
Inhibition of eNOS Partially Blunts the Beneficial Effects of Nebivolol on Angiotensin II-Induced Signaling in H9c2 Cardiomyoblasts


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture and Western Blotting
2.3. Quantitation of NADP+/NADPH
2.4. RNA Extraction and Real-Time Polymerase Chain Reaction
2.5. Measurement of Intracellular ROS
2.6. Measurement of Cell Surface Area
2.7. Statistical Analysis
3. Results
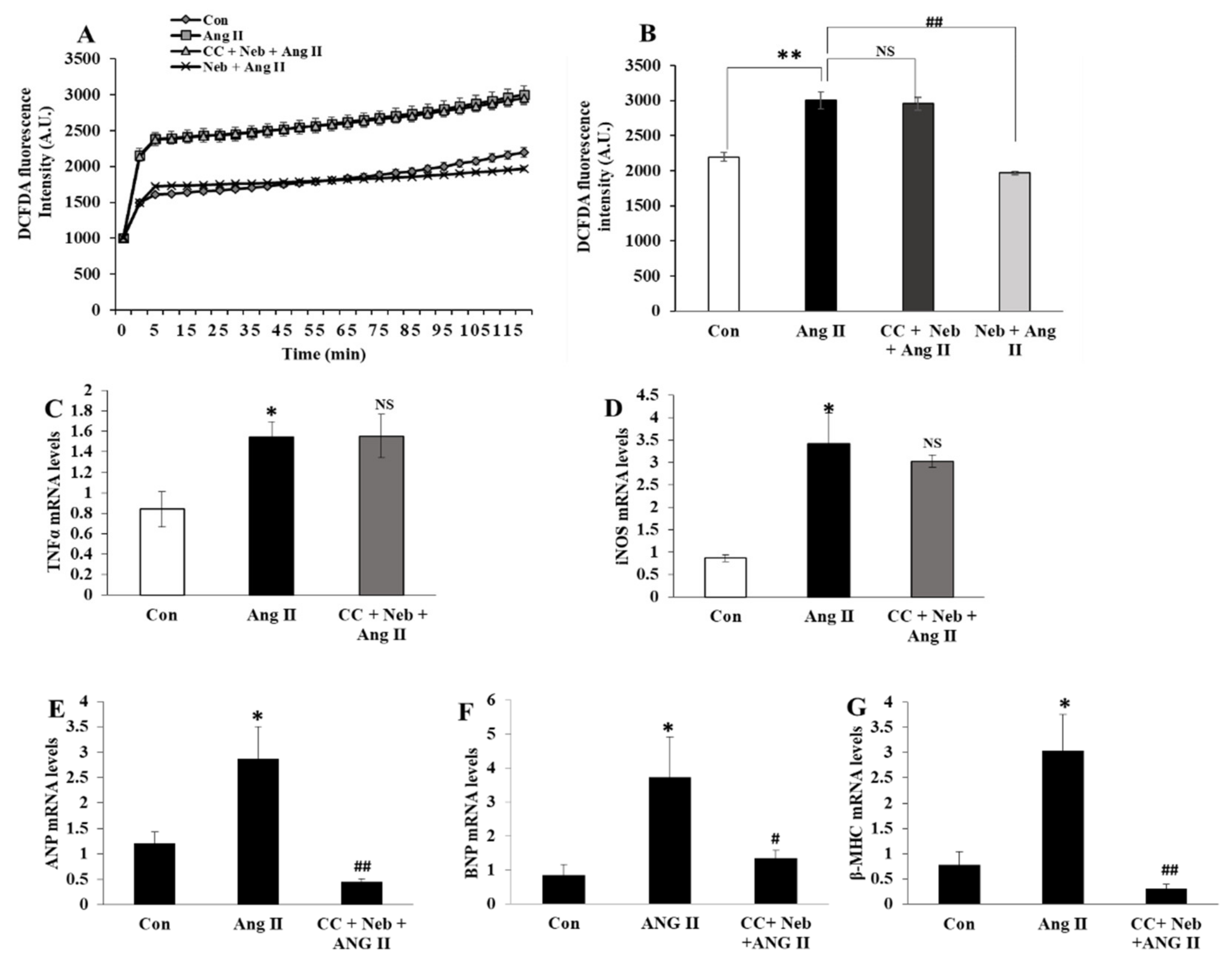
3.1. Nebivolol-Promoted NO Bioavailability Is Critical to Reduce Ang II-Triggered Oxidative Stress in H9c2 Cardiomyoblasts
3.2. Nebivolol-Mediated NO Production Is Not Critical to Reduce Ang II-Triggered Hypertrophy Response
3.3. Nebivolol-Mediated NO Bioavailability Is Not Required for Attenuation of Ang II-Induced Kinase Activation
3.4. Nebivolol-Mediated NO Bioavailability Is Essential for Suppression of Cardiac Proinflammatory Cytokines Induced by Ang II
3.5. Effects of Compound C plus Nebivolol Combination on Ang II-Triggered ROS and mRNA Levels of Inflammatory and Hypertrophy Markers
3.6. Effects of LNIO plus Nebivolol Combination on Ang II-Induced ROS Generation and mRNA Expressions of Inflammatory and Hypertrophic Genes
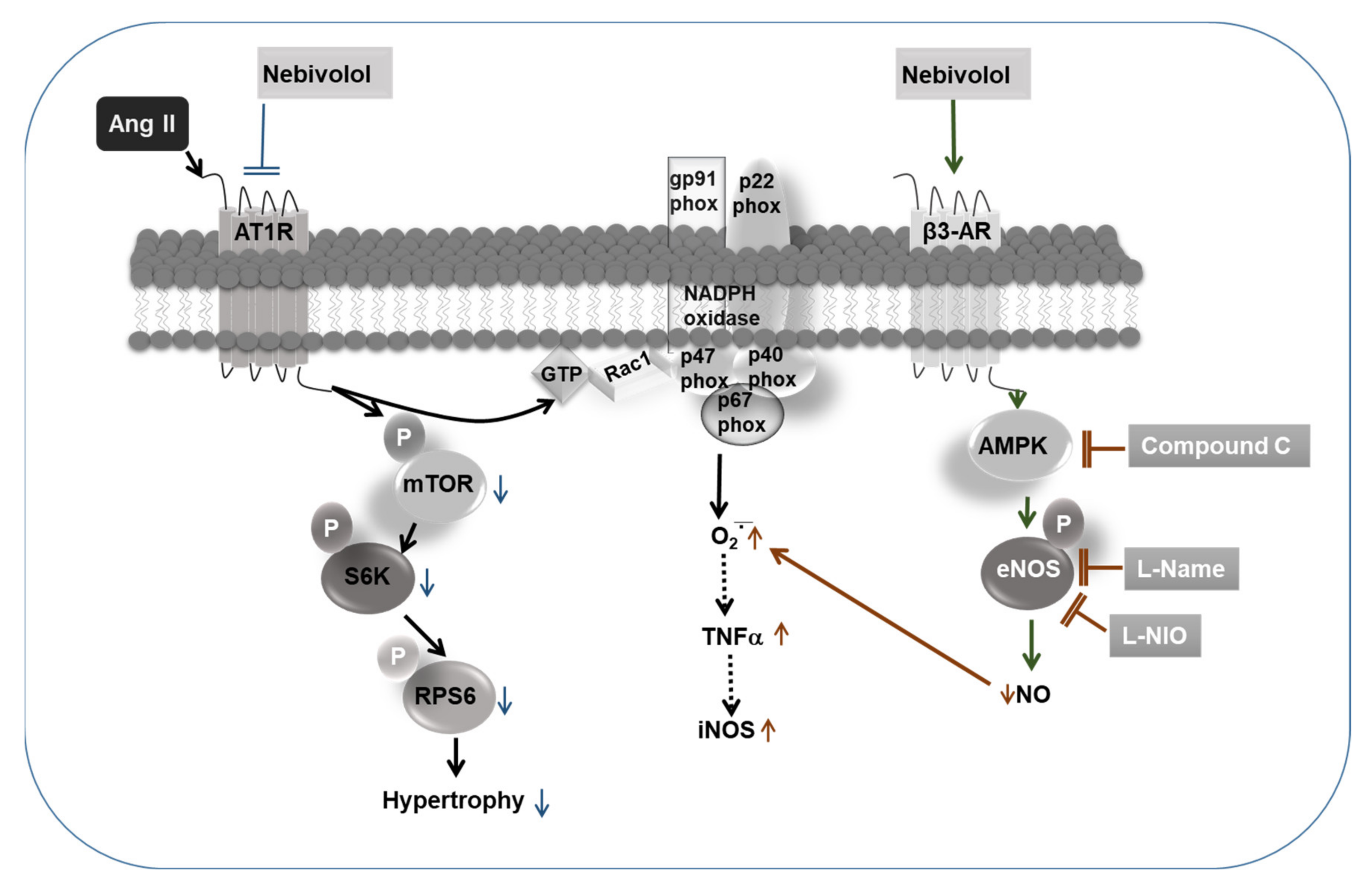
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Gul, R.; Demarco, V.G.; Sowers, J.R.; Whaley-Connell, A.; Pulakat, L. Regulation of Overnutrition-Induced Cardiac Inflammatory Mechanisms. Cardiorenal Med. 2012, 2, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Gul, R.; Kim, S.Y.; Park, K.H.; Kim, B.J.; Kim, S.J.; Im, M.J.; Kim, U.H. A novel signaling pathway of ADP-ribosyl cyclase activation by angiotensin II in adult rat cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H77–H88. [Google Scholar] [CrossRef] [PubMed]
- Gul, R.; Mahmood, A.; Luck, C.; Lum-Naihe, K.; Alfadda, A.A.; Speth, R.C.; Pulakat, L. Regulation of cardiac miR-208a, an inducer of obesity, by rapamycin and nebivolol. Obesisty 2015, 23, 2251–2259. