
Ulcerative Colitis Seems to Imply Oral Microbiome Dysbiosis

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Material and Methods
2.1. Study Design
2.2. DNA Extraction and Sequencing
2.3. Sequence Processing
2.4. Statistical Analysis
3. Results
3.1. Clinical Characteristics of the Participants
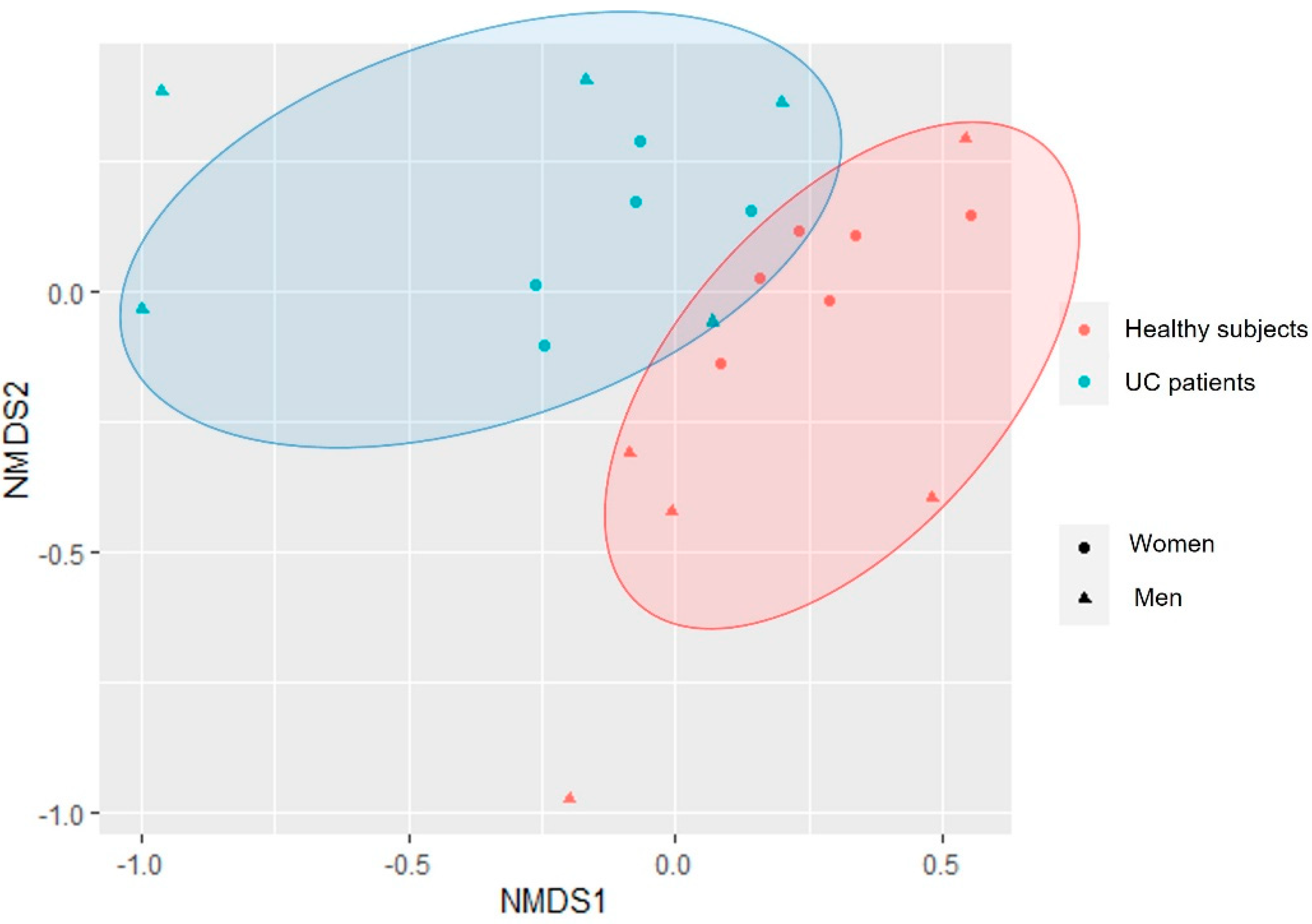
3.2. Changes in the Biodiversity Indices between UC Patients and Healthy Subjects
3.3. Taxonomic Profiles of the UC Patients and Healthy Subjects
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miyoshi, J.; Chang, E.B. The gut microbiota and inflammatory bowel diseases. Transl. Res. 2017, 179, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Docktor, M.J.; Paster, B.J.; Abramowicz, S.; Ingram, J.; Wang, Y.E.; Correll, M.; Jiang, H.; Cotton, S.L.; Kokaras, A.S.; Bousvaros, A. Alterations in diversity of the oral microbiome in pediatric inflammatory bowel disease. Inflamm. Bowel Dis. 2012, 18, 935–942. [Google Scholar] [CrossRef]
- Lucas López, R.; Grande Burgos, M.J.; Gálvez, A.; Pérez Pulido, R. The human gastrointestinal tract and oral microbiota in inflammatory bowel disease: A state of the science review. APMIS 2017, 125, 3–10. [Google Scholar] [CrossRef]
- Guo, X.Y.; Liu, X.J.; Hao, J.Y. Gut microbiota in ulcerative colitis: Insights on pathogenesis and treatment. J. Dig. Dis. 2020, 21, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Ribaldone, D.G.; Brigo, S.; Mangia, M.; Saracco, G.M.; Astegiano, M.; Pellicano, R. Oral manifestations of inflammatory bowel disease and the role of non-invasive surrogate markers of disease activity. Medicines 2020, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Vavricka, S.R.; Schoepfer, A.; Scharl, M.; Lakatos, P.L.M.D.; Navarini, A.; Rogler, G. Extraintestinal Manifestations of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1982–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lankarani, K.B.; Sivandzadeh, G.R.; Hassanpour, S. Oral manifestation in inflammatory bowel disease: A review. World J Gastroenterol. 2013, 19, 8571–8579. [Google Scholar] [CrossRef] [PubMed]
- Vasovic, M.; Gajovic, N.; Brajkovic, D.; Jovanovic, M.; Zdravkovaic, N.; Kanjevac, T. The relationship between the immune system and oral manifestations of inflammatory bowel disease: A review. Cent. Eur. J. Immunol. 2016, 41, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Elmaghrawy, K.; Hussey, S.; Moran, G.P. The oral microbiome in pediatric IBD: A source of pathobionts or biomarkers? Front. Pediatr. 2021, 8, 620254. [Google Scholar] [CrossRef]
- Rowland, M.; Fleming, P.; Bourke, B. Looking in the mouth for Crohn’s disease. Inflamm. Bowel Dis. 2010, 16, 332–337. [Google Scholar] [CrossRef]
- Harty, S.; Fleming, P.; Rowland, M.; Crushell, E.; McDermott, M.; Drumm, B.; Bourke, B. A prospective study of the oral manifestations of Crohn’s disease. Clin. Gastroenterol. Hepatol. 2005, 3, 886–891. [Google Scholar] [CrossRef]
- Pittock, S.; Drumm, B.; Fleming, P.; McDermott, M.; Imrie, C.; Flint, S.; Bourke, B. The oral cavity in Crohn’s disease. J. Pediatr. 2001, 138, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kayani, M.U.R.; Hong, L.; Zhang, C.; Zhong, J.; Wang, Z.; Chen, L. Dynamics of the salivary microbiome during different phases of Crohn’s Disease. Front. Cell. Infect. Microbiol. 2020, 10, 544704. [Google Scholar] [CrossRef]
