
The First Homozygote Mutation c.499G>T (Asp167Tyr) in the RPE65 Gene Encoding Retinoid Isomerohydrolase Causing Retinal Dystrophy

 ,
, {kind=link}
{kind=link}
Abstract
1. Introduction
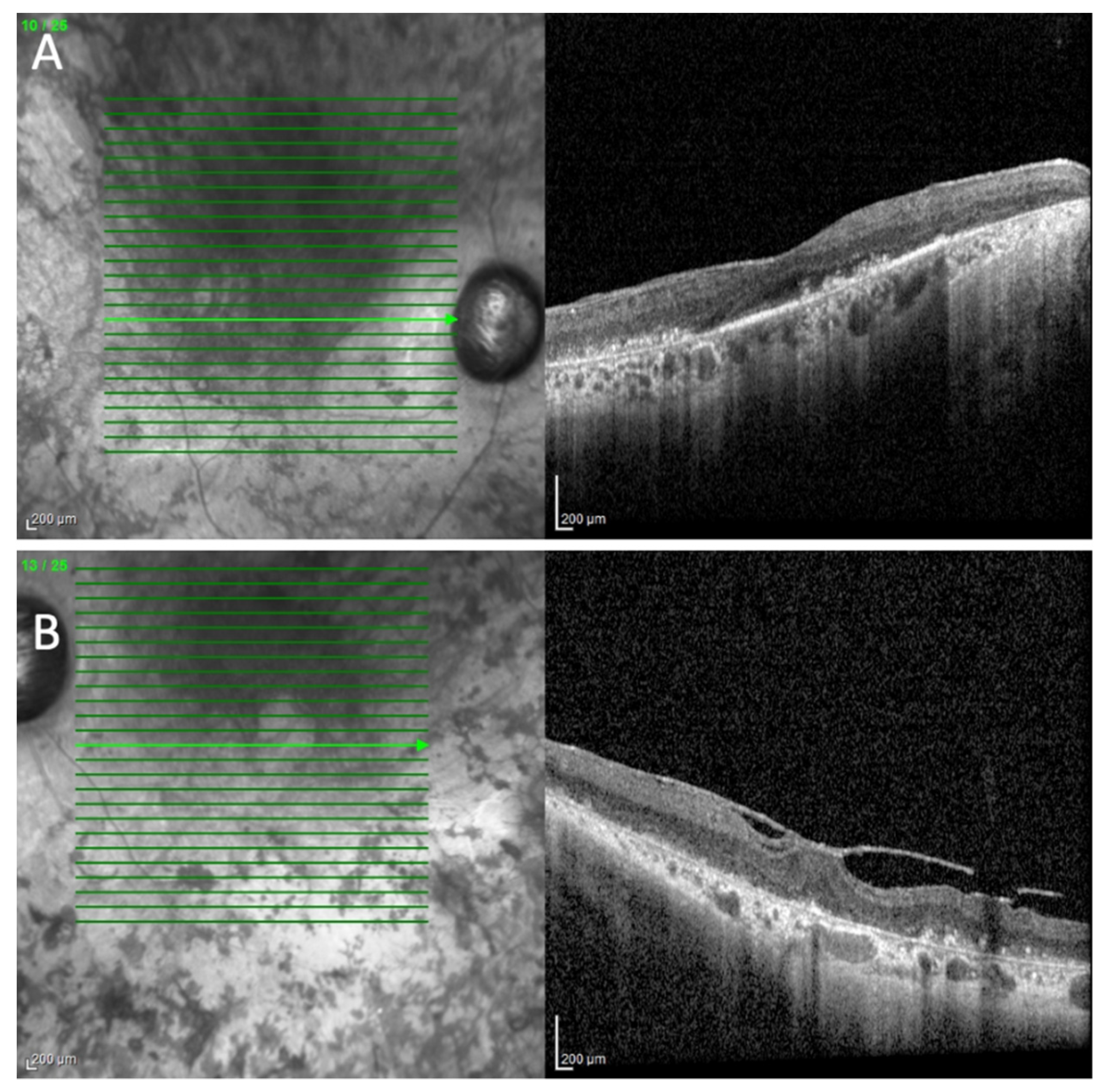
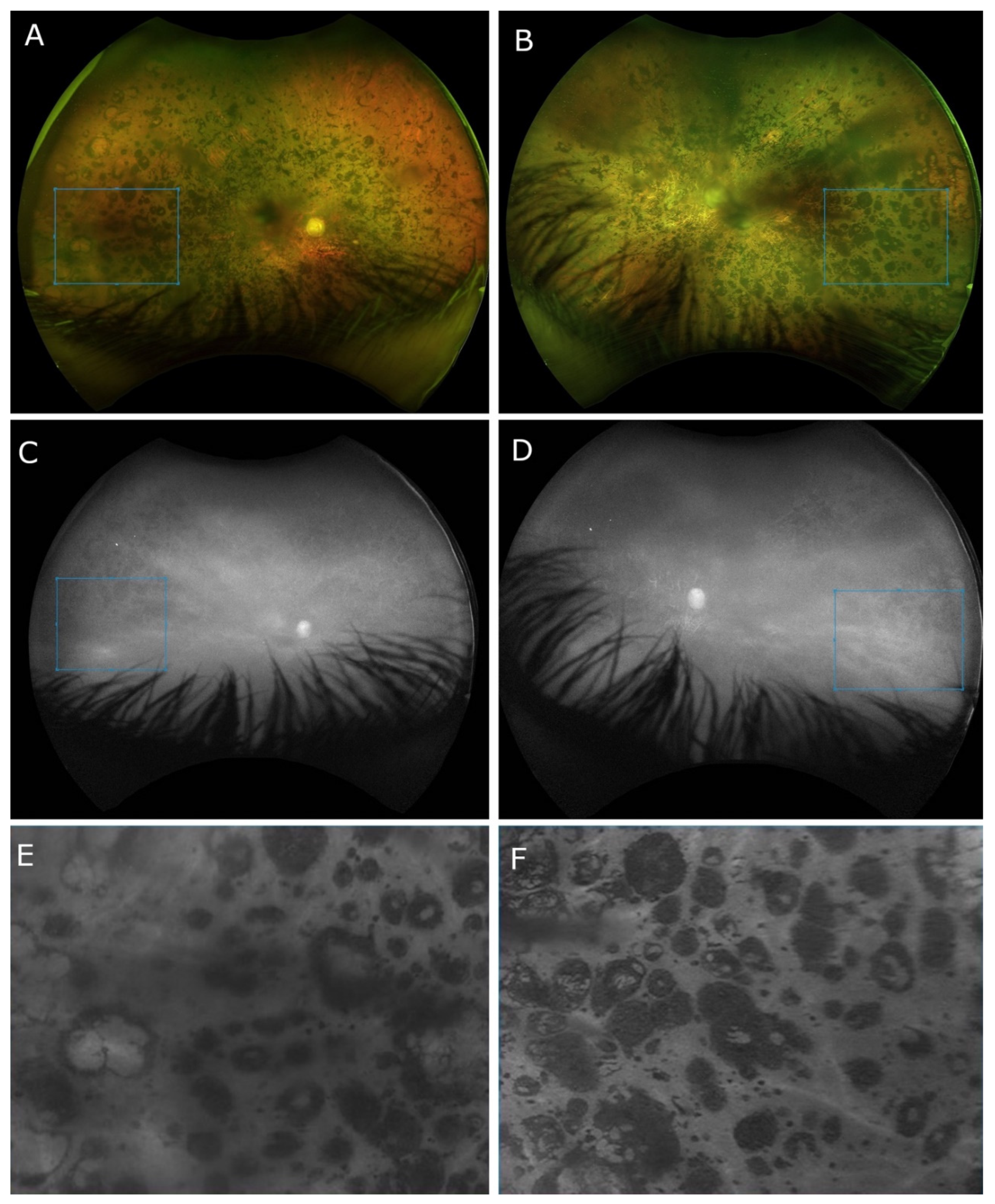
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- *180069 RETINOID ISOMEROHYDROLASE RPE65; RPE65. Available online: https://omim.org/entry/180069?search=rpe65&highlight=rpe65 (accessed on 31 October 2022).
- Thompson, D.A.; Gyürüs, P.; Fleischer, L.L.; Bingham, E.L.; McHenry, C.L.; Apfelstedt-Sylla, E.; Zrenner, E.; Lorenz, B.; Richards, J.E.; Jacobson, S.G.; et al. Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2000, 41, 4293–4299. [Google Scholar]
- The Human Gene Mutation Database. Available online: http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 31 October 2022).