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gul, R.; Park, J.H.; Kim, S.Y.; Jang, K.Y.; Chae, J.K.; Ko, J.K.; Kim, U.H. Inhibition of ADP-ribosyl cyclase attenuates angiotensin II-induced cardiac hypertrophy. Cardiovasc. Res. 2009, 81, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Gul, R.; Ramdas, M.; Mandavia, C.H.; Sowers, J.R.; Pulakat, L. RAS-Mediated Adaptive Mechanisms in Cardiovascular Tissues: Confounding Factors of RAS Blockade Therapy and Alternative Approaches. Cardiorenal Med. 2012, 2, 268–280. [Google Scholar] [CrossRef]
- Gul, R.; Shawl, A.I.; Kim, S.H.; Kim, U.H. Cooperative interaction between reactive oxygen species and Ca2+ signals contributes to angiotensin II-induced hypertrophy in adult rat cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H901–H909. [Google Scholar] [CrossRef]
- Sarkar, S.; Korolchuk, V.I.; Renna, M.; Imarisio, S.; Fleming, A.; Williams, A.; Garcia-Arencibia, M.; Rose, C.; Luo, S.; Underwood, B.R.; et al. Complex inhibitory effects of nitric oxide on autophagy. Mol. Cell 2011, 43, 19–32. [Google Scholar] [CrossRef]
- Cooper, S.A.; Whaley-Connell, A.; Habibi, J.; Wei, Y.; Lastra, G.; Manrique, C.; Stas, S.; Sowers, J.R. Renin-angiotensin-aldosterone system and oxidative stress in cardiovascular insulin resistance. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2009–H2023. [Google Scholar] [CrossRef]
- Moens, A.L.; Leyton-Mange, J.S.; Niu, X.; Yang, R.; Cingolani, O.; Arkenbout, E.K.; Champion, H.C.; Bedja, D.; Gabrielson, K.L.; Chen, J.; et al. Adverse ventricular remodeling and exacerbated NOS uncoupling from pressure-overload in mice lacking the beta3-adrenoreceptor. J. Mol. Cell. Cardiol. 2009, 47, 576–585. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef]
- Wang, D.; Yu, X.; Brecher, P. Nitric oxide inhibits angiotensin II-induced activation of the calcium-sensitive tyrosine kinase proline-rich tyrosine kinase 2 without affecting epidermal growth factor receptor transactivation. J. Biol. Chem. 1999, 274, 24342–24348. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, C.; Leblais, V.; Kobzik, L.; Trochu, J.N.; Khandoudi, N.; Bril, A.; Balligand, J.L.; Le Marec, H. The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J. Clin. Investig. 1998, 102, 1377–1384. [Google Scholar] [CrossRef]