- Kelsen, J.; Bittinger, K.; Pauly-Hubbard, H.; Posivak, L.; Grunberg, S.; Baldassano, R.; Lewis, J.D.; Wu, G.D.; Bushman, F.D. Alterations of the subgingival microbiota in pediatric Crohn’s disease studied longitudinally in discovery and validation cohorts. Inflamm. Bowel Dis. 2015, 21, 2797–2805. [Google Scholar] [CrossRef] [Green Version]
- Elmaghrawy, K. The oral mucosal microbiome in children with Crohn’s disease exhibits reduced biodiversity compared to healthy children, revealed by 16s profiling. J. Oral Microbiol. 2017, 9, 1325254. [Google Scholar] [CrossRef]
- Rautava, J.; Pinnell, L.J.; Vong, L.; Akseer, N.; Assa, A.; Sherman, P.M. Oral microbiome composition changes in mouse models of colitis. J. Gastroenterol. Hepatol. 2015, 30, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Szczeklik, K.; Owczarek, D.; Cibor, D.; Cześnikiewicz-Guzik, M.; Krzyściak, P.; Krawczyk, A.; Mach, T.; Karczewska, E.; Krzyściak, W. Relative homogeneity of oral bacterial flora in Crohn’s disease compared to ulcerative colitis and its connections with antioxidant defense-preliminary report. Folia Med. Cracov. 2019, LIX, 15–35. [Google Scholar]
- Xun, Z.; Zhang, Q.; Xu, T.; Chen, N.; Chen, F. Dysbiosis and ecotypes of the salivary microbiome associated with inflammatory bowel diseases and the assistance in diagnosis of diseases using oral bacterial profiles. Front. Microbiol. 2018, 9, 1136. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; Sankaran, K.; Fukuyama, J.A.; McMurdie, P.J.; Holmes, S.P. Bioconductor workflow for microbiome data analysis: From raw reads to community analyses. F1000Research 2016, 5, 1492. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.D.; Chuai, S.; Nessel, L.; Lichtenstein, G.R.; Aberra, F.N.; Ellenberg, J.H. Use of the non-invasive components of the Mayo score to assess clinical response in ulcerative colitis. Inflamm. Bowel Dis. 2008, 14, 1660–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaura, E.; Keijser, B.J.; Huse, S.M.; Crielaard, W. Defining the healthy ‘core microbiome’ of oral microbial communities. BMC Microbiol. 2009, 9, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakerska-Banaszak, O.; Tomczak, H.; Gabryel, M.; Baturo, A.; Wolko, L.; Michalak, M.; Malinska, N.; Mankowska-Wierzbicka, D.; Eder, P.; Dobrowolska, A.; et al. Dysbiosis of gut microbiota in Polish patients with ulcerative colitis: A pilot study. Sci. Rep. 2021, 11, 2166. [Google Scholar] [CrossRef]
- Byrd, K.; Gulati, A. The “gum–gut” axis in inflammatory bowel diseases: A hypothesis-driven review of associations and advances. Front. Immunol. 2021, 12, 620124. [Google Scholar] [CrossRef] [PubMed]
- Chandan, J.S.; Thomas, T. The impact of inflammatory bowel disease on oral health. Br. Dent. J. 2017, 222, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Greuter, T.; Bertoldo, F.; Rechner, R.; Straumann, A.; Biedermann, L.; Zeitz, J.; Misselwitz, B.; Scharl, M.; Rogler, G.; Safroneeva, E.; et al. Extraintestinal manifestations of pediatric inflammatory bowel disease: Prevalence, presentation, and anti-TNF treatment. J. Pediatr. Gastroenterol. Nutr. 2017, 65, 200–206. [Google Scholar] [CrossRef] [Green Version]
