
Identification of Putative Plant-Based ALR-2 Inhibitors to Treat Diabetic Peripheral Neuropathy

 ,
,  , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
2. Material and Methodology
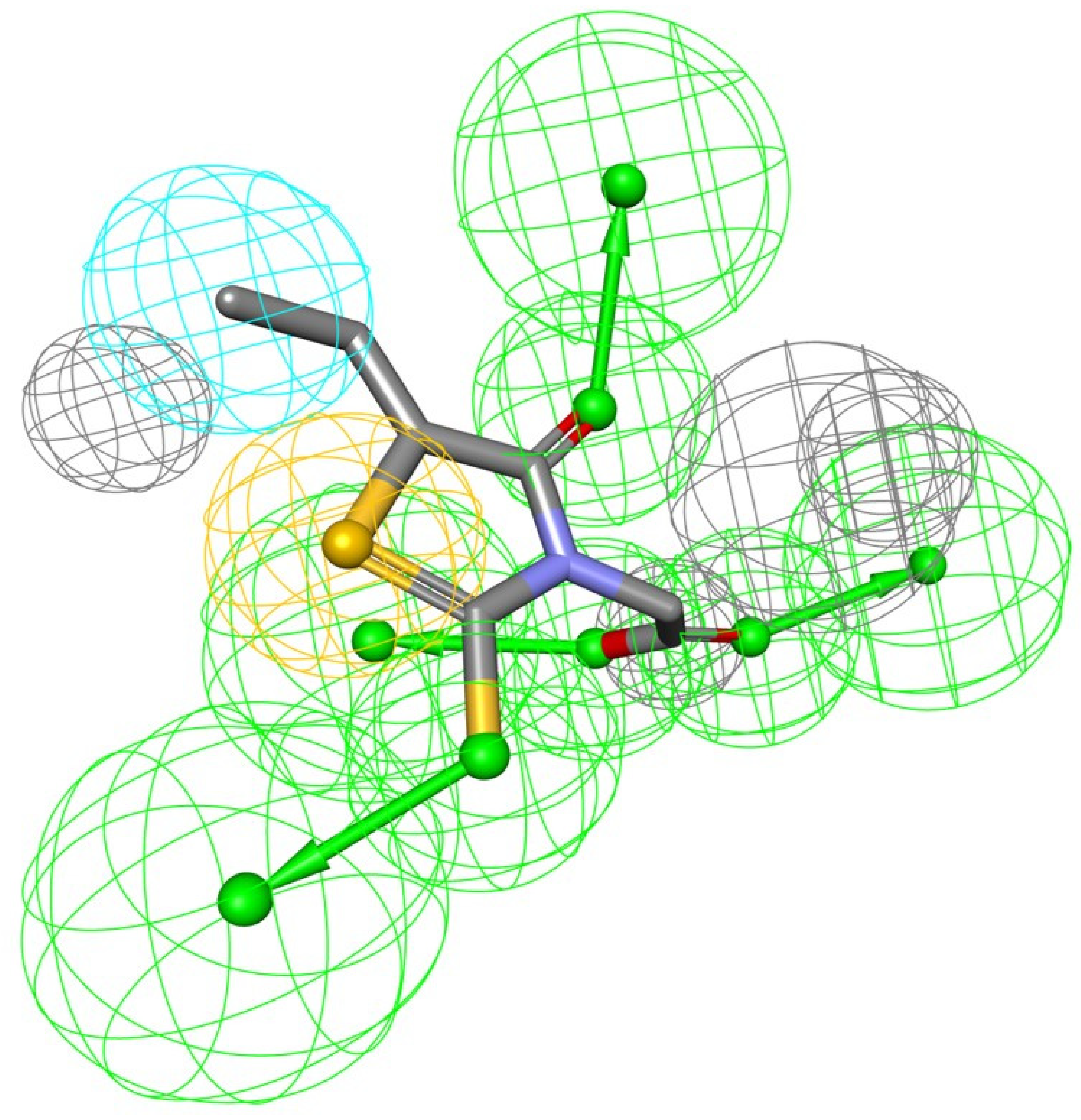
2.1. Structure-Based Pharmacophore Model Generation
2.2. Screening Hits and Enzyme Crystal Structure
2.3. Scoring the Screened Compounds
2.4. Drug Likeness and ADMET Analysis
2.5. Molecular Docking and Interaction Studies
3. Results and Discussions
3.1. Structure-Based Pharmacophore Model Generation
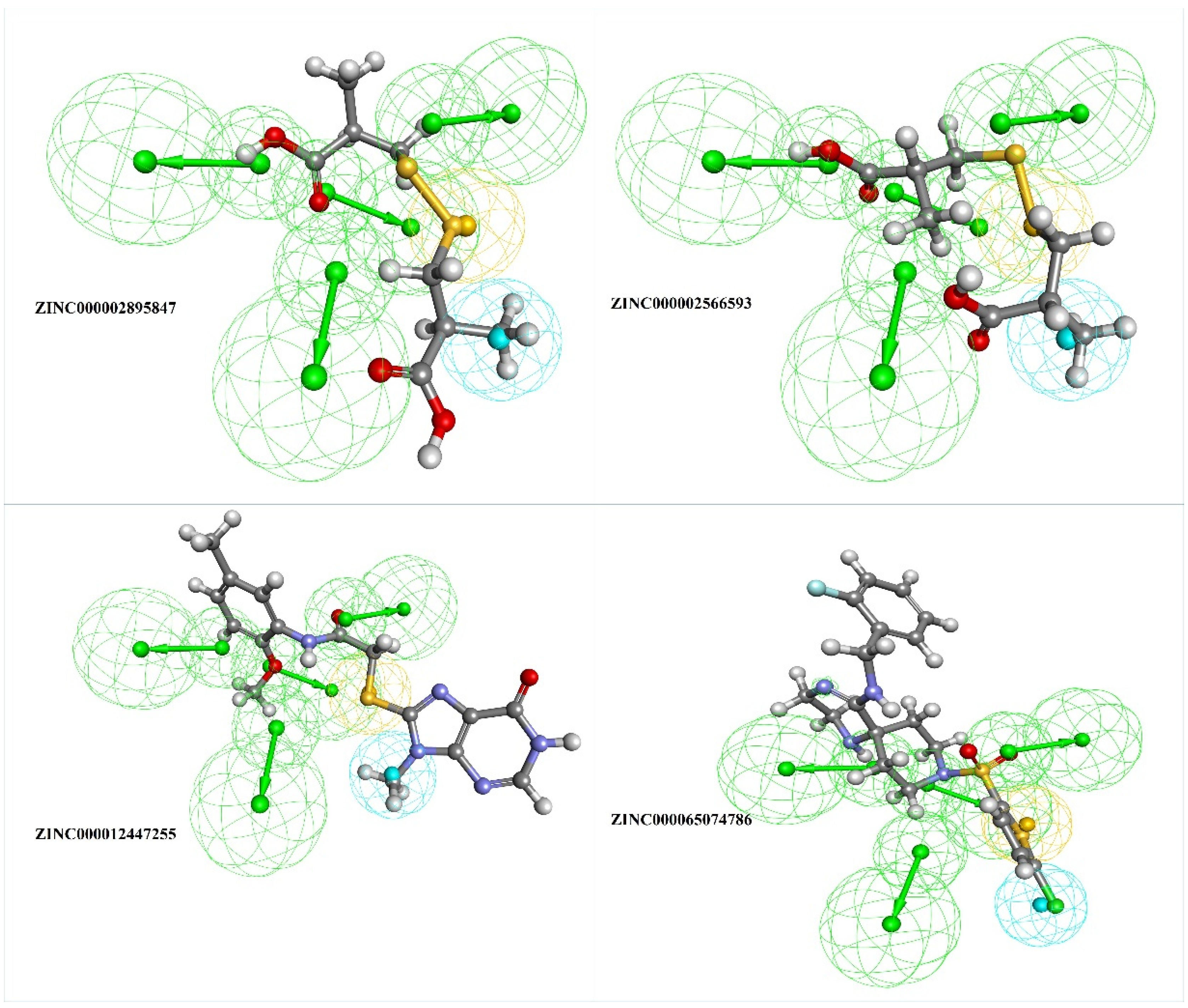
3.2. Screening and Scoring the Natural Compound Database
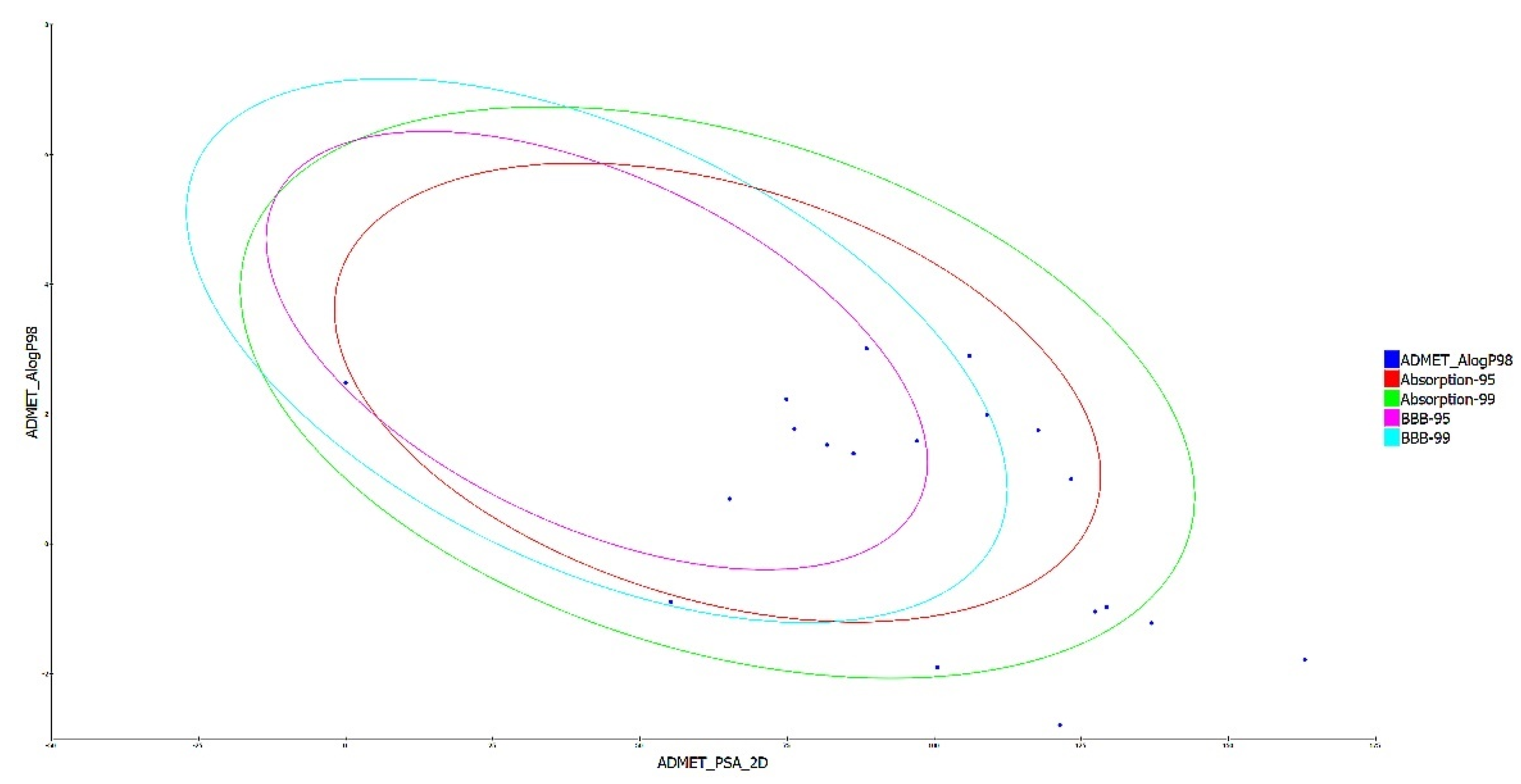
3.3. Drug Likeness and ADMET Analysis
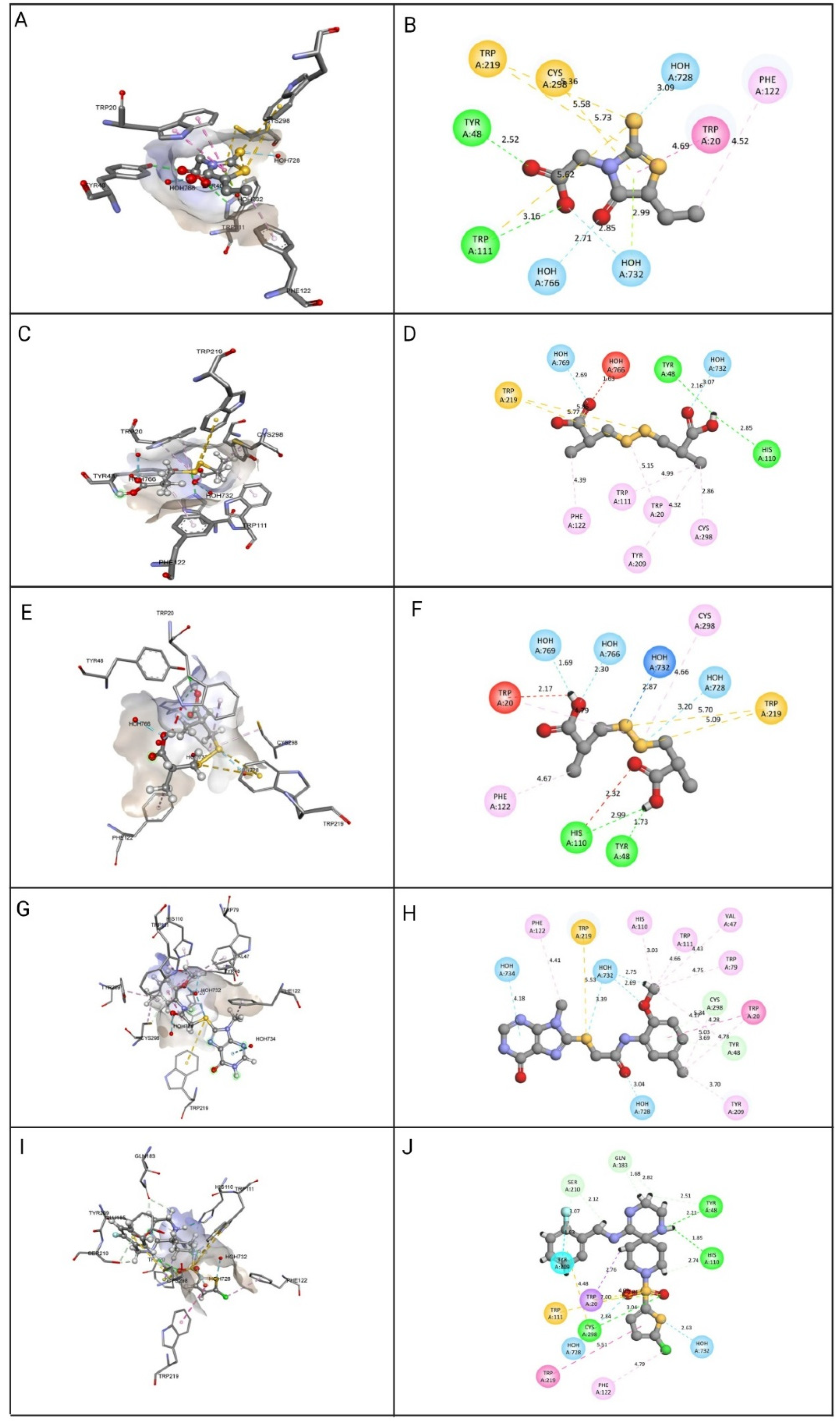
3.4. Molecular Docking and Interaction Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Naeem, Z. Burden of Diabetes Mellitus in Saudi Arabia. Int. J. Health Sci. 2015, 9, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-Y.; Li, Q.-R.; Wang, Z.; Zhou, W.; Fan, S.-R.; Ma, R.; Xue, L.; Yang, L.; Li, Y.-S.; Tan, H.-L.; et al. Epalrestat protects against diabetic peripheral neuropathy by alleviating oxidative stress and inhibiting polyol pathway. Neural Regen. Res. 2016, 11, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.; Liang, X. Chinese Medicine in Diabetic Peripheral Neuropathy: Experimental Research on Nerve Repair and Regeneration. Evid.-Based Complement. Altern. Med. 2012, 2012, 1–13. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, S.; Silakari, O. Virtual screening of epalrestat mimicking selective ALR2 inhibitors from natural product database: Auto pharmacophore, ADMET prediction and molecular dynamics approach. J. Biomol. Struct. Dyn. 2021, 1–19. [Google Scholar] [CrossRef]
- Morsi, M.; Maher, A.; Aboelmagd, O.; Johar, D.; Bernstein, L. A shared comparison of diabetes mellitus and neurodegenerative disorders. J. Cell. Biochem. 2017, 119, 1249–1256. [Google Scholar] [CrossRef]
- Singh, R.; Kishore, L.; Kaur, N. Diabetic peripheral neuropathy: Current perspective and future directions. Pharmacol. Res. 2014, 80, 21–35. [Google Scholar] [CrossRef]
- Vincent, A.M.; Calabek, B.; Roberts, L.; Feldman, E.L. Biology of diabetic neuropathy. Handb. Clin. Neurol. 2013, 115, 591–606. [Google Scholar] [CrossRef]
- Oates, P.J. Polyol pathway and diabetic peripheral neuropathy. Int. Rev. Neurobiol. 2002, 50, 325–392. [Google Scholar] [CrossRef]