- Aragon, J.P.; Condit, M.E.; Bhushan, S.; Predmore, B.L.; Patel, S.S.; Grinsfelder, D.B.; Gundewar, S.; Jha, S.; Calvert, J.W.; Barouch, L.A.; et al. Beta3-adrenoreceptor stimulation ameliorates myocardial ischemia-reperfusion injury via endothelial nitric oxide synthase and neuronal nitric oxide synthase activation. J. Am. Coll. Cardiol. 2011, 58, 2683–2691. [Google Scholar] [CrossRef] [PubMed]
- Barr, L.A.; Lambert, J.P.; Shimizu, Y.; Barouch, L.A.; Naqvi, N.; Calvert, J.W. Exercise training provides cardioprotection by activating and coupling endothelial nitric oxide synthase via a β(3)-adrenergic receptor-AMP-activated protein kinase signaling pathway. Med. Gas Res. 2017, 7, 1–8. [Google Scholar]
- Moens, A.L.; Yang, R.; Watts, V.L.; Barouch, L.A. Beta 3-adrenoreceptor regulation of nitric oxide in the cardiovascular system. J. Mol. Cell. Cardiol. 2010, 48, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Trappanese, D.M.; Liu, Y.; McCormick, R.C.; Cannavo, A.; Nanayakkara, G.; Baskharoun, M.M.; Jarrett, H.; Woitek, F.J.; Tillson, D.M.; Dillon, A.R.; et al. Chronic beta1-adrenergic blockade enhances myocardial beta3-adrenergic coupling with nitric oxide-cGMP signaling in a canine model of chronic volume overload: New insight into mechanisms of cardiac benefit with selective beta1-blocker therapy. Basic Res. Cardiol. 2015, 110, 456. [Google Scholar] [CrossRef]
- Zhang, Z.; Ding, L.; Jin, Z.; Gao, G.; Li, H.; Zhang, L.; Zhang, L.; Lu, X.; Hu, L.; Lu, B.; et al. Nebivolol protects against myocardial infarction injury via stimulation of beta 3-adrenergic receptors and nitric oxide signaling. PLoS ONE 2014, 9, e98179. [Google Scholar] [CrossRef]
- Oelze, M.; Daiber, A.; Brandes, R.P.; Hortmann, M.; Wenzel, P.; Hink, U.; Schulz, E.; Mollnau, H.; von Sandersleben, A.; Kleschyov, A.L.; et al. Nebivolol inhibits superoxide formation by NADPH oxidase and endothelial dysfunction in angiotensin II-treated rats. Hypertension 2006, 48, 677–684. [Google Scholar] [CrossRef]
- Maffei, A.; Di Pardo, A.; Carangi, R.; Carullo, P.; Poulet, R.; Gentile, M.T.; Vecchione, C.; Lembo, G. Nebivolol induces nitric oxide release in the heart through inducible nitric oxide synthase activation. Hypertension 2007, 50, 652–656. [Google Scholar] [CrossRef]