- Muhvić-Urek, M.; Tomac-Stojmenović, M.; Mijandrusic-Sincic, B. Oral pathology in inflammatory bowel disease. World J. Gastroenterol. 2016, 22, 5655–5667. [Google Scholar] [CrossRef]
- Rodrigues, E.; Laranjeira, N.; Nunes, G.; Roque-Ramos, L.; Vieira, A.; Fonseca, J. Are cariogenic bacteria the major risk factor to dental caries in patients with ulcerative colitis? Arq. Gastroenterol. 2019, 56, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Unno, T.; Kim, B.Y.; Park, M.S. Sex differences in gut microbiota. World J. Men’s Health 2020, 38, 48. [Google Scholar] [CrossRef] [PubMed]
- Son, H.J.; Kim, N.; Song, C.-H.; Nam, R.H.; Choi, S.I.; Kim, J.S.; Lee, D.H. Sex-related alterations of gut microbiota in the C57BL/6 mouse model of inflammatory bowel disease. J. Cancer Prev. 2019, 24, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Kozik, A.J.; Nakatsu, C.H.; Chun, H.; Jones-Hall, Y.L. Age, sex, and TNF associated differences in the gut microbiota of mice and their impact on acute TNBS colitis. Exp. Mol. Pathol. 2017, 103, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Fölsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004, 53, 685–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.W.; Sanderson, J.D.; Churcher, C.; Parkes, G.C.; Hudspith, B.N.; Rayment, N.; Brostoff, J.; Parkhill, J.; Dougan, G.; Petrovska, L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, L.; Ke, Y.; Zhao, H.; Wang, L.; Jia, C.; Liu, W.; Fu, Q.; Shi, M.; Cui, J.; Li, S. Role of colonic microbiota in the pathogenesis of ulcerative colitis. BMC Gastroenterol. 2019, 19, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vatn, S.; Carstens, A.; Kristoffersen, A.B.; Bergemalm, D.; Casén, C.; Moen, A.E.F.; Tannaes, T.M.; Lindstrøm, J.; Detlie, T.E.; Olbjørn, C.; et al. Faecal microbiota signatures of IBD and their relation to diagnosis, disease phenotype, inflammation, treatment escalation and anti-TNF response in a European Multicentre Study (IBD-Character). Scand. J. Gastroenterol. 2020, 55, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Said, H.S.; Suda, W.; Nakagome, S.; Chinen, H.; Oshima, K.; Kim, S.; Kimura, R.; Iraha, A.; Ishida, H.; Fujita, J.; et al. Dysbiosis of salivary microbiota in inflammatory bowel disease and its association with oral immunological biomarkers. DNA Res. 2014, 21, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deo, P.N.; Deshmukh, R. Oral microbiome: Unveiling the fundamentals. J. Oral Maxillofac. Pathol. 2019, 23, 122–128. [Google Scholar]
- Chen, H.; Jiang, W. Application of high-throughput sequencing in understanding human oral microbiome related with health and disease. Front. Microbiol. 2014, 5, 508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Tang, C.M.; Exley, R.M. Non-pathogenic Neisseria: Members of an abundant, multi-habitat, diverse genus. Microbiology 2015, 161, 1297–1312. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Garg, P.K.; Dubey, A.K. Insights into the human oral microbiome. Arch. Microbiol. 2018, 200, 525–540. [Google Scholar] [CrossRef]
- Mok, S.F.; Chinna, K.A.; Cheah, Y.K.; Ngeow, W.C.; Zain, R.B.; Yap, S.F.; Ong, H.K. The oral microbiome community variations associated with normal, potentially malignant disorders and malignant lesions of the oral cavity. Malays. J. Pathol. 2017, 39, 1–15. [Google Scholar] [PubMed]
