
A Novel Mutation in Sacsin, p.Val1335IIe, May Cause Late-Onset Sacsinopathy Due to Haploinsufficiency

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Case Report
2.2. Genetic Analysis
2.3. In Silico Analysis
2.4. Assessment of Protein Concentrations and mRNA Expression
2.5. Statistical Analysis
3. Results
3.1. Genetic Findings
3.2. Measurement of Protein and Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bouchard, J.P.; Barbeau, A.; Bouchard, R.; Bouchard, R.W. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can. J. Neurol. Sci. 1978, 5, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, J.P.; Barbeau, A.; Bouchard, R.; Bouchard, R.W. Electromyography and nerve conduction studies in Friedreich’s ataxia and autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Can. J. Neurol. Sci. 1979, 6, 185–189. [Google Scholar] [CrossRef]
- Xiromerisiou, G.; Dadouli, K.; Marogianni, C.; Provatas, A.; Ntellas, P.; Rikos, D.; Stathis, P.; Georgouli, D.; Loules, G.; Zamanakou, M.; et al. A novel homozygous SACS mutation identified by whole exome sequencing-genotype phenotype correlations of all published cases. J. Mol. Neurosci. 2020, 70, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Kwon, K.Y.; Huh, K.; Eun, B.L.; Yoo, H.W.; Kamsteeg, E.J.; Scheffer, H.; Koh, S.B. A Probable Korean Case of Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. Can. J. Neurol. Sci. 2015, 42, 271–273. [Google Scholar] [CrossRef]
- Bong, J.B.; Kim, S.W.; Lee, S.T.; Choi, J.R.; Shin, H.Y. Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. J. Korean Neurol. Assoc. 2019, 37, 69–72. [Google Scholar] [CrossRef]
- Kang, J.J.; Lee, H.M.; Ki, C.S.; Oh, S.Y. Ciliary Ganglioplegic Migraine Associated with SACS Mutation. J. Ophthalmol. Vis. Sci. 2021, 6, 1045. [Google Scholar]
- Vermeer, S.; van de Warrenburg, B.P.; Kamsteeg, E.J.; Brais, B.; Synofzik, M. ARSACS. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 9 December 2003. [Google Scholar]
