
Cucurbitacin E Exerts Anti-Proliferative Activity via Promoting p62-Dependent Apoptosis in Human Non-Small-Cell Lung Cancer A549 Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Cell Viability and Sulforhodamine B (SRB) Assay
2.3. Lentivirus System
2.4. Flow Cytometry
2.5. Western Blot
2.6. Statistics and Data Analysis
3. Results
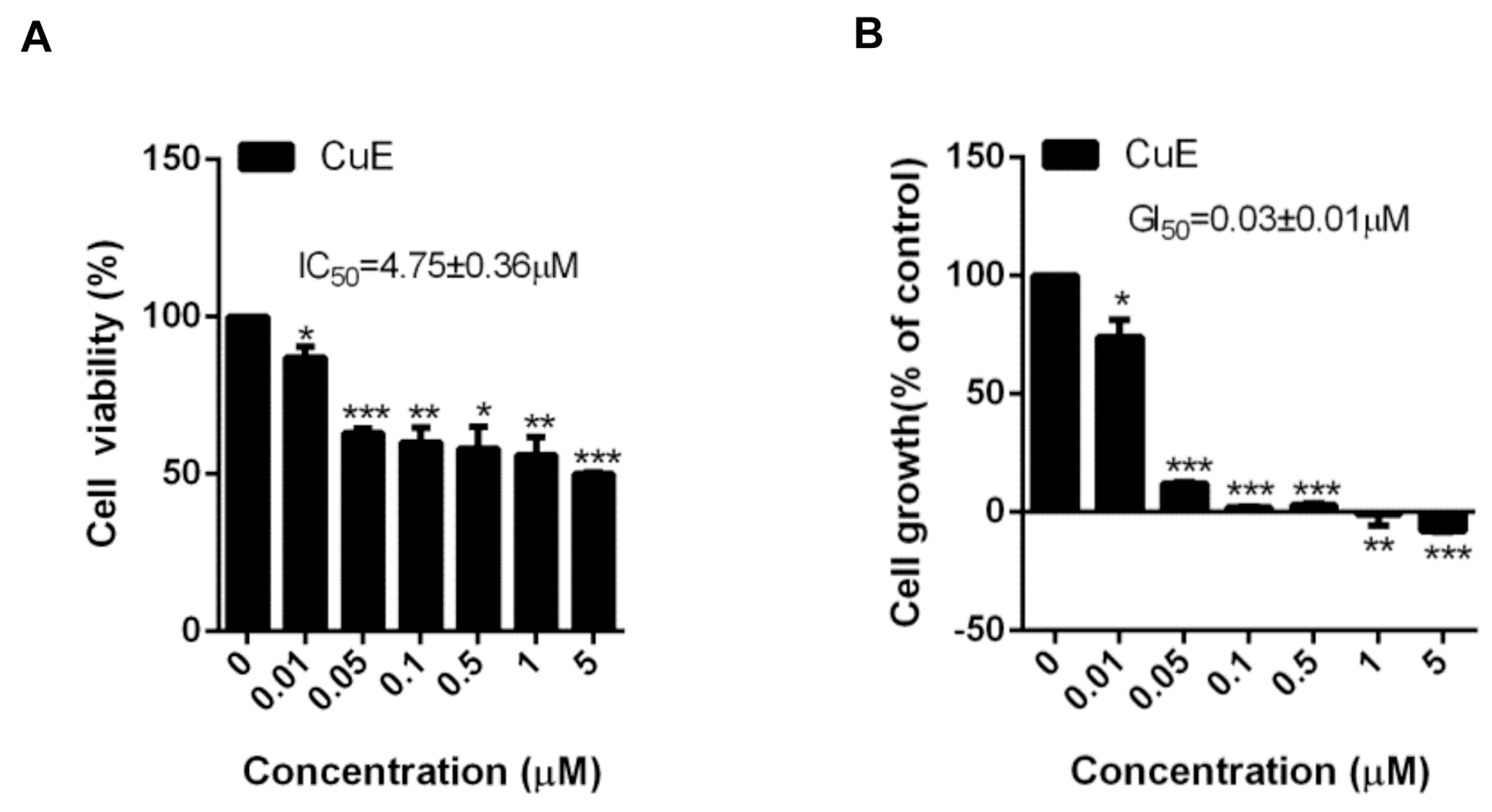
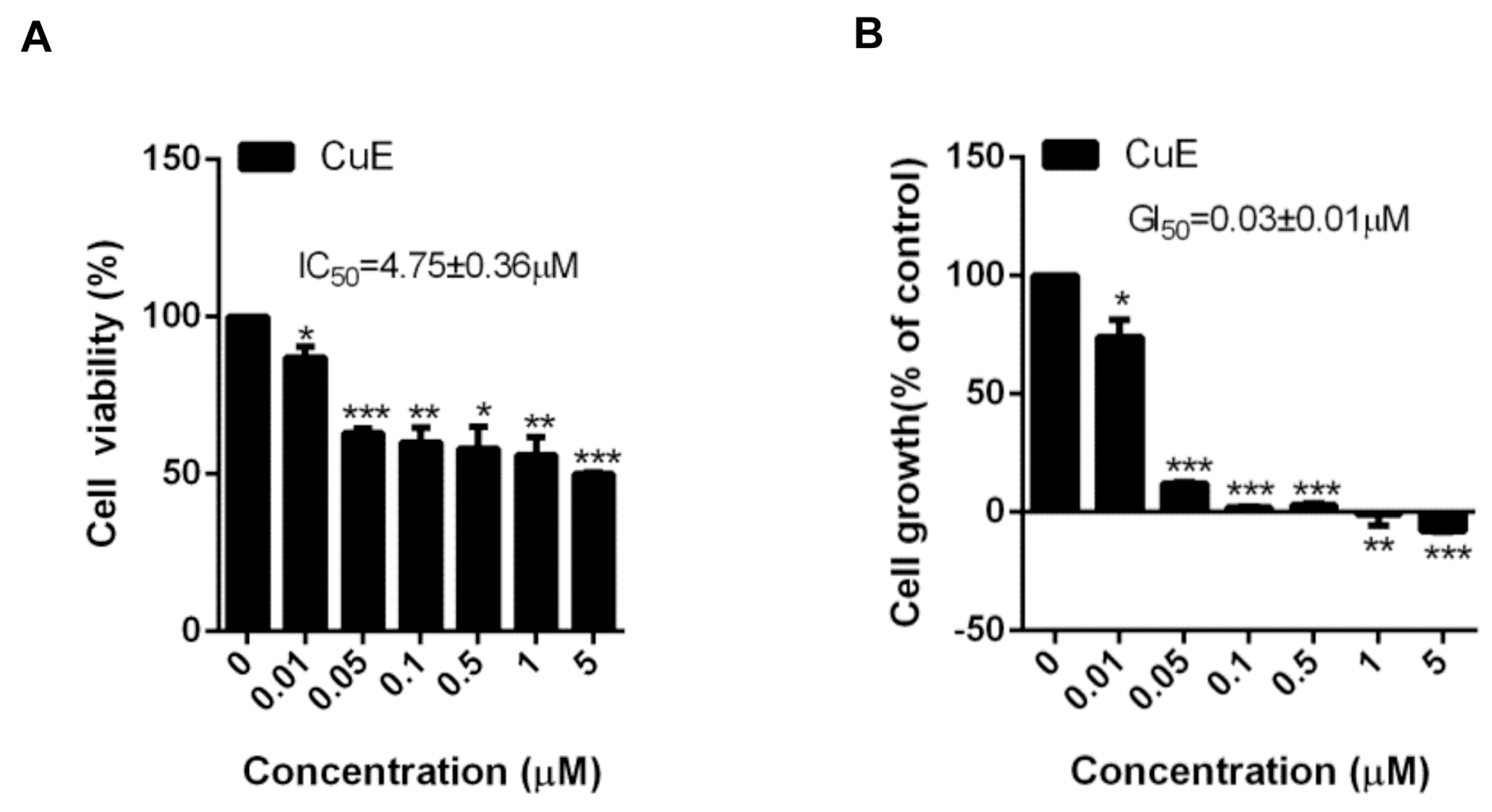
3.1. The Effects of Cu E on the Viability and Proliferation in A549 Cells
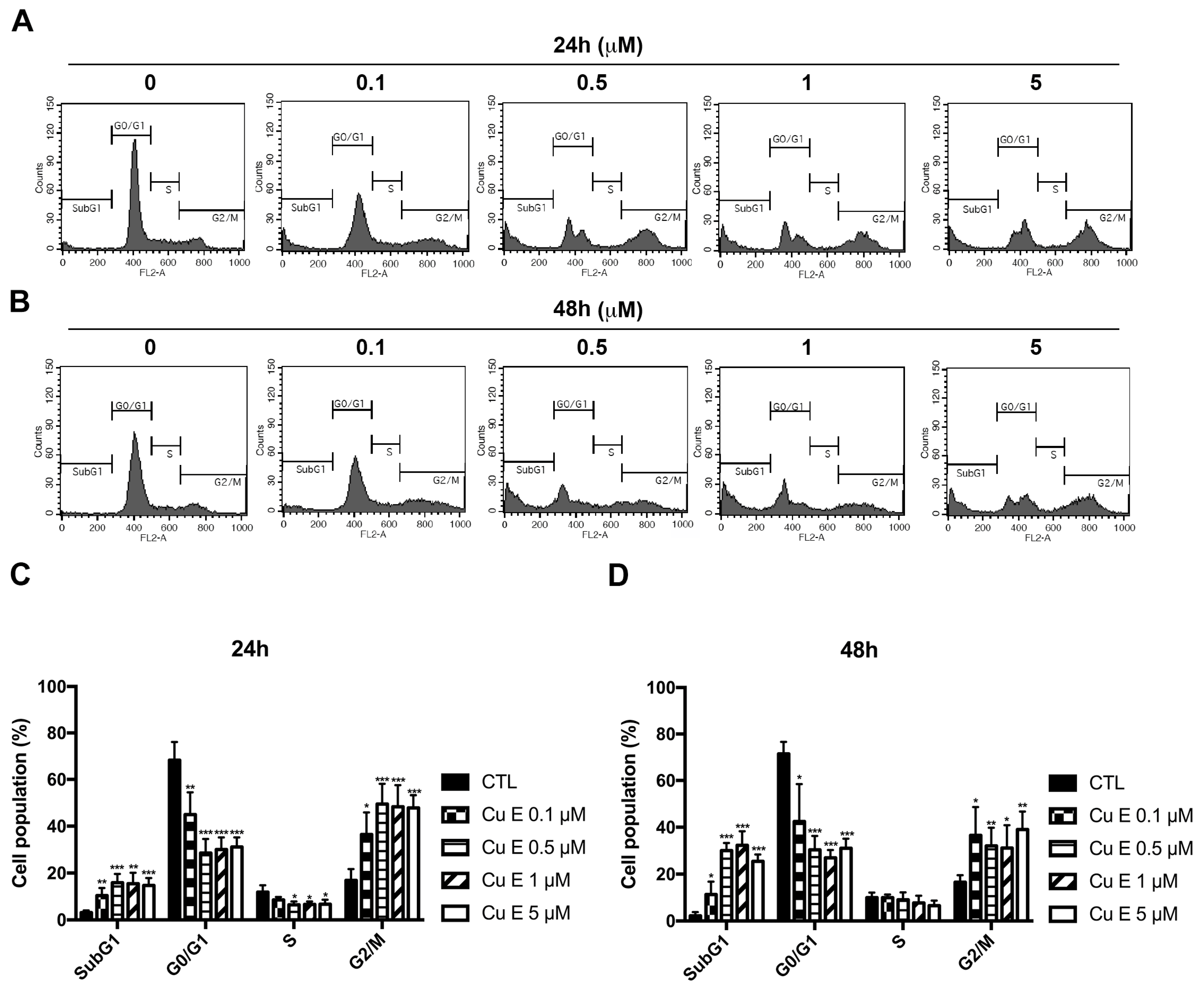
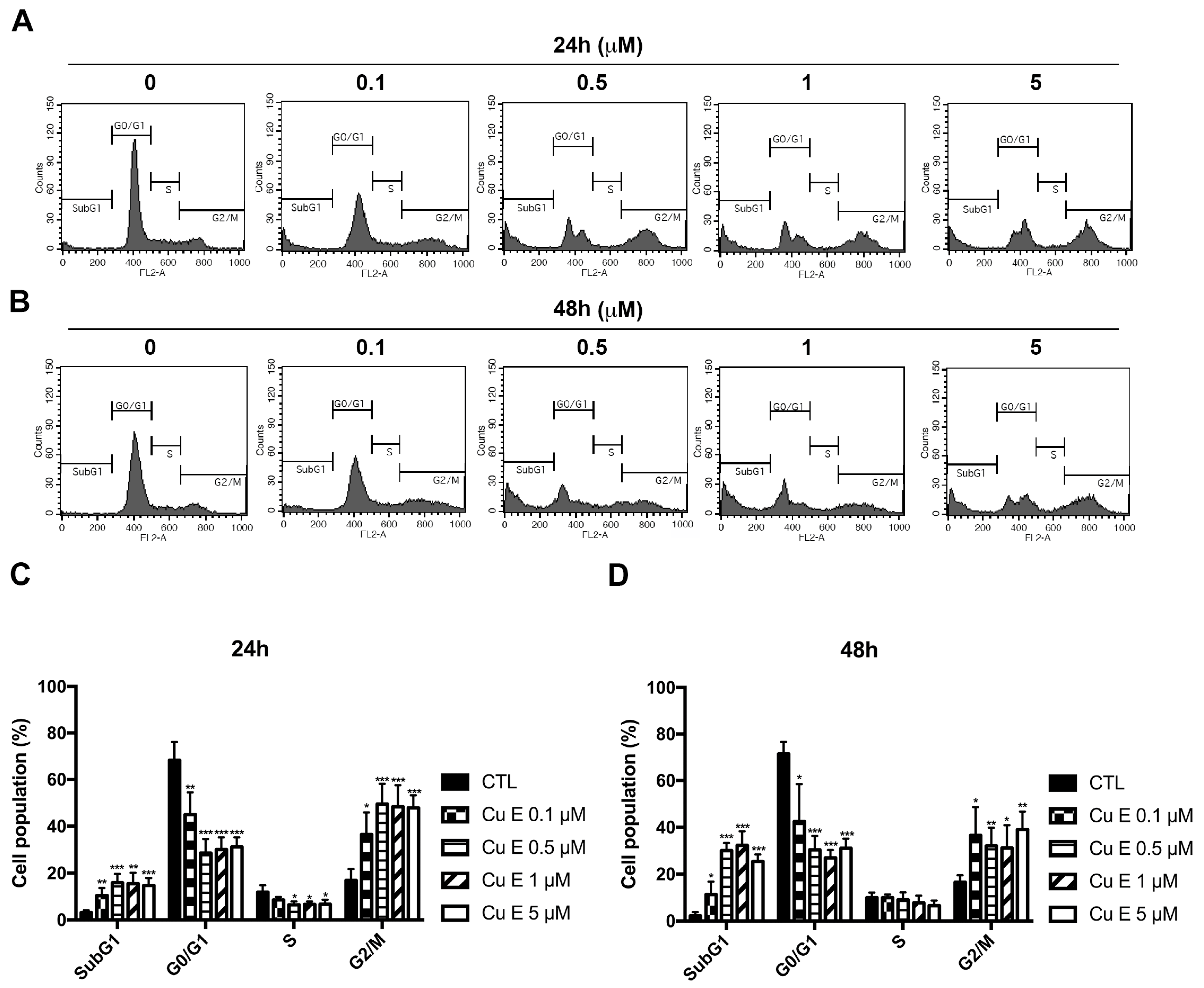
3.2. Cu E Increased Cell-Cycle Arrest at the G2/M and subG1 Phase in A549 Cells
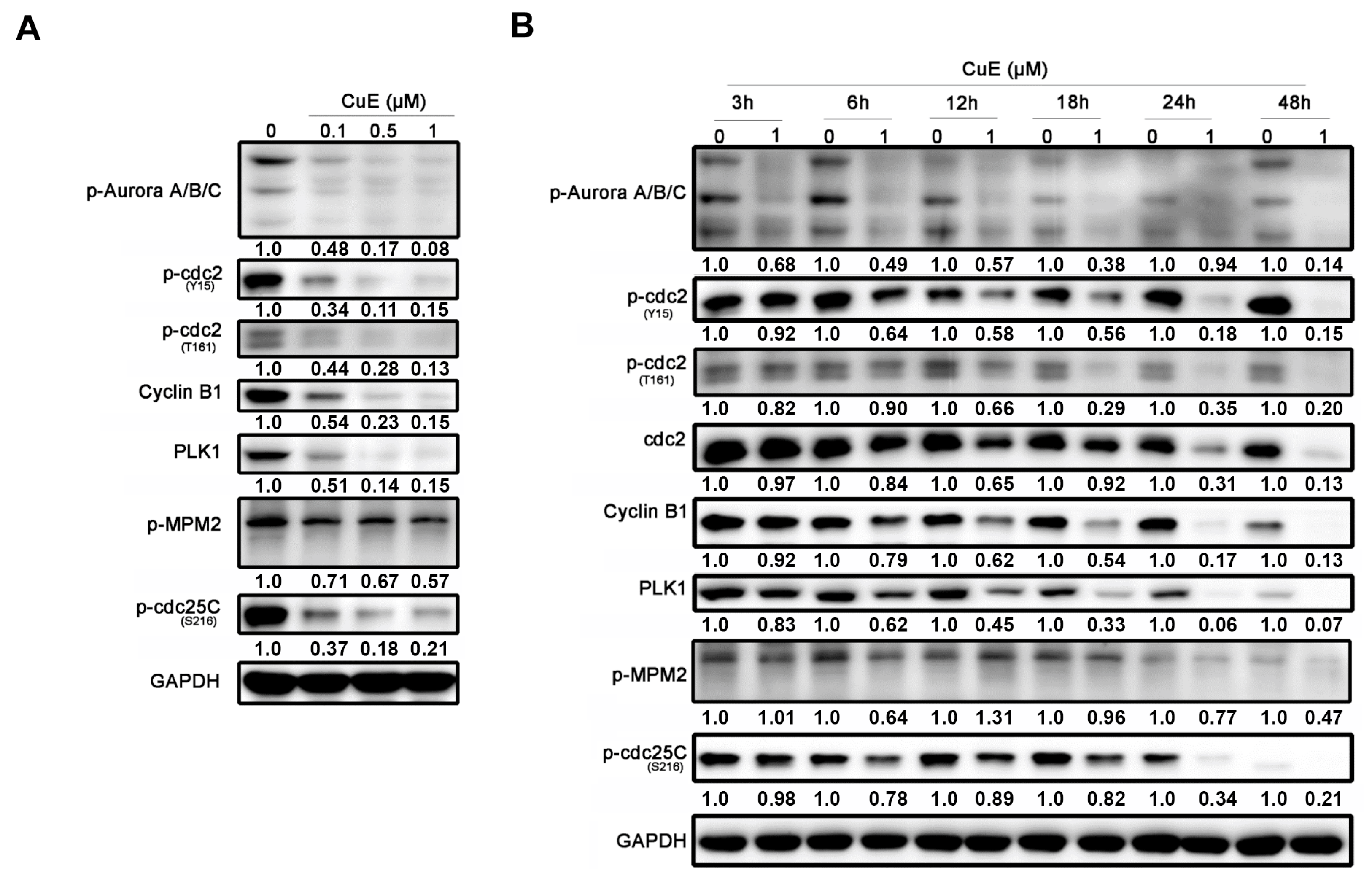
3.3. The Effects of Cu E on the Levels of Cell-Cycle Regulatory Proteins in A549 Cells
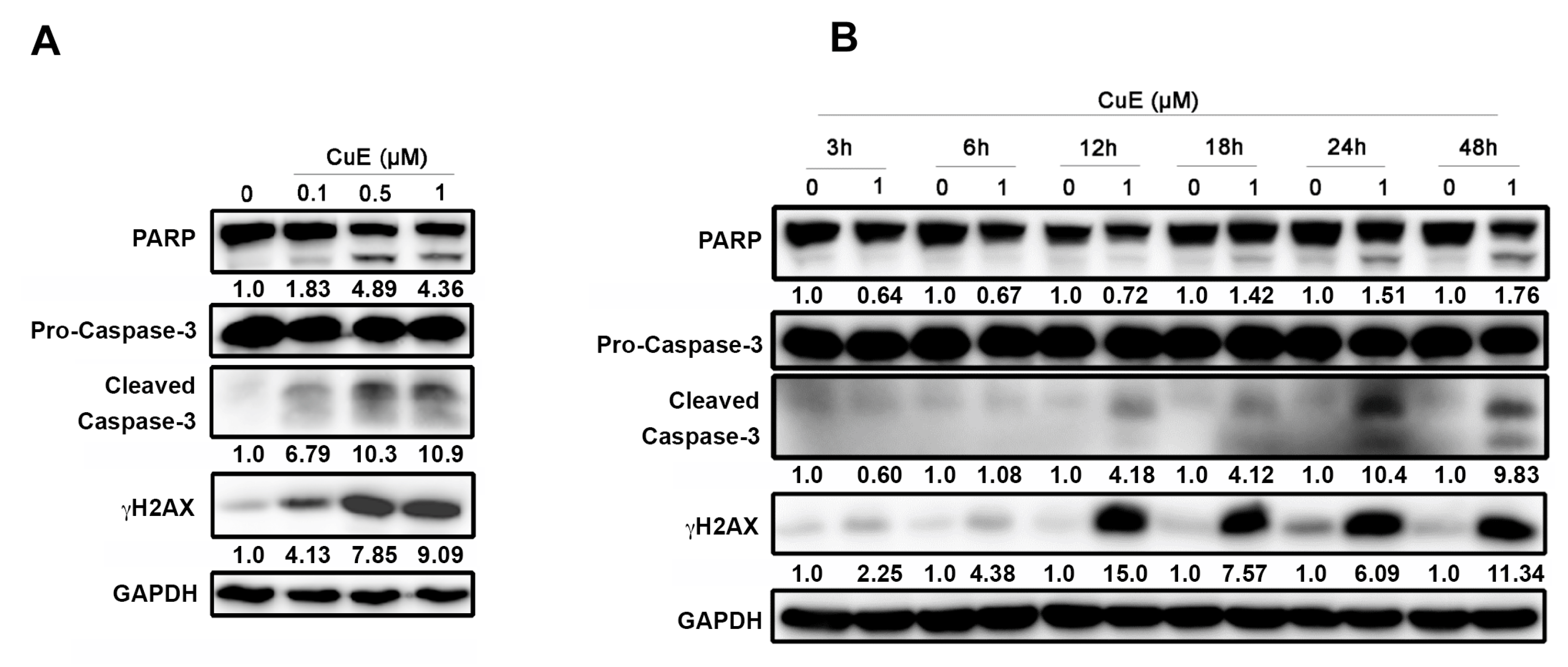
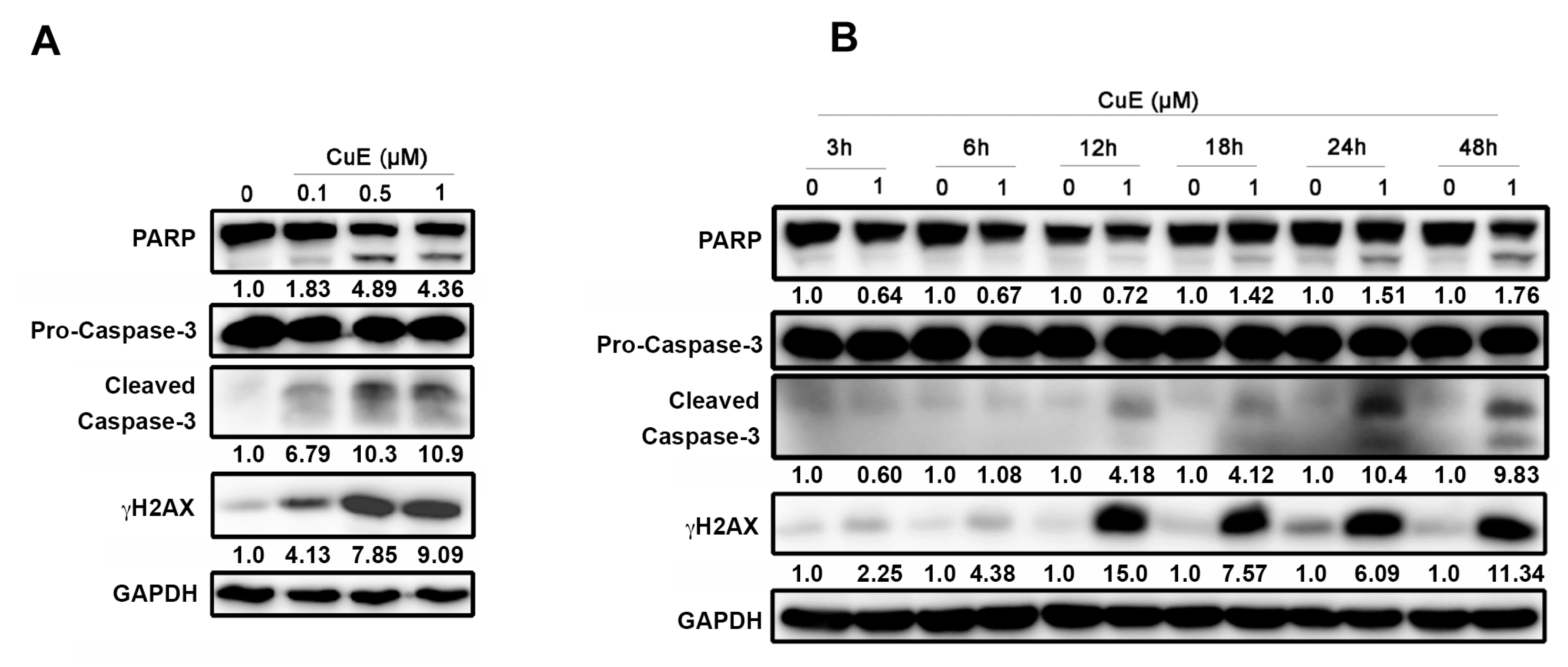
3.4. The Effects of Cu E on Apoptosis in A549 Cells
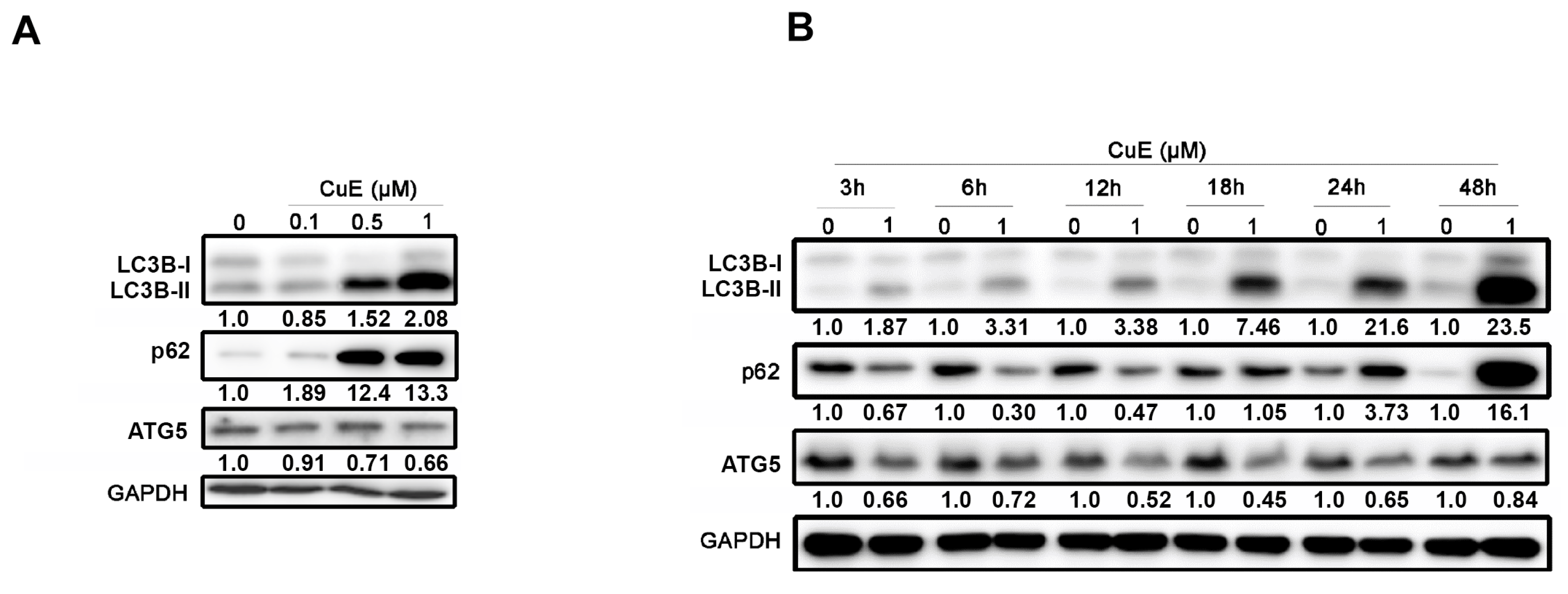
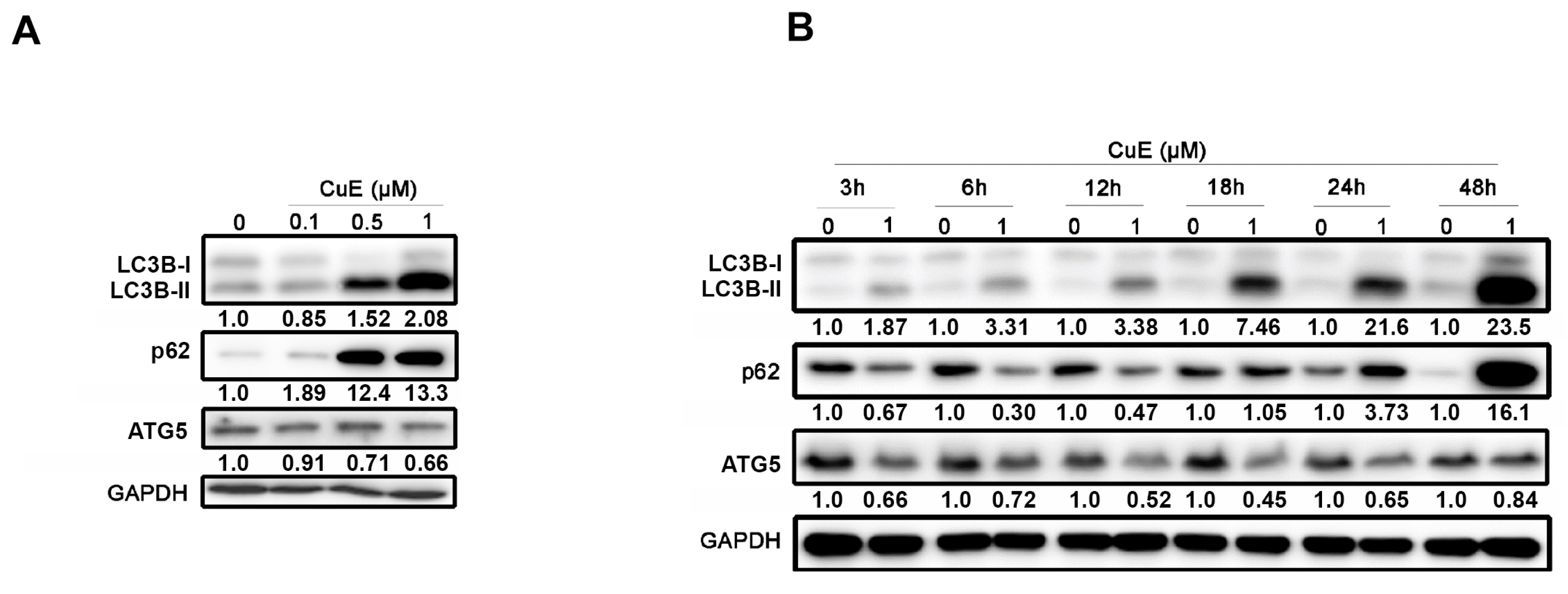
3.5. The Effects of Cu E on Autophagy in A549 Cells
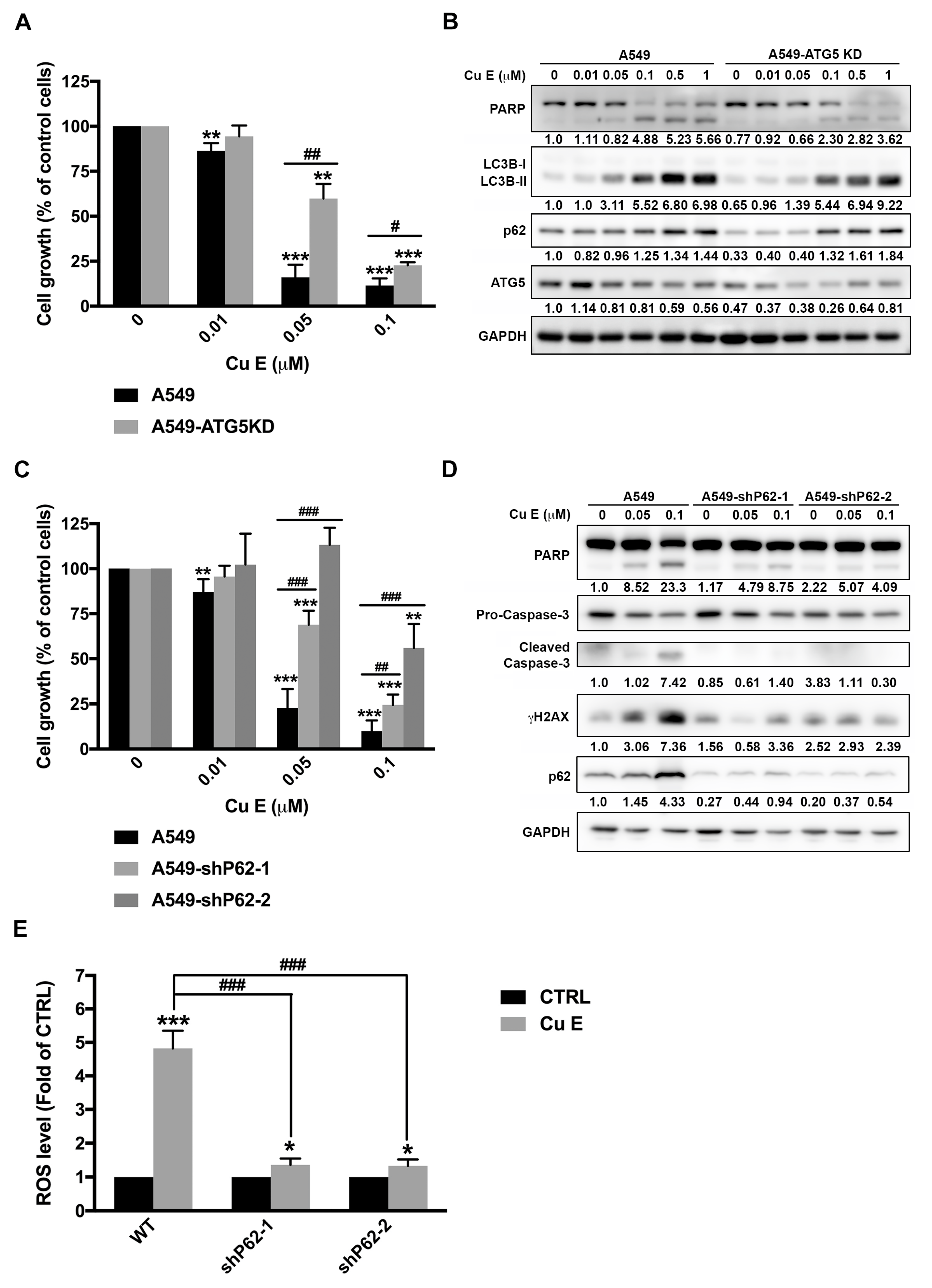
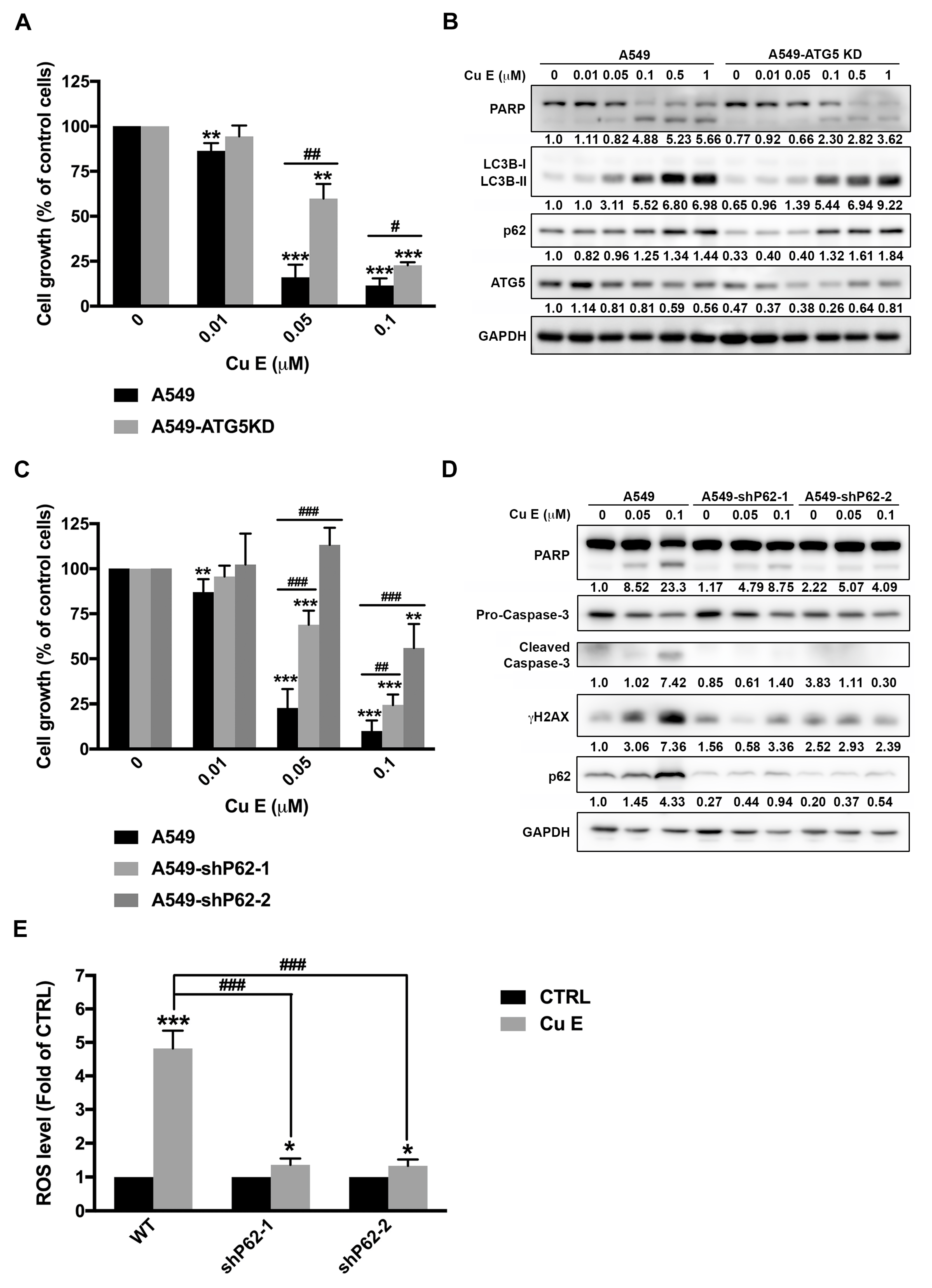
3.6. Cu E-Induced Incomplete Autophagy Is Crucial to ROS Production, DNA Damage, and Apoptosis in A549 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D. Lung Cancer Pathology: Current Concepts. Clin. Chest Med. 2020, 41, 67–85. [Google Scholar] [CrossRef] [PubMed]
- Kashima, J.; Kitadai, R.; Okuma, Y. Molecular and Morphological Profiling of Lung Cancer: A Foundation for “Next-Generation” Pathologists and Oncologists. Cancers 2019, 11, 599. [Google Scholar] [CrossRef]