- Xiao, C.; Ran, S.; Huang, Z.; Liang, J. Bacterial diversity and community structure of supragingival plaques in adults with dental health or caries revealed by 16S Pyrosequencing. Front. Microbiol. 2016, 7, 1145. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, I.; Verma, M.; Panda, M. Role of oral microbiome signatures in diagnosis and prognosis of oral cancer. Technol. Cancer Res. Treat. 2019, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Bayati, L.; Fasaei, B.N.; Merat, S.; Bahonar, A. Longitudinal analyses of gut-associated bacterial microbiota in ulcerative colitis patients. Arch. Iran. Med. 2018, 21, 578–584. [Google Scholar] [PubMed]
- Wlodarska, M.; Luo, C.; Kolde, R.; d’Hennezel, E.; Annand, J.W.; Heim, C.E.; Krastel, P.; Schmitt, E.K.; Omar, A.S.; Creasey, E.A.; et al. Indoleacrylic acid produced by commensal Peptostreptococcus species suppresses inflammation. Cell Host Microbe 2017, 22, 25–37.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zawadzki, P.J.; Perkowski, K.; Starościak, B.; Baltaza, W.; Padzik, M.; Pionkowski, K.; Chomicz, L. Identification of infectious microbiota from oral cavity environment of various population group patients as a preventive approach to human health risk factors. Ann. Agric. Environ. Med. 2016, 23, 566–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.Y.; Yeh, Y.M.; Yu, H.Y.; Chin, C.Y.; Hsu, C.W.; Liu, H.; Huang, P.J.; Hu, S.N.; Liao, C.T.; Chang, K.P.; et al. Oral microbiota community dynamics associated with oral squamous cell carcinoma staging. Front. Microbiol. 2018, 9, 862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirst, M.E.; Li, E.C.; Alfant, B.; Chi, Y.Y.; Walker, C.; Magnusson, I.; Wanga, G.P. Dysbiosis and alterations in predicted functions of the subgingival microbiome in chronic periodontitis. Appl. Environ. Microbiol. 2015, 81, 783–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.R.; Yu, W.-H.; Lakshmanan, A.; Wade, W.G. The Human Oral Microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knapp, S.; Brodal, C.; Peterson, J.; Qi, F.; Kreth, J.; Merritt, J. Natural competence is common among clinical isolates of Veillonella parvula and is useful for genetic manipulation of this key member of the oral microbiome. Front. Cell. Infect. Microbiol. 2017, 7, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashima, I.; Nakazawa, F. The influence of oral Veillonella species on biofilms formed by Streptococcus species. Anaerobe 2014, 28, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Belda-Ferre, P.; Alcaraz, L.D.; Cabrera-Rubio, R.R.; Romero, H.; Simón-Soro, A.; Pignatelli, M.; Mira, A. The oral metagenome in health and disease. ISME J. 2012, 6, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashima, I.; Nakazawa, F. Interaction between Streptococcus spp. and Veillonella tobetsuensis in the early stages of oral biofilm formation. J. Bacteriol. 2015, 197, 2104. [Google Scholar] [CrossRef] [Green Version]
- Pucino, V.; Certo, M.; Bulusu, V.; Cucchi, D.; Goldmann, K.; Pontarini, E.; Haas, R.; Smith, J.; Headland, S.E.; Blighe, K.; et al. Lactate buildup at the site of chronic inflammation promotes disease by inducing CD4+ T cell metabolic rewiring. Cell Metab. 2019, 30, 1055–1074.e8. [Google Scholar] [CrossRef] [Green Version]