- Ramirez, M.A.; Borja, N.L. Epalrestat: An Aldose Reductase Inhibitor for the Treatment of Diabetic Neuropathy. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2008, 28, 646–655. [Google Scholar] [CrossRef]
- Tesfaye, S.; Sloan, G. Diabetic Polyneuropathy–Advances in Diagnosis and Intervention Strategies. Eur. Endocrinol. 2020, 16, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Hotta, N.; Shigeta, Y.; Sakamoto, N.; Kikkawa, R. Effects of an aldose reductase inhibitor, epalrestat, on diabetic neuropathy. Clinical benefit and indication for the drug assessed from the results of a placebo-controlled double-blind study. Biomed. Pharmacother. 1995, 49, 269–277. [Google Scholar] [CrossRef]
- Hotta, N.; Akanuma, Y.; Kawamori, R.; Matsuoka, K.; Oka, Y.; Shichiri, M.; Toyota, T.; Nakashima, M.; Yoshimura, I.; Sakamoto, N.; et al. Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy: The 3-year, multicenter, comparative Aldose Reductase Inhibitor-Diabetes Complications Trial. Diabetes Care 2006, 29, 1538–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thangapandian, S.; John, S.; Sakkiah, S.; Lee, K.W. Ligand and structure based pharmacophore modeling to facilitate novel histone deacetylase 8 inhibitor design. Eur. J. Med. Chem. 2010, 45, 4409–4417. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2020, 49, D437–D451. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, H.; Zhao, Y.; Li, Z.; Chen, S.; Zhai, J.; Chen, Y.; Xie, W.; Wang, Z.; Li, Q.; et al. Inhibitor selectivity between aldo-keto reductase superfamily members AKR1B10 and AKR1B1: Role of Trp112 (Trp111). FEBS Lett. 2013, 587, 3681–3686. [Google Scholar] [CrossRef] [Green Version]
- Greenidge, P.A.; Carlsson, B.; Bladh, A.L.-G.; Gillner, M. Pharmacophores Incorporating Numerous Excluded Volumes Defined by X-ray Crystallographic Structure in Three-Dimensional Database Searching: Application to the Thyroid Hormone Receptor. J. Med. Chem. 1998, 41, 2503–2512. [Google Scholar] [CrossRef]
- Pilon, A.; Valli, M.; Dametto, A.C.; Pinto, M.E.F.; Freire, R.T.; Castro-Gamboa, I.; Andricopulo, A.D.; Bolzani, V.S. NuBBEDB: An updated database to uncover chemical and biological information from Brazilian biodiversity. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Böhm, H.-J. LUDI: Rule-based automatic design of new substituents for enzyme inhibitor leads. J. Comput. Mol. Des. 1992, 6, 593–606. [Google Scholar] [CrossRef]