- Breckpot, J.; Takiyama, Y.; Thienpont, B.; Van Vooren, S.; Vermeesch, J.R.; Ortibus, E.; Devriendt, K. A novel genomic disorder: A deletion of the SACS gene leading to spastic ataxia of Charlevoix-Saguenay. Eur. J. Hum. Genet. 2008, 16, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, W.M.; Egger, J.I.; Ahmed, A.I.; Kremer, B.P.; Vermeer, S.; van de Warrenburg, B.P. Cerebellar cognitive affective syndrome and autosomal recessive spastic ataxia of charlevoix-saguenay: A report of two male sibs. Psychopathology 2012, 45, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Soehn, A.S.; Gburek-Augustat, J.; Schicks, J.; Karle, K.N.; Schüle, R.; Haack, T.B.; Schöning, M.; Biskup, S.; Rudnik-Schöneborn, S.; et al. Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): Expanding the genetic, clinical and imaging spectrum. Orphanet J. Rare Dis. 2013, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, D.A.; Michael, G.J.; Vermeulen, E.G.; Prodromou, N.V.; Webb, T.R.; Gallo, J.M.; Cheetham, M.E.; Nicoll, W.S.; Blatch, G.L.; Chapple, J.P. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum. Mol. Genet. 2009, 18, 1556–1565. [Google Scholar] [CrossRef]
- Muchowski, P.J.; Wacker, J.L. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 2005, 6, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Dabbaghizadeh, A.; Paré, A.; Cheng-Boivin, Z.; Dagher, R.; Minotti, S.; Dicaire, M.J.; Brais, B.; Young, J.C.; Durham, H.D.; Gentil, B.J. The J domain of sacsin disrupts intermediate filament assembly. Int. J. Mol. Sci. 2022, 23, 15742. [Google Scholar] [CrossRef]
- Pyle, A.; Griffin, H.; Duff, J.; Bennett, S.; Zwolinski, S.; Smertenko, T.; Yu-Wai Man, P.; Santibanez-Koref, M.; Horvath, R.; Chinnery, P.F. Late-onset sacsinopathy diagnosed by exome sequencing and comparative genomic hybridization. J. Neurogenet. 2013, 27, 176–182. [Google Scholar] [CrossRef]
- Cheng, H.L.; Shao, Y.R.; Dong, Y.; Dong, H.L.; Yang, L.; Ma, Y.; Shen, Y.; Wu, Z.Y. Genetic spectrum and clinical features in a cohort of Chinese patients with autosomal recessive cerebellar ataxias. Transl. Neurodegener. 2021, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, F.A.; Canafoglia, L.; Aljaafari, D.; Muona, M.; Lehesjoki, A.E.; Berkovic, S.F.; Franceschetti, S.; Andrade, D.M. Progressive myoclonus epilepsy associated with SACS gene mutations. Neurol. Genet. 2016, 2, e83. [Google Scholar] [CrossRef] [PubMed]