- McLean, J.S.; Fansler, S.J.; Majors, P.D.; McAteer, K.; Allen, L.Z.; Shirtliff, M.E.; Lux, R.; Shi, W. Identifying low ph active and lactate-utilizing taxa within oral microbiome communities from healthy children using stable isotope probing techniques. PLoS ONE 2012, 7, e32219. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.W. Fusobacterium nucleatum: A commensal-turned pathogen. Curr. Opin. Microbiol. 2015, 23, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Huh, J.W.; Roh, T.Y. Opportunistic detection of Fusobacterium nucleatum as a marker for the early gut microbial dysbiosis. BMC Microbiol. 2020, 20, 208. [Google Scholar] [CrossRef]
- Schirmer, M.; Denson, L.; Vlamakis, H.; Franzosa, E.A.; Thomas, S.; Gotman, N.M.; Rufo, P.; Baker, S.S.; Sauer, C.; Markowitz, J.; et al. Compositional and temporal changes in the gut microbiome of pediatric ulcerative colitis patients are linked to disease course. Cell Host Microbe 2018, 24, 600–610.e4. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Hong, X.L.; Sun, T.T.; Huang, X.W.; Wang, J.L.; Xiong, H. Fusobacterium nucleatum exacerbates colitis by damaging epithelial barriers and inducing aberrant inflammation. J. Dig. Dis. 2020, 21, 385–398. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Cao, P.; Su, W.; Zhan, N.; Dong, W. Fusobacterium nucleatum facilitates ulcerative colitis through activating IL-17F signaling to NF-κB via the upregulation of CARD3 expression. J. Pathol. 2020, 250, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Tai, S.; Tsukahara, T.; Inoue, R. Fusobacterium nucleatum impedes remission of colitis in a mouse model. Biosci. Biotechnol. Biochem. 2021, 85, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.A.; Garrett, W.S. Fusobacterium nucleatum—Symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 2019, 17, 156–166. [Google Scholar] [CrossRef]
- Bajer, L.; Kverka, M.; Kostovcik, M.; Macinga, P.; Dvorak, J.; Stehlikova, Z.; Brezina, J.; Wohl, P.; Spicak, J.; Drastich, P. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J. Gastroenterol. 2017, 23, 4548–4558. [Google Scholar] [CrossRef] [PubMed]
- Mar, J.S.; Lamere, B.J.; Lin, D.L.; Levan, S.; Nazareth, M.; Mahadevan, U.; Lynch, S.V. Disease severity and immune activity relate to distinct interkingdom gut microbiome states in ethnically distinct ulcerative colitis patients. MBio 2016, 7, e01072-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinakaran, V.; Mandape, S.N.; Shuba, K.; Pratap, S.; Sakhare, S.S.; Tabatabai, M.A.; Smoot, D.T.; Farmer-Dixon, C.M.; Kesavalu, L.N.; Adunyah, S.E.; et al. Identification of specific oral and gut pathogens in full thickness colon of colitis patients: Implications for colon motility. Front. Microbiol. 2019, 9, 3220. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; Amand, A.L.S.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, I.; Ullah, N.; Zha, L.; Bai, Y.; Khan, A.; Zhao, T.; Che, T.; Zhang, C. Alteration of gut microbiota in inflammatory bowel disease (IBD): Cause or consequence? IBD treatment targeting the gut microbiome. Pathogens 2019, 8, 126. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.S.; Hayward, M.R.; Coelho, L.P.; Li, S.S.; Costea, P.I.; Voigt, A.Y.; Wirbel, J.; Maistrenko, O.M.; Alves, R.J.; Bergsten, E.; et al. Extensive transmission of microbes along the gastrointestinal tract. Elife 2019, 12, e42693. [Google Scholar] [CrossRef]