- Barillari, C.; Marcou, G.; Rognan, D. Hot-Spots-Guided Receptor-Based Pharmacophores (HS-Pharm): A Knowledge-Based Approach to Identify Ligand-Anchoring Atoms in Protein Cavities and Prioritize Structure-Based Pharmacophores. J. Chem. Inf. Model. 2008, 48, 1396–1410. [Google Scholar] [CrossRef]
- Saeed, M.; Saeed, A.; Alam, M.J.; Alreshidi, M. (Receptor-Based Pharmacophore Modeling in the Search for Natural Products for COVID-19 Mpro. Molecules 2021, 26, 1549. [Google Scholar] [CrossRef] [PubMed]
- Tasleem, M.; Alrehaily, A.; Almeleebia, T.M.; Alshahrani, M.Y.; Ahmad, I.; Asiri, M.; Alabdallah, N.M.; Saeed, M. Investigation of Antidepressant Properties of Yohimbine by Employing Structure-Based Computational Assessments. Curr. Issues Mol. Biol. 2021, 43, 127. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.; Shoaib, A.; Tasleem, M.; Alabdallah, N.; Alam, J.; Asmar, Z.; Jamal, Q.; Bardakci, F.; Alqahtani, S.; Ansari, I.; et al. Assessment of Antidiabetic Activity of the Shikonin by Allosteric Inhibition of Protein-Tyrosine Phosphatase 1B (PTP1B) Using State of Art: An In Silico and In Vitro Tactics. Molecules 2021, 26, 3996. [Google Scholar] [CrossRef]
- Wu, G.; Robertson, D.H.; Brooks, C.L.; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Akhter, M.; Tasleem, M.; Alam, M.M.; Ali, S. In silico approach for bioremediation of arsenic by structure prediction and docking studies of arsenite oxidase from Pseudomonas stutzeri TS44. Int. Biodeterior. Biodegradation 2017, 122, 82–91. [Google Scholar] [CrossRef]
- Tasleem, M.; Ishrat, R.; Islam, A.; Ahmad, F. Hassan Structural Characterization, Homology Modeling and Docking Studies of ARG674 Mutation in MyH8 Gene Associated with Trismus-Pseudocamptodactyly Syndrome. Lett. Drug Des. Discov. 2014, 11, 1177–1187. [Google Scholar] [CrossRef]
- Rajendran, B.K.; Suresh, M.X.; Bhaskaran, S.P.; Harshitha, Y.; Gaur, U.; Kwok, H.F. Pharmacoinformatic Approach to Explore the Antidote Potential of Phytochemicals on Bungarotoxin from Indian Krait, Bungarus caeruleus. Comput. Struct. Biotechnol. J. 2018, 16, 450–461. [Google Scholar] [CrossRef]
- Pu, Q.; Han, Z.; Li, X.; Li, Q.; Li, Y. Designing and screening of fluoroquinolone substitutes using combined in silico approaches: Biological metabolism–bioconcentration bilateral selection and their mechanism analyses. Green Chem. 2022, 24, 3778–3793. [Google Scholar] [CrossRef]