- Tan, A.C.; Tan, D.S.W. Targeted Therapies for Lung Cancer Patients with Oncogenic Driver Molecular Alterations. J. Clin. Oncol. 2022, 40, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Asao, T.; Takahashi, F.; Takahashi, K. Resistance to molecularly targeted therapy in non-small-cell lung cancer. Respir. Investig. 2019, 57, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H. Molecular basis of drug resistance: Epidermal growth factor receptor tyrosine kinase inhibitors and anaplastic lymphoma kinase inhibitors. Tuberc. Respir. Dis. 2013, 75, 188–198. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Mushtaq, S.; Abbasi, B.H.; Uzair, B.; Abbasi, R. Natural products as reservoirs of novel therapeutic agents. EXCLI J. 2018, 17, 420–451. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Song, L.; Hua, S. Perspectives and controversies regarding the use of natural products for the treatment of lung cancer. Cancer Med. 2021, 10, 2396–2422. [Google Scholar] [CrossRef] [PubMed]
- Umar, A.; Dunn, B.K.; Greenwald, P. Future directions in cancer prevention. Nat. Rev. Cancer 2012, 12, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Haddad, R.I.; Shin, D.M. Recent advances in head and neck cancer. N. Engl. J. Med. 2008, 359, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Glade, M.J. Food, nutrition, and the prevention of cancer: A global perspective. American Institute for Cancer Research/World Cancer Research Fund, American Institute for Cancer Research, 1997. Nutrition 1999, 15, 523–526. [Google Scholar] [PubMed]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Surh, Y.J. Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer 2003, 3, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, C.; Vuillemin, A.T.; Demarchi, M.; Bazan, F.; Chaigneau, L.; Pivot, X. Ixabepilone: A new active chemotherapy in the treatment of breast cancer. Women’s Health 2009, 5, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Swami, U.; Chaudhary, I.; Ghalib, M.H.; Goel, S. Eribulin—A review of preclinical and clinical studies. Crit. Rev. Oncol. Hematol. 2012, 81, 163–184. [Google Scholar] [CrossRef]