- Olsen, I.; Yamazaki, K. Can oral bacteria affect the microbiome of the gut? J. Oral Microbiol. 2019, 11, 1586422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abed, J.; Maalouf, N.; Manson, A.L.; Earl, A.M.; Parhi, L.; Emgård, J.; Klutstein, M.; Tayeb, S.; Almogy, G.; Atlan, K.A.; et al. Colon cancer-associated Fusobacterium nucleatum may originate from the oral cavity and reach colon tumors via the circulatory system. Front. Cell. Infect. Microbiol. 2020, 10, 400. [Google Scholar] [CrossRef] [PubMed]
- Cueva, C.; Silva, M.; Pinillos, I.; Bartolomé, B.; Moreno-Arribas, M.V. Interplay between dietary polyphenols and oral and gut microbiota in the development of colorectal cancer. Nutrients 2020, 12, 625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atarashi, K.; Suda, W.; Luo, C.; Kawaguchi, T.; Motoo, I.; Narushima, S.; Kiguchi, Y.; Yasuma, K.; Watanabe, E.; Tanoue, T.; et al. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science 2017, 358, 359–365. [Google Scholar] [CrossRef]
- Kobayashi, R.; Ogawa, Y.; Hashizume-Takizawa, T.; Kurita-Ochiai, T. Oral bacteria affect the gut microbiome and intestinal immunity. Pathog. Dis. 2020, 78, ftaa024. [Google Scholar] [CrossRef] [PubMed]
- Khor, B.; Snow, M.; Herrman, E.; Ray, N.; Mansukhani, K.; Patel, K.A.; Said-al-naief, N.; Maier, T.; Machida, C.A. Interconnections between the oral and gut microbiomes: Reversal of microbial dysbiosis and the balance between systemic health and disease. Microorganisms 2021, 9, 496. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Taxonomy | Mean ± SD Healthy Subjects | Mean ± SD UC Patients | p-Value |
|---|---|---|---|
| Phylum | |||
| Bacteroidetes | 31.46 ± 10.18 | 33.88 ± 5.46 | 0.704 |
| Firmicutes | 41.90 ± 5.56 | 44.89 ± 6.88 | 0.511 |
| Proteobacteria | 11.49 ± 7.47 | 6.24 ± 4.35 | 0.072 # |
| Patescibacteria | 2.30 ± 1.69 | 3.75 ± 2.55 | 0.314 |
| Fusobacteria | 6.56 ± 3.68 | 5.29 ± 3.26 | 0.468 |
| Actinobacteria | 5.64 ± 3.57 | 4.77 ± 2.20 | 0.704 |
| Epsilonbacteraeota | 0.50 ± 0.33 | 0.69 ± 0.35 | 0.426 |
| Family | |||
| Actinomycetaceae | 1.41 ± 0.71 | 1.3 ± 1.22 | 0.349 |
| Atopobiaceae | 1.08 ± 0.56 | 0.67 ± 0.66 | 0.084 # |
| Campylobacteraceae | 0.69 ± 0.35 | 0.50 ± 0.33 | 0.426 |
| Carnobacteriaceae | 1.27 ± 0.69 | 1.22 ± 0.55 | 0.972 |
| Erysipelotrichaceae | 0.54 ± 0.23 | 0.50 ± 0.53 | 0.217 |
| Family_XI | 1.06 ± 0.42 | 1.90 ± 1.30 | 0.132 |
| Family_XIII | 1.16 ± 0.89 | 0.35 ± 0.31 | 0.024 * |
| Flavobacteriaceae | 0.68 ± 0.45 | 1.35 ± 1.31 | 0.511 |
| Fusobacteriaceae | 2.48 ± 1.47 | 2.96 ± 2.49 | 0.972 |
| Lachnospiraceae | 3.38 ± 1.45 | 2.00 ± 1.33 | 0.061 # |
| Leptotrichiaceae | 2.81 ± 2.18 | 3.60 ± 2.58 | 0.511 |
| Micrococcaceae | 2.20 ± 1.65 | 3.30 ± 2.14 | 0.251 |
| Neisseriaceae | 2.19 ± 1.58 | 6.29 ± 5.14 | 0.019 * |
| Pasteurellaceae | 3.90 ± 2.83 | 5.00 ± 2.94 | 0.349 |
| Peptostreptococcaceae | 2.54 ± 4.02 | 0.38 ± 0.46 | 0.044 * |
| Porphyromonadaceae | 5.92 ± 5.15 | 2.93 ± 2.83 | 0.223 |