- Shukla, A.; Sharma, P.; Prakash, O.; Singh, M.; Kalani, K.; Khan, F.; Bawankule, D.U.; Luqman, S.; Srivastava, S.K. QSAR and Docking Studies on Capsazepine Derivatives for Immunomodulatory and Anti-Inflammatory Activity. PLoS ONE 2014, 9, e100797. [Google Scholar] [CrossRef] [Green Version]
- Fatima, S.; Gupta, P.; Sharma, S.; Sharma, A.; Agarwal, S.M. ADMET profiling of geographically diverse phytochemical using chemoinformatic tools. Futur. Med. Chem. 2020, 12, 69–87. [Google Scholar] [CrossRef]
- Chandra, A.; Chaudhary, M.; Qamar, I.; Singh, N.; Nain, V. In silico identification and validation of natural antiviral compounds as potential inhibitors of SARS-CoV-2 methyltransferase. J. Biomol. Struct. Dyn. 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided-Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Patil, R.; Das, S.; Stanley, A.; Yadav, L.; Sudhakar, A.; Varma, A.K. Optimized Hydrophobic Interactions and Hydrogen Bonding at the Target-Ligand Interface Leads the Pathways of Drug-Designing. PLoS ONE 2010, 5, e12029. [Google Scholar] [CrossRef]
- Panigrahi, S.K. Strong and weak hydrogen bonds in protein-ligand complexes of kinases: A comparative study. Amino Acids 2008, 34, 617–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, M.; Bharatam, P.V. Importance of hydrophobic parameters in identifying appropriate pose of CYP substrates in cytochromes. Eur. J. Med. Chem. 2014, 71, 15–23. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Zinc ID | Pharmprint Frequency | Absolute Energy | Relative Energy |
|---|---|---|---|---|
| 1. | ZINC000000839520 | 17,776 | 121.29 | 6.96097 |
| 2. | ZINC000002566593 | 16,450 | 168.778 | 16.3736 |
| 3. | ZINC000002895847 | 16,228 | 22.9699 | 13.843 |
| 4. | ZINC000004221776 | 12,899 | 22.1846 | 9.94709 |
| 5. | ZINC000004558041 | 10,411 | 123.539 | 14.3409 |
| 6. | ZINC000005189601 | 7480 | 53.2018 | 5.80273 |
| 7. | ZINC000005410978 | 7028 | 54.9265 | 2.95975 |
| 8. | ZINC000012447255 | 6756 | 75.4233 | 12.904 |
| 9. | ZINC000012483342 | 6741 | 44.9922 | 7.5057 |
| 10. | ZINC000013378346 | 5475 | 25.5627 | 14.7066 |
| 11. | ZINC000013380451 | 1254 | 40.2457 | 17.0317 |
| 12. | ZINC000018164733 | 1057 | 123.565 | 12.2268 |
| 13. | ZINC000038281168 | 1001 | 58.6725 | 8.66919 |
| 14. | ZINC000059585866 | 989 | 37.7033 | 1.78389 |