- Zhou, X.; Ma, L.; Habibi, J.; Whaley-Connell, A.; Hayden, M.R.; Tilmon, R.D.; Brown, A.N.; Kim, J.A.; Demarco, V.G.; Sowers, J.R. Nebivolol improves diastolic dysfunction and myocardial remodeling through reductions in oxidative stress in the Zucker obese rat. Hypertension 2010, 55, 880–888. [Google Scholar] [CrossRef]
- Togliatto, G.; Lombardo, G.; Brizzi, M.F. The Future Challenge of Reactive Oxygen Species (ROS) in Hypertension: From Bench to Bed Side. Int. J. Mol. Sci. 2017, 18, 1988. [Google Scholar] [CrossRef] [PubMed]
- Gul, R.; Alsalman, N.; Bazighifan, A.; Alfadda, A.A. Comparative beneficial effects of nebivolol and nebivolol/valsartan combination against mitochondrial dysfunction in angiotensin II-induced pathology in H9c2 cardiomyoblasts. J. Pharm. Pharm. 2021, 73, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Jung, Y.; Nam, M.; Sun Kang, M.; Lee, M.K.; Cho, Y.; Choi, E.-K.; Hwang, G.-S.; Soo Kim, H. Angiotensin II affects inflammation mechanisms via AMPK-related signalling pathways in HL-1 atrial myocytes. Sci. Rep. 2017, 7, 10328. [Google Scholar] [CrossRef] [PubMed]
- Broeders, M.A.; Doevendans, P.A.; Bekkers, B.C.; Bronsaer, R.; van Gorsel, E.; Heemskerk, J.W.; Egbrink, M.G.; van Breda, E.; Reneman, R.S.; van Der Zee, R. Nebivolol: A third-generation beta-blocker that augments vascular nitric oxide release: Endothelial beta(2)-adrenergic receptor-mediated nitric oxide production. Circulation 2000, 102, 677–684. [Google Scholar] [CrossRef]
- Gao, Y.; Vanhoutte, P.M. Nebivolol: An endothelium-friendly selective beta1-adrenoceptor blocker. J. Cardiovasc. Pharm. 2012, 59, 16–21. [Google Scholar] [CrossRef]
- Ma, L.; Gul, R.; Habibi, J.; Yang, M.; Pulakat, L.; Whaley-Connell, A.; Ferrario, C.M.; Sowers, J.R. Nebivolol improves diastolic dysfunction and myocardial remodeling through reductions in oxidative stress in the transgenic (mRen2) rat. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H2341–H2351. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, F.; Liu, Y.; Yin, S.; Pang, X.; Li, Z.; Wei, Z. Nebivolol alleviates aortic remodeling through eNOS upregulation and inhibition of oxidative stress in l-NAME-induced hypertensive rats. Clin. Exp. Hypertens. 2017, 39, 628–639. [Google Scholar] [CrossRef]