- Chen, X.; Bao, J.; Guo, J.; Ding, Q.; Lu, J.; Huang, M.; Wang, Y. Biological activities and potential molecular targets of cucurbitacins: A focus on cancer. Anticancer Drugs 2012, 23, 777–787. [Google Scholar] [CrossRef]
- Alghasham, A.A. Cucurbitacins—A promising target for cancer therapy. Int. J. Health Sci. 2013, 7, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sharma, B.; Sharma, U.; Parashar, G.; Parashar, N.C.; Rani, I.; Ramniwas, S.; Kaur, S.; Haque, S.; Tuli, H.S. Apoptotic and antimetastatic effect of cucurbitacins in cancer: Recent trends and advancement. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2023, 396, 1867–1878. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, G.A.; Ibrahim, S.R.M.; El-Agamy, D.S.; Elsaed, W.M.; Sirwi, A.; Asfour, H.Z.; Koshak, A.E.; Elhady, S.S. Cucurbitacin E glucoside alleviates concanavalin A-induced hepatitis through enhancing SIRT1/Nrf2/HO-1 and inhibiting NF-kB/NLRP3 signaling pathways. J. Ethnopharmacol. 2022, 292, 115223. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Xu, L.H.; He, J.; Ouyang, D.Y.; He, X.H. Cucurbitacin E exhibits anti-inflammatory effect in RAW 264.7 cells via suppression of NF-kappaB nuclear translocation. Inflamm. Res. 2013, 62, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Kumar, M.; Devi, S.; Kabra, A. The Mechanisms of Cucurbitacin E as a Neuroprotective and Memory-Enhancing Agent in a Cerebral Hypoperfusion Rat Model: Attenuation of Oxidative Stress, Inflammation, and Excitotoxicity. Front. Pharmacol. 2021, 12, 794933. [Google Scholar] [CrossRef]