| 15. | ZINC000059586551 | 983 | 59.5097 | 6.99921 |
| 16. | ZINC000065074786 | 961 | 76.1116 | 1.88067 |
| 17. | ZINC000065259848 | 953 | 27.6677 | 7.31742 |
| 18. | ZINC000069482290 | 916 | 15.8298 | 5.27962 |
| 19. | ZINC000070455381 | 805 | 25.6468 | 8.67805 |
| 20. | ZINC000070686641 | 724 | 70.307 | 13.1053 |
| 21. | ZINC000070707266 | 720 | 33.4486 | 13.6762 |
| 22. | ZINC000084394823 | 703 | 62.7607 | 19.8907 |
| 23. | ZINC000085507556 | 661 | 14.4589 | 2.11996 |
| 24. | ZINC000085531653 | 660 | 27.514 | 8.40651 |
| 25. | ZINC000085593748 | 580 | 93.4406 | 18.6682 |
| 26. | ZINC000085809059 | 570 | 40.9 | 13.4514 |
| 27. | ZINC000097944195 | 562 | 130.569 | 9.30741 |
| 28. | ZINC000098022974 | 510 | 50.6495 | 12.9341 |
| 29. | ZINC000100393110 | 486 | 20.0607 | 9.37799 |
| 30. | ZINC000103571159 | 341 | 18.6231 | 13.8256 |
| 31. | ZINC000150349570 | 336 | 48.9624 | 5.99486 |
| 32. | ZINC000245238479 | 289 | 60.8105 | 5.31395 |
| 33. | ZINC000245296023 | 160 | 25.2688 | 10.6235 |
| 34. | ZINC000247764628 | 89 | 61.4826 | 17.7796 |
| S. No. | ZINC ID | Ludi_3 |
|---|---|---|
| 1. | ZINC000097944195 | 1328 |
| 2. | ZINC000070707266 | 1307 |
| 3. | ZINC000247764628 | 1280 |
| 4. | ZINC000012447255 | 1160 |
| 5. | ZINC000004558041 | 1013 |
| 6. | ZINC000100393110 | 989 |
| 7. | ZINC000065074786 | 964 |
| 8. | ZINC000004221776 | 879 |
| 9. | ZINC000000839520 | 875 |
| 10. | ZINC000070455381 | 832 |
| 11. | ZINC000085593748 | 801 |
| 12. | ZINC000245296023 | 799 |
| 13. | ZINC000245238479 | 783 |
| 14. | ZINC000098022974 | 747 |
| 15. | ZINC000253499410 | 719 |
| 16. | ZINC000012483342 | 705 |
| 17. | ZINC000065259848 | 655 |
| 18. | ZINC000103571159 | 590 |
| 19. | ZINC000248015717 | 523 |
| 20. | ZINC1533688 (Epalrestat) | 490 |
| 21. | ZINC000085531653 | 456 |
| 22. | ZINC000070686641 | 433 |
| 23. | ZINC000059586551 | 427 |
| 24. | ZINC000038281168 | 404 |
| 25. | ZINC000059585866 | 403 |
| 26. | ZINC000013380451 | 367 |
| 27. | ZINC000084394823 | 360 |
| 28. | ZINC000069482290 | 349 |
| 29. | ZINC000013378346 | 324 |
| 30. | ZINC000085809059 | 299 |
| 31. | ZINC000002895847 | 289 |
| 32. | ZINC000002566593 | 282 |
| 33. | ZINC000005410978 | 264 |
| 34. | ZINC000005189601 | 264 |
| 35. | ZINC000018164733 | 237 |
| S. No. | ZINC ID | HBA | HBD | MW | ALogP | RoT | PSA |
| 1. | ZINC000103571159 | 8 | 4 | 394.416 | −1.043 | 1 | 132.28 |