- Mercanoglu, G.; Semen, O. Nitric oxide mediated the effects of nebivolol in cardiorenal syndrome. Iran. J. Basic Med. Sci. 2019, 22, 1314–1324. [Google Scholar]
- Ozakca, I. Antihypertrophic Effects of Nebivolol on Neonatal Cardiomyocyte Hypertrophy Models. J. Cardiovasc. Pharm. 2019, 73, 155–164. [Google Scholar] [CrossRef]
- Sharma, N.M.; Zheng, H.; Li, Y.F.; Patel, K.P. Nitric oxide inhibits the expression of AT1 receptors in neurons. Am. J. Physiol. Cell Physiol. 2012, 302, C1162–C1173. [Google Scholar] [CrossRef]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New Insights into the Role of mTOR Signaling in the Cardiovascular System. Circ. Res. 2018, 122, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Murakami, H.; Murakami, R.; Kambe, F.; Cao, X.; Takahashi, R.; Asai, T.; Hirai, T.; Numaguchi, Y.; Okumura, K.; Seo, H.; et al. Fenofibrate activates AMPK and increases eNOS phosphorylation in HUVEC. Biochem. Biophys. Res. Commun. 2006, 341, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Deruy, E.; Gelinas, R.; Beauloye, C.; Esfahani, H.; Michel, L.Y.M.; Dessy, C.; Bertrand, L.; Balligand, J.L. Beta 3 adrenoreceptors protect from hypertrophic remodelling through AMP-activated protein kinase and autophagy. ESC Heart Fail. 2020, 7, 920–932. [Google Scholar] [CrossRef] [PubMed]
- Callis, T.E.; Pandya, K.; Seok, H.Y.; Tang, R.H.; Tatsuguchi, M.; Huang, Z.P.; Chen, J.F.; Deng, Z.; Gunn, B.; Shumate, J.; et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Investig. 2009, 119, 2772–2786. [Google Scholar] [CrossRef]
- Mariappan, N.; Elks, C.M.; Haque, M.; Francis, J. Interaction of TNF with angiotensin II contributes to mitochondrial oxidative stress and cardiac damage in rats. PLoS ONE 2012, 7, e46568. [Google Scholar] [CrossRef]
- Granger, D.N.; Vowinkel, T.; Petnehazy, T. Modulation of the inflammatory response in cardiovascular disease. Hypertension 2004, 43, 924–931. [Google Scholar] [CrossRef]