- Varela, C.; Melim, C.; Neves, B.G.; Sharifi-Rad, J.; Calina, D.; Mamurova, A.; Cabral, C. Cucurbitacins as potential anticancer agents: New insights on molecular mechanisms. J. Transl. Med. 2022, 20, 630. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.W.; Yang, J.S.; Lin, M.W.; Chen, P.Y.; Chiou, S.M.; Chueh, F.S.; Lan, Y.H.; Pai, S.J.; Tsuzuki, M.; Ho, W.J.; et al. Cucurbitacin E Induces G(2)/M Phase Arrest through STAT3/p53/p21 Signaling and Provokes Apoptosis via Fas/CD95 and Mitochondria-Dependent Pathways in Human Bladder Cancer T24 Cells. Evid. Based Complement. Altern. Med. 2012, 2012, 952762. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, H.; Guo, Q.; Liu, T.; Jiang, Y.; Zhao, M.; Zeng, K.; Tu, P. Cucurbitacin E Inhibits Huh7 Hepatoma Carcinoma Cell Proliferation and Metastasis via Suppressing MAPKs and JAK/STAT3 Pathways. Molecules 2020, 25, 560. [Google Scholar] [CrossRef]
- Zhang, L.; Liang, H.; Xin, Y. Cucurbitacin E inhibits esophageal carcinoma cell proliferation, migration, and invasion by suppressing Rac1 expression through PI3K/AKT/mTOR pathway. Anticancer Drugs 2020, 31, 847–855. [Google Scholar] [CrossRef]
- Yang, P.; Lian, Q.; Fu, R.; Ding, G.B.; Amin, S.; Li, Z.; Li, Z. Cucurbitacin E Triggers Cellular Senescence in Colon Cancer Cells via Regulating the miR-371b-5p/TFAP4 Signaling Pathway. J. Agric. Food Chem. 2022, 70, 2936–2947. [Google Scholar] [CrossRef]
- Zha, Q.B.; Zhang, X.Y.; Lin, Q.R.; Xu, L.H.; Zhao, G.X.; Pan, H.; Zhou, D.; Ouyang, D.Y.; Liu, Z.H.; He, X.H. Cucurbitacin E Induces Autophagy via Downregulating mTORC1 Signaling and Upregulating AMPK Activity. PLoS ONE 2015, 10, e0124355. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Lu, B.; Zhang, X.; Zhang, J.; Lai, L.; Li, D.; Wu, Y.; Song, Y.; Luo, J.; Pang, X.; et al. Cucurbitacin E, a tetracyclic triterpenes compound from Chinese medicine, inhibits tumor angiogenesis through VEGFR2-mediated Jak2-STAT3 signaling pathway. Carcinogenesis 2010, 31, 2097–2104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, J.; Dong, Y.; Zhai, D.; Lai, L.; Dai, F.; Deng, H.; Chen, Y.; Liu, M.; Yi, Z. Cucurbitacin E inhibits breast tumor metastasis by suppressing cell migration and invasion. Breast Cancer Res. Treat. 2012, 135, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.; Kim, J. Autophagy: An Essential Degradation Program for Cellular Homeostasis and Life. Cells 2018, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Noda, N.N.; Inagaki, F. Mechanisms of Autophagy. Annu. Rev. Biophys. 2015, 44, 101–122. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Sui, H.; Zhang, Q.; Wang, P.; Wang, F. Cucurbitacin E Induces Autophagy-Involved Apoptosis in Intestinal Epithelial Cells. Front. Physiol. 2020, 11, 1020. [Google Scholar] [CrossRef]
- Ma, G.; Luo, W.; Lu, J.; Ma, D.L.; Leung, C.H.; Wang, Y.; Chen, X. Cucurbitacin E induces caspase-dependent apoptosis and protective autophagy mediated by ROS in lung cancer cells. Chem. Biol. Interact. 2016, 253, 1–9. [Google Scholar] [CrossRef]
- Zhang, J.; Aray, B.; Zhang, Y.; Bai, Y.; Yuan, T.; Ding, S.; Xue, Y.; Huang, X.; Li, Z. Synergistic effect of cucurbitacin E and myricetin on Anti-Non-Small cell lung cancer: Molecular mechanism and therapeutic potential. Phytomedicine 2023, 111, 154619. [Google Scholar] [CrossRef]