| 2. | ZINC000084394823 | 6 | 5 | 180.156 | −2.791 | 5 | 118.22 |
| 3. | ZINC000245238479 | 7 | 2 | 382.448 | 1.389 | 8 | 86.61 |
| 4. | ZINC000018164733 | 5 | 4 | 164.156 | −1.903 | 4 | 97.99 |
| 5. | ZINC000100393110 | 7 | 2 | 446.56 | 2.897 | 9 | 113.71 |
| 6. | ZINC000000839520 | 8 | 3 | 376.43 | 1.797 | 6 | 132.99 |
| 7. | ZINC000012447255 | 8 | 2 | 359.403 | 1.586 | 5 | 122.91 |
| 8. | ZINC000013378346 | 0 | 0 | 238.457 | 2.483 | 7 | 101.2 |
| 9. | ZINC000002895847 | 4 | 2 | 238.324 | 1.77 | 7 | 125.2 |
| 10. | ZINC000065259848 | 5 | 2 | 328.45 | 1.476 | 3 | 117.08 |
| 11. | ZINC000248015717 | 7 | 3 | 374.449 | 0.996 | 4 | 146.43 |
| 12. | ZINC000253499410 | 6 | 2 | 507.706 | 3.533 | 5 | 144.46 |
| 13. | ZINC000004558041 | 9 | 2 | 381.404 | 1.747 | 7 | 144.1 |
| 14. | ZINC000004221776 | 9 | 6 | 375.421 | −1.836 | 6 | 195.43 |
| 15. | ZINC000065074786 | 6 | 2 | 456.985 | 1.764 | 5 | 110.42 |
| 16. | ZINC000002566593 | 4 | 2 | 238.324 | 1.77 | 7 | 125.2 |
| 17. | ZINC000005410978 | 6 | 6 | 254.327 | −5.934 | 8 | 177.23 |
| 18. | ZINC000012483342 | 4 | 3 | 253.317 | 0.693 | 2 | 90.01 |
| 19. | ZINC000098022974 | 11 | 0 | 382.367 | −1.218 | 5 | 154.63 |
| 20. | ZINC1533688 (Epalrestat) | 4 | 0 | 220.289 | −0.279 | 3 | 112.04 |
| ZINC ID | Solubility | BBB Level | CYP 2D6 | Hepatotoxic | Absorption | PPB | AlogP98 | PSA 2D | BBB |
|---|---|---|---|---|---|---|---|---|---|
| ZINC000103571159 | 4 | 4 | FALSE | TRUE | 1 | FALSE | −1.043 | 127.353 | - |
| ZINC000084394823 | 5 | 4 | FALSE | FALSE | 3 | FALSE | −2.791 | 121.378 | - |
| ZINC000245238479 | 3 | 3 | FALSE | FALSE | 0 | FALSE | 1.388 | 86.281 | −1.09 |
| ZINC000018164733 | 5 | 4 | FALSE | FALSE | 1 | FALSE | −1.903 | 100.562 | - |
| ZINC000100393110 | 2 | 4 | FALSE | FALSE | 0 | FALSE | 2.897 | 105.998 | - |
| ZINC000000839520 | 3 | 3 | FALSE | FALSE | 0 | FALSE | 1.991 | 109.004 | −1.26 |
| ZINC000012447255 | 3 | 3 | FALSE | TRUE | 0 | FALSE | 1.586 | 97.084 | −1.2 |
| ZINC000013378346 | 3 | 1 | FALSE | TRUE | 1 | FALSE | 2.483 | 0 | 0.61 |
| ZINC000002895847 | 4 | 3 | FALSE | FALSE | 0 | FALSE | 1.77 | 76.232 | −0.81 |
| ZINC000065259848 | 3 | 3 | FALSE | TRUE | 0 | TRUE | 1.526 | 81.794 | −0.97 |
| ZINC000248015717 | 3 | 4 | FALSE | FALSE | 0 | FALSE | 0.996 | 123.278 | - |
| ZINC000253499410 | 2 | 3 | FALSE | FALSE | 0 | FALSE | 3.009 | 88.515 | −0.62 |