- Srikajon, J.; Pitakpatapee, Y.; Limwongse, C.; Chirapapaisan, N.; Srivanitchapoom, P. Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS) in a Thai Patient: The Classic Clinical Manifestations, Funduscopic Feature, and Brain Imaging Findings with a Novel Mutation in the SACS Gene. Tremor Other Hyperkinetic Mov. 2020, 10, 1. [Google Scholar] [CrossRef]
- Haga, R.; Miki, Y.; Funamizu, Y.; Kon, T.; Suzuki, C.; Ueno, T.; Nishijima, H.; Arai, A.; Tomiyama, M.; Shimazaki, H.; et al. Novel compound heterozygous mutations of the SACS gene in autosomal recessive spastic ataxia of Charlevoix-Saguenay. Clin. Neurol. Neurosurg. 2012, 114, 746–747. [Google Scholar] [CrossRef]
- Larivière, R.; Sgarioto, N.; Márquez, B.T.; Gaudet, R.; Choquet, K.; McKinney, R.A.; Watt, A.J.; Brais, B. Sacs R272C missense homozygous mice develop an ataxia phenotype. Mol. Brain 2019, 12, 19. [Google Scholar] [CrossRef]
- Engert, J.C.; Bérubé, P.; Mercier, J.; Doré, C.; Lepage, P.; Ge, B.; Bouchard, J.P.; Mathieu, J.; Melançon, S.B.; Schalling, M.; et al. ARSACS, a spastic ataxia common in northeastern Québec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat. Genet. 2000, 24, 120–125. [Google Scholar] [CrossRef]
- Thiffault, I.; Dicaire, M.J.; Tetreault, M.; Huang, K.N.; Demers-Lamarche, J.; Bernard, G.; Duquette, A.; Larivière, R.; Gehring, K.; Montpetit, A.; et al. Diversity of ARSACS mutations in French-Canadians. Can. J. Neurol. Sci. 2013, 40, 61–66. [Google Scholar] [CrossRef]
- Longo, F.; De Ritis, D.; Miluzio, A.; Fraticelli, D.; Baets, J.; Scarlato, M.; Santorelli, F.M.; Biffo, S.; Maltecca, F. Assessment of Sacsin Turnover in Patients With ARSACS: Implications for Molecular Diagnosis and Pathogenesis. Neurology 2021, 97, e2315–e2327. [Google Scholar] [CrossRef]
- Del Bondio, A.; Longo, F.; De Ritis, D.; Spirito, E.; Podini, P.; Brais, B.; Bachi, A.; Quattrini, A.; Maltecca, F. Restoring calcium homeostasis in Purkinje cells arrests neurodegeneration and neuroinflammation in the ARSACS mouse model. JCI Insight 2023, 8, e163576. [Google Scholar] [CrossRef] [PubMed]
- Lariviere, R.; Gaudet, R.; Gentil, B.J.; Girard, M.; Conte, T.C.; Minotti, S.; Leclerc-Desaulniers, K.; Gehring, K.; McKinney, R.A.; Shoubridge, E.A.; et al. Sacs knockout mice present pathophysiological defects underlying autosomal recessive spastic ataxia of Charlevoix-Saguenay. Hum. Mol. Genet. 2015, 24, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Baets, J.; Deconinck, T.; Smets, K.; Goossens, D.; Van den Bergh, P.; Dahan, K.; Schmedding, E.; Santens, P.; Rasic, V.M.; Van Damme, P.; et al. Mutations in SACS cause atypical and late-onset forms of ARSACS. Neurology 2010, 75, 1181–1188. [Google Scholar] [CrossRef] [PubMed]