- Chang, C.H.; Lin, B.J.; Chen, C.H.; Nguyen, N.L.; Hsieh, T.H.; Su, J.H.; Chen, M.C. Stellettin B Induces Cell Death in Bladder Cancer Via Activating the Autophagy/DAPK2/Apoptosis Signaling Cascade. Mar. Drugs 2023, 21, 73. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Lin, Y.C.; Liao, Y.H.; Liou, J.P.; Chen, C.H. MPT0G612, a Novel HDAC6 Inhibitor, Induces Apoptosis and Suppresses IFN-gamma-Induced Programmed Death-Ligand 1 in Human Colorectal Carcinoma Cells. Cancers 2019, 11, 1617. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pan, Y.Y.; Zhang, Y. Synergistic interaction between sorafenib and gemcitabine in EGFR-TKI-sensitive and EGFR-TKI-resistant human lung cancer cell lines. Oncol. Lett. 2013, 5, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Kroemer, G. Cyclin-dependent kinase-1: Linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. 2002, 9, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Joukov, V.; De Nicolo, A. Aurora-PLK1 cascades as key signaling modules in the regulation of mitosis. Sci. Signal. 2018, 11, eaar4195. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)1. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Liu, Y.M.; Pan, S.L.; Liu, Y.R.; Liou, J.P.; Yen, Y. Trichlorobenzene-substituted azaaryl compounds as novel FGFR inhibitors exhibiting potent antitumor activity in bladder cancer cells in vitro and in vivo. Oncotarget 2016, 7, 26374–26387. [Google Scholar] [CrossRef] [PubMed]
- Kongara, S.; Karantza, V. The interplay between autophagy and ROS in tumorigenesis. Front. Oncol. 2012, 2, 171. [Google Scholar] [CrossRef]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef]
- Chen, C.H.; Changou, C.A.; Hsieh, T.H.; Lee, Y.C.; Chu, C.Y.; Hsu, K.C.; Wang, H.C.; Lin, Y.C.; Lo, Y.N.; Liu, Y.R.; et al. Dual Inhibition of PIK3C3 and FGFR as a New Therapeutic Approach to Treat Bladder Cancer. Clin. Cancer Res. 2018, 24, 1176–1189. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, H.; Huang, S.; Chu, Q. KRAS Mutations in Solid Tumors: Characteristics, Current Therapeutic Strategy, and Potential Treatment Exploration. J. Clin. Med. 2023, 12, 709. [Google Scholar] [CrossRef]