| ZINC000004558041 | 3 | 4 | FALSE | TRUE | 0 | FALSE | 1.747 | 117.664 | - |
| ZINC000004221776 | 4 | 4 | FALSE | TRUE | 3 | FALSE | −1.786 | 162.994 | - |
| ZINC000065074786 | 2 | 3 | FALSE | TRUE | 0 | TRUE | 2.229 | 74.897 | −0.65 |
| ZINC000002566593 | 4 | 3 | FALSE | FALSE | 0 | FALSE | 1.77 | 76.232 | −0.81 |
| ZINC000005410978 | 4 | 4 | FALSE | FALSE | 1 | FALSE | −0.976 | 129.312 | - |
| ZINC000012483342 | 3 | 3 | FALSE | TRUE | 0 | FALSE | 0.693 | 65.215 | −0.97 |
| ZINC000098022974 | 4 | 4 | FALSE | TRUE | 2 | FALSE | −1.219 | 136.9 | - |
| ZINC1533688 | 4 | 3 | FALSE | TRUE | 1 | FALSE | −0.894 | 55.254 | −1.30 |
| ZINC ID | ZINC000065259848 | ZINC000065074786 | ZINC000012447255 | ZINC000245238479 | ZINC000012483342 | ZINC000253499410 | ZINC000002895847 | ZINC000002566593 | Epalrestat |
| Rat Female FDA | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen | Single-Carcinogen | Multi-Carcinogen | Multi-Carcinogen | Multi-Carcinogen |
| Rat Male FDA | Multi-Carcinogen | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen | Multi-Carcinogen |
| TD50 Mouse (mg/kg body weight/day) | 59.487 | 12.1147 | 101.749 | 13.831 | 53.7732 | 4.79071 | 107.426 | 107.426 | 82.6684 |
| TD50 Rat (mg/kg body weight/day) | 110.02 | 22.3253 | 11.4233 | 4.03207 | 5.3266 | 0.357734 | 95.4177 | 95.4177 | 30.0162 |
| Ames Prediction | Non-Mutagen | Non-Mutagen | Non-Mutagen | Non-Mutagen | Non-Mutagen | Non-Mutagen | Non-Mutagen | Non-Mutagen | Non-Mutagen |
| DTP Prediction | Non-Toxic | Toxic | Non-Toxic | Toxic | Toxic | Toxic | Non-Toxic | Non-Toxic | Toxic |
| Rat Oral LD50 (g/kg body weight) | 0.55412 | 0.536551 | 5.11761 | 5.11263 | 0.19364 | 0.569967 | 1.06368 | 1.06368 | 2.71011 |
| Rat inhalation LC50 (mg/m3/h) | 317.172 | 739.959 | 6232.27 | 1761.33 | 519.752 | 245.134 | 2315.21 | 2315.21 | 5330.78 |
| Skin Irritancy | Mild | None | None | None | None | Mild | None | None | Mild |
| Ocular Irritancy | Moderate | Moderate | Mild | None | Severe | Severe | Moderate | Moderate | Mild |
| Aerobic Biodegradability | Non-Degradable | Non-Degradable | Non-Degradable | - | Non-Degradable | Degradable | Degradable | Degradable | Degradable |
| Fathead Minnow LC50 (g/L) | 0.03 | 0.02 | 0.02 | - | 0.28 | 0 | 0.01 | 0.01 | 0.12 |
| Daphnia EC50 (mg/L) | 0.69 | 4.81 | 16.57 | - | 4.6 | 0.27 | 44.84 | 44.84 | 21.28 |
| Compound | Intra-Molecular Interactions | -CDOCKER Energy | -CDOCKER Interaction Energy |