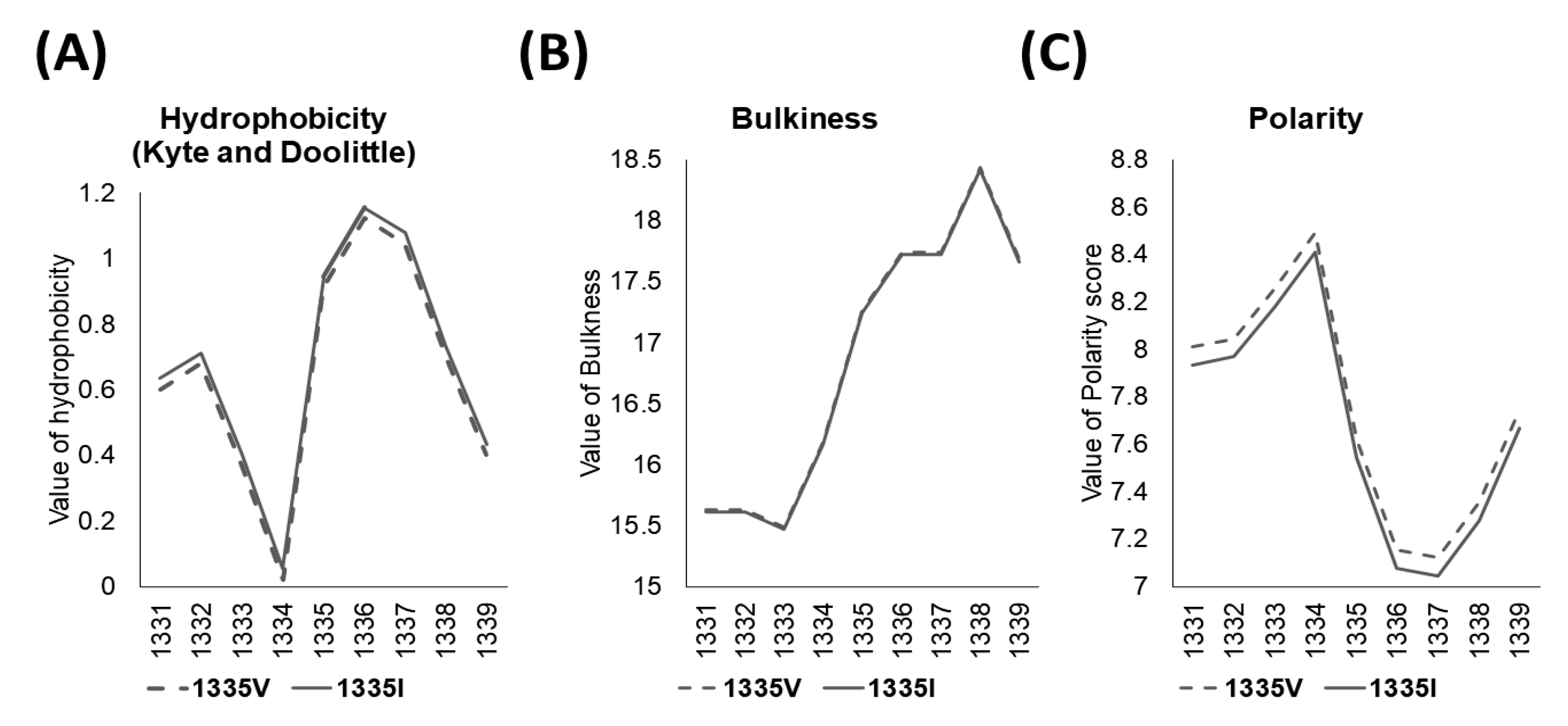
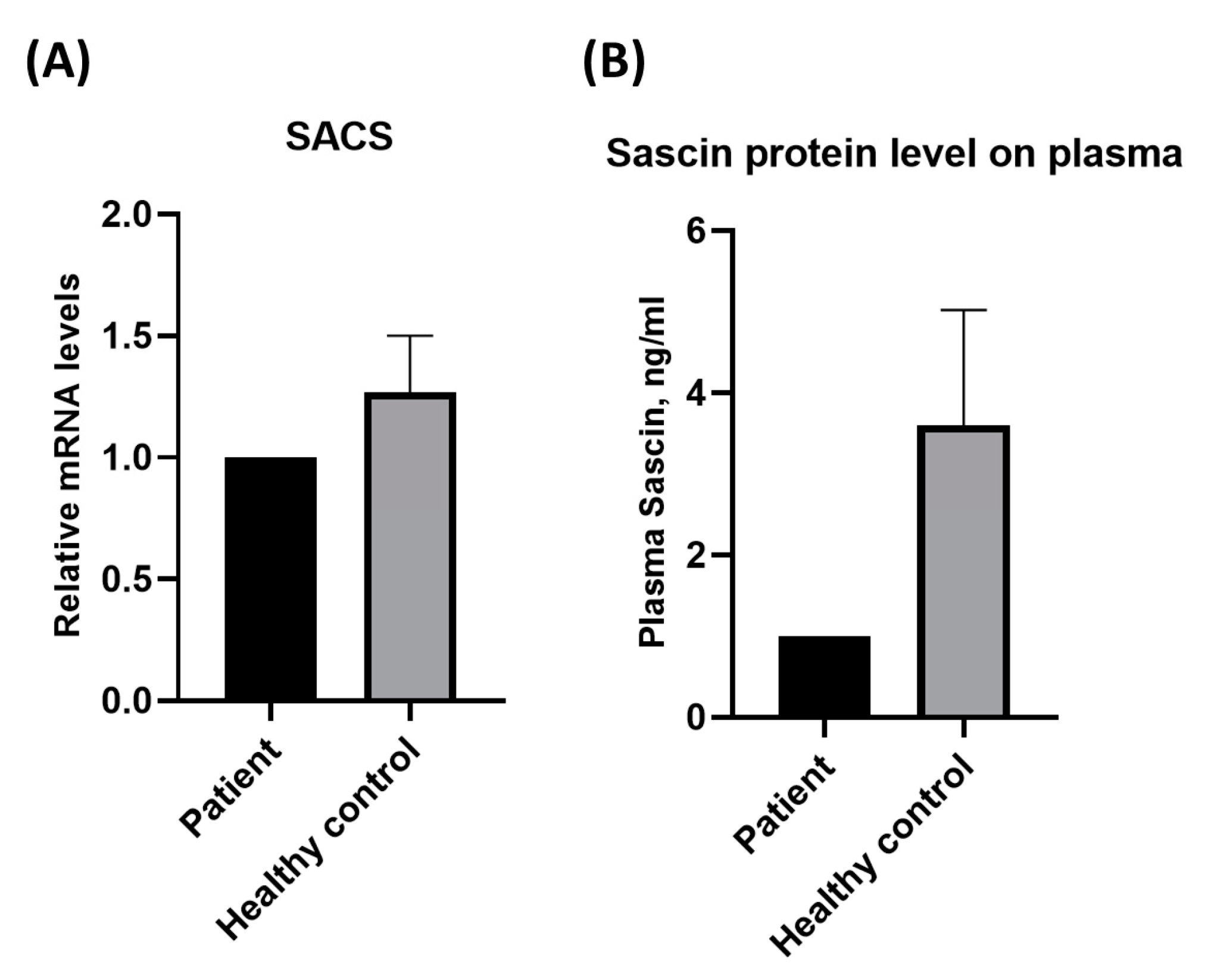

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age/Sex | Age of Onset | Clinical Symptoms at Presentation | Cerebellar Ataxia | Spasticity | Polyneuropathy | Muscle Weakness/Atrophy | Ambulation | Brain MRI | Cervical Spine MRI |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 51/M | 51 | Weakness in the left distal upper limb | Yes | Yes | No | Progressed from left to both sides | Wheelchair-bound since age 53 | Arachnoid cyst, otherwise normal | Central canal stenosis in C3/4 |
| Predictive Algorithms | Score | Judgment |
|---|---|---|
| MutationTaster | 1.000 | Disease-causing |
| Polyphen-2 (Hum VAR) | 0.858 | Probably damaging |
| PROVEAN | −0.322 | Neutral |
| SIFT | 0.12 | Tolerated |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Ryoo, N.; Park, Y.H.; Bagyinszky, E.; An, S.S.A.; Kim, S. A Novel Mutation in Sacsin, p.Val1335IIe, May Cause Late-Onset Sacsinopathy Due to Haploinsufficiency. Curr. Issues Mol. Biol. 2023, 45, 9917-9925. https://doi.org/10.3390/cimb45120619
Kim D, Ryoo N, Park YH, Bagyinszky E, An SSA, Kim S. A Novel Mutation in Sacsin, p.Val1335IIe, May Cause Late-Onset Sacsinopathy Due to Haploinsufficiency. Current Issues in Molecular Biology. 2023; 45(12):9917-9925. https://doi.org/10.3390/cimb45120619
Chicago/Turabian StyleKim, Danyeong, Nayoung Ryoo, Young Ho Park, Eva Bagyinszky, Seong Soo Alexander An, and SangYun Kim. 2023. "A Novel Mutation in Sacsin, p.Val1335IIe, May Cause Late-Onset Sacsinopathy Due to Haploinsufficiency" Current Issues in Molecular Biology 45, no. 12: 9917-9925. https://doi.org/10.3390/cimb45120619
APA StyleKim, D., Ryoo, N., Park, Y. H., Bagyinszky, E., An, S. S. A., & Kim, S. (2023). A Novel Mutation in Sacsin, p.Val1335IIe, May Cause Late-Onset Sacsinopathy Due to Haploinsufficiency. Current Issues in Molecular Biology, 45(12), 9917-9925. https://doi.org/10.3390/cimb45120619