| Prevotellaceae | 26.75 ± 8.24 | 26.76 ± 10.43 | 1 |
| Ruminococcaceae | 0.59 ± 0.42 | 0.24 ± 0.22 | 0.066 # |
| Saccharimonadaceae | 2.84 ± 1.98 | 2.02 ± 1.77 | 0.426 |
| Streptococcaceae | 11.77 ± 4.98 | 12.29 ± 6.20 | 0.863 |
| Veillonellaceae | 21.86 ± 7.26 | 22.24 ± 7.52 | 0.917 |
| Genus | |||
| Actinomyces | 1.40 ± 0.72 | 1.28 ± 1.20 | 0.387 |
| Alloprevotella | 3.00 ± 1.53 | 3.15 ± 2.10 | 0.918 |
| Atopobium | 1.07 ± 0.56 | 0.67 ± 0.66 | 0.084 # |
| Campylobacter | 0.69 ± 0.35 | 0.50 ± 0.33 | 0.426 |
| Capnocytophaga | 0.68 ± 0.45 | 1.35 ± 1.31 | 0.511 |
| Fusobacterium | 2.48 ± 1.47 | 2.96 ± 2.49 | 0.972 |
| Gemella | 1.06 ± 0.42 | 1.90 ± 1.30 | 0.132 |
| Granulicatella | 1.27 ± 0.69 | 1.22 ± 0.55 | 0.972 |
| Haemophilus | 3.73 ± 2.78 | 4.66 ± 2.74 | 0.386 |
| Lachnoanaerobaculum | 0.95 ± 0.60 | 0.33 ± 0.19 | 0.018 * |
| Leptotrichia | 2.78 ± 2.15 | 3.60 ± 2.58 | 0.511 |
| Megasphaera | 1.65 ± 1.57 | 1.06 ± 1.02 | 0.573 |
| Neisseria | 2.01 ± 1.54 | 5.66 ± 5.15 | 0.034 * |
| Oribacterium | 1.25 ± 0.73 | 0.96 ± 0.69 | 0.511 |
| Peptostreptococcus | 2.19 ± 3.85 | 0.34 ± 0.43 | 0.084 # |
| Porphyromonas | 5.92 ± 5.15 | 2.93 ± 2.83 | 0.223 |
| Prevotella | 4.18 ± 2.39 | 3.48 ± 2.27 | 0.756 |
| Prevotella_6 | 1.20 ± 0.75 | 0.93 ± 0.86 | 0.314 |
| Prevotella_7 | 18.1 ± 8.54 | 18.83 ± 9.34 | 0.917 |
| Rothia | 2.20 ± 1.65 | 3.30 ± 2.14 | 0.251 |
| Ruminococcaceae_UCG-014 | 0.58 ± 0.42 | 0.24 ± 0.22 | 0.066 # |
| Selenomonas_3 | 0.88 ± 0.84 | 0.67 ± 0.71 | 0.459 |
| Solobacterium | 0.52 ± 0.24 | 0.50 ± 0.53 | 0.245 |
| Stomatobaculum | 0.59 ± 0.37 | 0.24 ± 0.18 | 0.035 * |
| Streptococcus | 11.77 ± 4.98 | 12.29 ± 6.2 | 0.863 |
| Veillonella | 18.89 ± 7.41 | 20.18 ± 7.8 | 0.704 |
| ASV Code | Species | Mean ± SD Healthy Subjects | Mean ± SD UC Patients | p-Value |
|---|---|---|---|---|
| ASV4 | Veillonella parvula | ND | 0.136 ± 0.296 | - |
| ASV14 | Veillonella parvula | ND | 0.152 ± 0.316 | - |
| ASV39 | Fusobacterium nucleatum | ND | 0.068 ± 0.118 | - |
| ASV219 | Prevotella NA | ND | 0.01 ± 0.019 | - |
| ASV754 | Haemophilus parainfluenzae | 0.771 ± 0.896 | ND | - |
| ASV756 | Saccharimonadaceae family NA | 0.742 ± 0.591 | ND | - |
| ASV758 | Rothia mucilaginosa | 0.567 ± 0.626 | 0.162 ± 0.326 | 3.65 × 10−2 |
| ASV762 | Veillonella rogosae | 0.704 ± 1.004 | ND | - |
| ASV763 | Porphyromonas NA | 0.530 ± 0.823 | ND | - |
| ASV765 | Veillonella atypica | 0.690 ± 0.469 | ND | - |
| ASV773 | Prevotella histicola | 0.519 ± 0.573 | ND | - |
| ASV778 | Veillonella rogosae | 0.356 ± 0.440 | ND | - |
| ASV781 | Prevotella melaninogenica | 0.390 ± 0.978 | ND | - |
| ASV787 | Rothia mucilaginosa | 0.246 ± 0.434 | 1.419 ± 1.304 | 2.72 × 10−2 |
| ASV792 | Porphyromonas NA | 0.270 ± 0.271 | ND | - |
| ASV795 | Peptostreptococcus stomatis | 0.793 ± 1.565 | ND | - |
| ASV797 | Streptococcus salivarius | 0.453 ± 0.736 | ND | - |
| ASV809 | Prevotella salivae | 0.245 ± 0.225 | ND | - |
| ASV814 | Ruminococcaceae_UCG-014 NA | 0.247 ± 0.224 | 0.071 ± 0.103 | 3.43 × 10−2 |
| ASV816 | Prevotella jejuni | 0.269 ± 0.381 | ND | - |
| ASV821 | Oribacterium sinus | 0.199 ± 0.225 | ND | - |
| ASV834 | Eubacterium sulci | 0.264 ± 0.270 | ND | - |
| ASV842 | Prevotella nanceiensis | 0.139 ± 0.323 | ND | - |