- Kong, Y.; Chen, J.; Zhou, Z.; Xia, H.; Qiu, M.H.; Chen, C. Cucurbitacin E induces cell cycle G2/M phase arrest and apoptosis in triple negative breast cancer. PLoS ONE 2014, 9, e103760. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.C.; Hsu, Y.C.; Tsai, C.C. The effects of cucurbitacin E on GADD45beta-trigger G2/M arrest and JNK-independent pathway in brain cancer cells. J. Cell. Mol. Med. 2019, 23, 3512–3519. [Google Scholar] [CrossRef]
- Wang, X.; Tanaka, M.; Peixoto, H.S.; Wink, M. Cucurbitacins: Elucidation of their interactions with the cytoskeleton. PeerJ 2017, 5, e3357. [Google Scholar] [CrossRef] [PubMed]
- Dey, P. Aneuploidy and malignancy: An unsolved equation. J. Clin. Pathol. 2004, 57, 1245–1249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Cao, S.; Qiu, F.; Kang, N. Incomplete autophagy: Trouble is a friend. Med. Res. Rev. 2022, 42, 1545–1587. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Wu, S.; Wang, X.; Zhu, G.; Chen, X.; Ding, Y.; Jiang, W. Cucurbitacin I induces pro-death autophagy in A549 cells via the ERK-mTOR-STAT3 signaling pathway. J. Cell. Biochem. 2018, 119, 6104–6112. [Google Scholar] [CrossRef] [PubMed]
- Arel-Dubeau, A.M.; Longpre, F.; Bournival, J.; Tremblay, C.; Demers-Lamarche, J.; Haskova, P.; Attard, E.; Germain, M.; Martinoli, M.G. Cucurbitacin E has neuroprotective properties and autophagic modulating activities on dopaminergic neurons. Oxidative Med. Cell. Longev. 2014, 2014, 425496. [Google Scholar] [CrossRef]
- Kast, D.J.; Dominguez, R. The Cytoskeleton-Autophagy Connection. Curr. Biol. 2017, 27, R318–R326. [Google Scholar] [CrossRef]