- Tian, M.; Yuan, Y.C.; Li, J.Y.; Gionfriddo, M.R.; Huang, R.C. Tumor necrosis factor-α and its role as a mediator in myocardial infarction: A brief review. Chronic Dis. Transl. Med. 2015, 1, 18–26. [Google Scholar] [CrossRef]
- Csont, T.; Viappiani, S.; Sawicka, J.; Slee, S.; Altarejos, J.Y.; Batinić-Haberle, I.; Schulz, R. The involvement of superoxide and iNOS-derived NO in cardiac dysfunction induced by pro-inflammatory cytokines. J. Mol. Cell. Cardiol. 2005, 39, 833–840. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward and Reverse Primers 5′-3′ |
|---|---|
| ANP | AAAGCAAACTGAGGGCTCTGCTCG |
| TTCGGTACCGGAAGCTGTTGCA | |
| BNP | ACAATCCACGATGCAGAAGC |
| CGCCGATCCGGTCTATCTTC | |
| β-MHC | ATCTACAGCGGGTGAAGCAG |
| CAGGTTAGCCTTGGCCTTGA | |
| TNF-α | CACTCAGGCATCGACATTCG |
| CACCGGCAAGGATTCCAA | |
| NOX2 | TGAATCTCAGGCCAATCACTTT |
| AAT GGTCTTGAACTCGTTATCCC | |
| iNOS | CGGCCACCAGCTTCTTCA |
| TGCTTACAGGTCTACGTTCAAGACAT | |
| β-actin | CAA CGT CAC ACT TCA TGA TGG A |
| ATG CCC CGA GGC TCT CTT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gul, R.; Alsalman, N.; Alfadda, A.A. Inhibition of eNOS Partially Blunts the Beneficial Effects of Nebivolol on Angiotensin II-Induced Signaling in H9c2 Cardiomyoblasts. Curr. Issues Mol. Biol. 2022, 44, 2139-2152. https://doi.org/10.3390/cimb44050144
Gul R, Alsalman N, Alfadda AA. Inhibition of eNOS Partially Blunts the Beneficial Effects of Nebivolol on Angiotensin II-Induced Signaling in H9c2 Cardiomyoblasts. Current Issues in Molecular Biology. 2022; 44(5):2139-2152. https://doi.org/10.3390/cimb44050144
Chicago/Turabian StyleGul, Rukhsana, Nouf Alsalman, and Assim A. Alfadda. 2022. "Inhibition of eNOS Partially Blunts the Beneficial Effects of Nebivolol on Angiotensin II-Induced Signaling in H9c2 Cardiomyoblasts" Current Issues in Molecular Biology 44, no. 5: 2139-2152. https://doi.org/10.3390/cimb44050144
APA StyleGul, R., Alsalman, N., & Alfadda, A. A. (2022). Inhibition of eNOS Partially Blunts the Beneficial Effects of Nebivolol on Angiotensin II-Induced Signaling in H9c2 Cardiomyoblasts. Current Issues in Molecular Biology, 44(5), 2139-2152. https://doi.org/10.3390/cimb44050144