. [Google Scholar] [CrossRef]
- Iacovelli, R.; Modica, D.; Palazzo, A.; Trenta, P.; Piesco, G.; Cortesi, E. Clinical outcome and prognostic factors in renal medullary carcinoma: A pooled analysis from 18 years of medical literature. Can. Urol. Assoc. J. 2015, 3–4, E172–E177. [Google Scholar] [CrossRef]
- Wiele, A.J.; Surasi, D.S.; Rao, P.; Sircar, K.; Su, X.; Bathala, T.K.; Shah, A.Y.; Jonasch, E.; Cataldo, V.D.; Genovese, G.; et al. Efficacy and Safety of Bevacizumab Plus Erlotinib in Patients with Renal Medullary Carcinoma. Cancers 2021, 9, 2170. [Google Scholar] [CrossRef]
- Forrest, S.J.; Al-Ibraheemi, A.; Doan, D.; Ward, A.; Clinton, C.M.; Putra, J.; Pinches, R.S.; Kadoch, C.; Chi, S.N.; DuBois, S.G.; et al. Genomic and Immunologic Characterization of INI1-Deficient Pediatric Cancers. Clin. Cancer Res. 2020, 12, 2882–2890. [Google Scholar] [CrossRef]
- Ngo, C.; Postel-Vinay, S. Immunotherapy for SMARCB1-Deficient Sarcomas: Current Evidence and Future Developments. Biomedicines 2022, 3, 650. [Google Scholar] [CrossRef]


{kind=link}
| Molecularly Defined Renal Cell Carcinoma Types | TFE3-Rearranged Renal Cell Carcinomas | TFEB-Altered Renal Cell Carcinomas | Elongin C (ELOC, Formerly TCEB1)-Mutated Renal Cell Carcinoma | Fumarate Hydratase-Deficient Renal Cell Carcinoma | Succinate Dehydrogenase-Deficient Renal Cell Carcinoma | ALK-Rearranged Renal Cell Carcinomas | SMARCB1-Deficient Renal Medullary Carcinoma |
|---|---|---|---|---|---|---|---|
| Mutated genes | Transcription factor binding to IGHM enhancer 3 (TFE3) | Transcription factor EB (TFEB) | Elongin C (ELOC) | Fumarate hydratase (FH) gene | Succinate dehydrogenase (SDH) | Anaplastic lymphoma kinase (ALK) | Subfamily B member 1 (SMARCB1) |
| Location of genes | Xp11.23 | 6p21 | 8q21.11 | 1q43 | SDHA: 5p15 SDHB: lp35-p36.1 SDHC: 1q21 SDHD: 11q23 | 2p23 | 22q11.2 |
| Prevalence age | Childhood | Childhood | Middle and old age | Adult | All ages | Childhood | Teenage |
| Clinical Syndromes | None | None | None | Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) | SDH-deficient tumor syndrome | None | Rhabdoid tumor predisposition syndrome; familial schwannomatosis syndrome |
| Chaperone genes | ASPL, PRCC, SFPQ, CLTC, PARP14, RBM10, NONO, MED15 | MALAT1, CLTC, KHDRBS2, CADM2 | None | None | None | VCL, TPM3, EML4, STRN, HOOK1 | None |
| Mode of inheritance | Dominant inheritance | Dominant inheritance | Dominant inheritance | Dominant inheritance | Dominant inheritance | Dominant inheritance | Dominant inheritance |
| Morphological characteristics | Transparent eosinophils; papillary architecture and psammoma bodies under the microscope | TFEB-translocated RCC: the biphasic growth pattern consisting of large and small tumor cells; smaller cells around the basement membrane-like structures; extensive hyalinization; papillary architecture; clear cell morphology. TFEB-amplified RCC: above pattern was less common | A clear cellular morphology under the microscope; thick fibromuscular bands; branching glandular vesicular; tubular structures | The papillary type or solid, tubulocystic, sieve-like type; abundant eosinophilic granulocytes, perinuclear halo | Cuboidal tumor cells, nested or tubular growth pattern. Characteristic morphology: the presence of vesicles or flocculent inclusions in the cytoplasm | ALK-rearranged RCC with VCL as a fusion gene: sickle-cell trait; eosinophilic granulocytic stroma; cytoplasmic lumen. Other ALK-rearranged RCC: similar to PRCC; consist of abundant intracellular and extracellular mucins; eosinophilic granuloplasm | At a high grade at the time of detection; infiltrative growth; sieve or reticular appearance |
| Ancillary test (IHC, FISH) | Positive: PAX8 (100%); TFE3 (95%); CD10 (89%); achromatase (82%). Negative: cytokeratin 7 (CK7); carbonic anhydrase 9 (CA9); GATA3 | Positive: histone K; Melan-A TFEB-amplified RCC: diffusely or patchily positive when tested for TFEB levels | Positive: CK7; ELOC; CA9; CD10; ELOC in the nucleus. | Positive: PAX8; succinate dehydrogenase B abnormal succinate semicarbonate (2SC)S-(2-succino)-cysteine. Negative: FH; CK7; TFE3 | Positive: PAX8; epithelial membrane antigen (EMA). Negative: SDHB; CK7; CD117; histone K; TFE3; HMB45. SDHA-deficient RCC showed negativity for SDHA | Positive: PAX7; CK10; AMACR; CD3; cytokeratin; ALK. Negative: carbonic anhydrase IX; TFE45; histone enzyme K; Melan A; HMB45 | Negative: SMARCB1 |
| Oncological behavior and prognosis | May develop metastases within 20–30 years after diagnosis | TFEB-amplified RCC had higher tumor aggressiveness than TFEB-rearranged tumors. The 5-year survival rate for TFEB-amplified RCC was 48% | Has an aggressive oncological behavior | Have highly staged or distant metastases when diagnosed | Most cases are low grade and have a good prognosis with a low probability of metastasis | ALK-rearranged RCC with VCL as a fusion gene: no recurrence or distant metastasis. Other ALK-rearranged RCC: more aggressive clinical course | Often found at an advanced stage or with distant metastases; highly aggressive nature of the tumor. Average overall survival: 6–8 months |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, X.; Tan, C.; Zhu, G. Clinical Characteristics of Molecularly Defined Renal Cell Carcinomas. Curr. Issues Mol. Biol. 2023, 45, 4763-4777. https://doi.org/10.3390/cimb45060303
Hu X, Tan C, Zhu G. Clinical Characteristics of Molecularly Defined Renal Cell Carcinomas. Current Issues in Molecular Biology. 2023; 45(6):4763-4777. https://doi.org/10.3390/cimb45060303
Chicago/Turabian StyleHu, Xinfeng, Congzhu Tan, and Guodong Zhu. 2023. "Clinical Characteristics of Molecularly Defined Renal Cell Carcinomas" Current Issues in Molecular Biology 45, no. 6: 4763-4777. https://doi.org/10.3390/cimb45060303
APA StyleHu, X., Tan, C., & Zhu, G. (2023). Clinical Characteristics of Molecularly Defined Renal Cell Carcinomas. Current Issues in Molecular Biology, 45(6), 4763-4777. https://doi.org/10.3390/cimb45060303