- Trisciuoglio, D.; Degrassi, F. The Tubulin Code and Tubulin-Modifying Enzymes in Autophagy and Cancer. Cancers 2021, 14, 6. [Google Scholar] [CrossRef]
- Sorensen, P.M.; Iacob, R.E.; Fritzsche, M.; Engen, J.R.; Brieher, W.M.; Charras, G.; Eggert, U.S. The natural product cucurbitacin E inhibits depolymerization of actin filaments. ACS Chem. Biol. 2012, 7, 1502–1508. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, H.-L.; Lin, B.-J.; Lin, Y.-C.; Tu, C.-C.; Nguyen, N.-L.; Wang, C.-C.; Chen, M.-C.; Chen, C.-H. Cucurbitacin E Exerts Anti-Proliferative Activity via Promoting p62-Dependent Apoptosis in Human Non-Small-Cell Lung Cancer A549 Cells. Curr. Issues Mol. Biol. 2023, 45, 8138-8151. https://doi.org/10.3390/cimb45100514
Hsu H-L, Lin B-J, Lin Y-C, Tu C-C, Nguyen N-L, Wang C-C, Chen M-C, Chen C-H. Cucurbitacin E Exerts Anti-Proliferative Activity via Promoting p62-Dependent Apoptosis in Human Non-Small-Cell Lung Cancer A549 Cells. Current Issues in Molecular Biology. 2023; 45(10):8138-8151. https://doi.org/10.3390/cimb45100514
Chicago/Turabian StyleHsu, Han-Lin, Bo-Jyun Lin, Yu-Chen Lin, Chih-Chieh Tu, Nham-Linh Nguyen, Ching-Chiung Wang, Mei-Chuan Chen, and Chun-Han Chen. 2023. "Cucurbitacin E Exerts Anti-Proliferative Activity via Promoting p62-Dependent Apoptosis in Human Non-Small-Cell Lung Cancer A549 Cells" Current Issues in Molecular Biology 45, no. 10: 8138-8151. https://doi.org/10.3390/cimb45100514
APA StyleHsu, H.-L., Lin, B.-J., Lin, Y.-C., Tu, C.-C., Nguyen, N.-L., Wang, C.-C., Chen, M.-C., & Chen, C.-H. (2023). Cucurbitacin E Exerts Anti-Proliferative Activity via Promoting p62-Dependent Apoptosis in Human Non-Small-Cell Lung Cancer A549 Cells. Current Issues in Molecular Biology, 45(10), 8138-8151. https://doi.org/10.3390/cimb45100514