| ASV852 | Gemella sanguinis | 0.121 ± 0.101 | ND | - |
| ASV855 | Porphyromonas pasteri | 0.170 ± 0.290 | ND | - |
| ASV857 | Alloprevotella NA | 0.302 ± 0.687 | ND | - |
| ASV859 | Prevotella nanceiensis | 0.121 ± 0.216 | ND | - |
| ASV861 | Bergeyella NA | 0.093 ± 0.042 | 0.18 ± 0.115 | 2.42 × 10−2 |
| ASV865 | Absconditabacteriales_SR1 order NA | 0.162 ± 0.223 | ND | - |
| ASV875 | Leptotrichia NA | 0.191 ± 0.378 | ND | - |
| ASV878 | Campylobacter concisus | 0.105 ± 0.071 | ND | - |
| ASV883 | Lachnoanaerobaculum gingivalis | 0.125 ± 0.144 | ND | - |
| ASV893 | Oribacterium parvum | 0.115 ± 0.135 | ND | - |
| ASV894 | Granulicatella elegans | 0.043 ± 0.093 | ND | - |
| ASV896 | Parvimonas micra | 0.059 ± 0.089 | ND | - |
| ASV901 | Rothia dentocariosa | 0.082 ± 0.123 | ND | - |
| ASV905 | Ruminococcaceae_UCG-014 NA | 0.089 ± 0.096 | ND | - |
| ASV906 | Leptotrichia NA | 0.099 ± 0.142 | ND | - |
| ASV920 | Streptococcus sanguinis | 0.068 ± 0.060 | ND | - |
| ASV964 | Prevotella melaninogenica | 0.098 ± 0.204 | ND | - |
| ASV967 | Stomatobaculum longum | 0.097 ± 0.177 | ND | - |
| ASV979 | Catonella morbi | 0.043 ± 0.064 | ND | - |
| ASV1001 | Oribacterium asaccharolyticum | 0.028 ± 0.040 | ND | - |
| ASV1010 | Lachnospiraceae family NA | 0.064 ± 0.084 | 0.013 ± 0.029 | 3.02 × 10−2 |
| ASV1015 | Treponema_2 NA | 0.032 ± 0.045 | ND | - |
| ASV1050 | Eubacterium yurii | 0.058 ± 0.137 | ND | - |
| ASV1101 | Capnocytophaga NA | 0.026 ± 0.040 | ND | - |
| ASV1153 | Campylobacter gracilis | 0.043 ± 0.089 | ND | - |
| ASV1158 | Capnocytophaga sputigena | 0.039 ± 0.070 | ND | - |
| ASV1161 | Selenomonas_3 NA | 0.027 ± 0.038 | ND | - |
| ASV1329 | Streptococcus sanguinis | 0.032 ± 0.071 | ND | - |
| ASV1363 | Prevotella NA | 0.003 ± 0.007 | ND | - |
| ASV1405 | Treponema refringens | 0.015 ± 0.025 | ND | - |
| ASV1414 | Streptococcus gordonii | 0.012 ± 0.023 | ND | - |
| ASV1473 | Butyrivibrio_2 NA | 0.015 ± 0.035 | ND | - |
| ASV1569 | Candidatus_Saccharimonas NA | 0.013 ± 0.021 | ND | - |
| ASV1600 | Fretibacterium NA | 0.024 ± 0.052 | ND | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molinero, N.; Taladrid, D.; Zorraquín-Peña, I.; de Celis, M.; Belda, I.; Mira, A.; Bartolomé, B.; Moreno-Arribas, M.V. Ulcerative Colitis Seems to Imply Oral Microbiome Dysbiosis. Curr. Issues Mol. Biol. 2022, 44, 1513-1527. https://doi.org/10.3390/cimb44040103
Molinero N, Taladrid D, Zorraquín-Peña I, de Celis M, Belda I, Mira A, Bartolomé B, Moreno-Arribas MV. Ulcerative Colitis Seems to Imply Oral Microbiome Dysbiosis. Current Issues in Molecular Biology. 2022; 44(4):1513-1527. https://doi.org/10.3390/cimb44040103
Chicago/Turabian StyleMolinero, Natalia, Diego Taladrid, Irene Zorraquín-Peña, Miguel de Celis, Ignacio Belda, Alex Mira, Begoña Bartolomé, and M. Victoria Moreno-Arribas. 2022. "Ulcerative Colitis Seems to Imply Oral Microbiome Dysbiosis" Current Issues in Molecular Biology 44, no. 4: 1513-1527. https://doi.org/10.3390/cimb44040103
APA StyleMolinero, N., Taladrid, D., Zorraquín-Peña, I., de Celis, M., Belda, I., Mira, A., Bartolomé, B., & Moreno-Arribas, M. V. (2022). Ulcerative Colitis Seems to Imply Oral Microbiome Dysbiosis. Current Issues in Molecular Biology, 44(4), 1513-1527. https://doi.org/10.3390/cimb44040103