|---|---|---|---|
| Epalrestat | Hydrogen Bond: HOH728, HOH732, HOH766, Tyr48, Trp111. Other: Cys298, Trp111, Trp219. Hydrophobic: HOH732, Trp20, Phe122. | 13.4173 | 25.039 |
| ZINC000002895847 | Hydrogen Bond: HOH728, HOH769, Tyr48, His110. Other: Trp219. Hydrophobic: Cys298, Trp20, Trp111, Phe122, Phe209. | 32.4309 | 29.8047 |
| ZINC000002566593 | Hydrogen Bond: HOH732, HOH728, HOH766, HOH769, Tyr48, His110. Other: Trp219. Hydrophobic: Cys298, Trp20, Phe122. | 30.5462 | 26.683 |
| ZINC000012447255 | Hydrogen Bond: HOH732, HOH728, HOH734, HOH732, Tyr48, Cys298. Other: Trp219. Hydrophobic: Trp20, Tyr48, Val47, Cys298, Trp79, His110, Trp111, Phe122, Tyr209. | 26.5493 | 47.9781 |
| ZINC000065074786 | Hydrogen Bond: HOH732, HOH728, Tyr48, Cys298, his110, Gln183, Tyr48, Ser210. Other: Trp20, Cys298, Trp111. Hydrophobic: Trp20, Trp219, Phe122. | 13.6588 | 32.9367 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saeed, M.; Tasleem, M.; Shoib, A.; Kausar, M.A.; Sulieman, A.M.E.; Alabdallah, N.M.; El Asmar, Z.; Abdelgadir, A.; Al-Shammary, A.; Alam, M.J.; et al. Identification of Putative Plant-Based ALR-2 Inhibitors to Treat Diabetic Peripheral Neuropathy. Curr. Issues Mol. Biol. 2022, 44, 2825-2841. https://doi.org/10.3390/cimb44070194
Saeed M, Tasleem M, Shoib A, Kausar MA, Sulieman AME, Alabdallah NM, El Asmar Z, Abdelgadir A, Al-Shammary A, Alam MJ, et al. Identification of Putative Plant-Based ALR-2 Inhibitors to Treat Diabetic Peripheral Neuropathy. Current Issues in Molecular Biology. 2022; 44(7):2825-2841. https://doi.org/10.3390/cimb44070194
Chicago/Turabian StyleSaeed, Mohd, Munazzah Tasleem, Ambreen Shoib, Mohd Adnan Kausar, Abdel Moneim E. Sulieman, Nadiyah M. Alabdallah, Zeina El Asmar, Abdelmuhsin Abdelgadir, Asma Al-Shammary, Md Jahoor Alam, and et al. 2022. "Identification of Putative Plant-Based ALR-2 Inhibitors to Treat Diabetic Peripheral Neuropathy" Current Issues in Molecular Biology 44, no. 7: 2825-2841. https://doi.org/10.3390/cimb44070194
APA StyleSaeed, M., Tasleem, M., Shoib, A., Kausar, M. A., Sulieman, A. M. E., Alabdallah, N. M., El Asmar, Z., Abdelgadir, A., Al-Shammary, A., Alam, M. J., Badroui, R., & Zahin, M. (2022). Identification of Putative Plant-Based ALR-2 Inhibitors to Treat Diabetic Peripheral Neuropathy. Current Issues in Molecular Biology, 44(7), 2825-2841. https://doi.org/10.3390/cimb44070194