- Redmond, T.M.; Poliakov, E.; Yu, S.; Tsai, J.-Y.; Lu, Z.; Gentleman, S. Mutation of key residues of RPE65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc. Natl. Acad. Sci. USA 2005, 102, 13658–13663. [Google Scholar] [CrossRef]
- Chen, Y.; Moiseyev, G.; Takahashi, Y.; Ma, J.-X. Impacts of two point mutations of RPE65 from Leber’s congenital amaurosis on the stability, subcellular localization and isomerohydrolase activity of RPE65. FEBS Lett. 2006, 580, 4200–4204. [Google Scholar] [CrossRef][Green Version]
- German Society of Ophthalmology (Deutsche Ophthalmologische Gesellschaft, DOG); German Retina Society e.V. (Retinologische Gesellschaft e. V., RG); Professional Association of German Ophthalmologists (Berufsverband der Augenärzte Deutschlands e. V., BVA). Statement of the DOG, the RG, and the BVA on the therapeutic use of voretigene neparvovec (LuxturnaTM) in ophthalmology. English version: January 2019. Ophthalmologe 2020, 117, 16–24. [Google Scholar] [CrossRef]
- Tsin, A.; Betts-Obregon, B.; Grigsby, J. Visual cycle proteins: Structure, function, and roles in human retinal disease. J. Biol. Chem. 2018, 293, 13016–13021. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.H.; Suh, S.; Sander, C.L.; Hernandez, C.J.O.; Bulman, E.R.; Khadka, N.; Dong, Z.; Shi, W.; Palczewski, K.; Kiser, P.D. Insights into the pathogenesis of dominant retinitis pigmentosa associated with a D477G mutation in RPE65. Hum. Mol. Genet. 2018, 27, 2225–2243. [Google Scholar] [CrossRef]
- Kiser, P.D.; Golczak, M.; Lodowski, D.T.; Chance, M.R.; Palczewski, K. Crystal structure of native RPE65, the retinoid isomerase of the visual cycle. Proc. Natl. Acad. Sci. USA 2009, 106, 17325–17330. [Google Scholar] [CrossRef]
- De Baets, G.; Van Doorn, L.; Rousseau, F.; Schymkowitz, J. Increased Aggregation Is More Frequently Associated to Human Disease-Associated Mutations Than to Neutral Polymorphisms. PLoS Comput. Biol. 2015, 11, e1004374. [Google Scholar] [CrossRef]
- Reumers, J.; Maurer-Stroh, S.; Schymkowitz, J.; Rousseau, F. Protein sequences encode safeguards against aggregation. Hum. Mutat. 2009, 30, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Genome Aggregation Database (gnomAD). Available online: https://gnomad.broadinstitute.org/variant/1-68906680-C-A?dataset=gnomad_r2_1 (accessed on 1 November 2022).
- Simonelli, F.; Ziviello, C.; Testa, F.; Rossi, S.; Fazzi, E.; Bianchi, P.E.; Fossarello, M.; Signorini, S.; Bertone, C.; Galantuomo, S.; et al. Clinical and molecular genetics of Leber’s congenital amaurosis: A multicenter study of Italian patients. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4284–4290. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, J.W.B.; Smith, A.J.; Barker, S.S.; Robbie, S.; Henderson, R.; Balaggan, K.; Viswanathan, A.; Holder, G.E.; Stockman, A.; Tyler, N.; et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N. Engl. J. Med. 2008, 358, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Ripamonti, C.; Henning, G.B.; Ali, R.R.; Bainbridge, J.W.; Robbie, S.J.; Sundaram, V.; Luong, V.A.; van den Born, L.I.; Casteels, I.; de Ravel, T.J.L.; et al. Nature of the visual loss in observers with Leber’s congenital amaurosis caused by specific mutations in RPE65. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6817–6828. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Henderson, R.H.; Waseem, N.; Searle, R.; van der Spuy, J.; Russell-Eggitt, I.; Bhattacharya, S.S.; Thompson, D.A.; Holder, G.E.; Cheetham, M.E.; Webster, A.R.; et al. An assessment of the apex microarray technology in genotyping patients with Leber congenital amaurosis and early-onset severe retinal dystrophy. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5684–5689. [Google Scholar] [CrossRef][Green Version]
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/98873/ (accessed on 1 November 2022).
- Chung, D.C.; Bertelsen, M.; Lorenz, B.; Pennesi, M.E.; Leroy, B.P.; Hamel, C.P.; Pierce, E.; Sallum, J.; Larsen, M.; Stieger, K.; et al. The Natural History of Inherited Retinal Dystrophy Due to Biallelic Mutations in the RPE65 Gene. Am. J. Ophthalmol. 2019, 199, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.-J.; Wang, D.-D.; Li, J.-K.; Hu, F.-Y.; Xu, P.; Chen, F.; Qi, Y.-H.; Liu, W.; Li, W.; Zhang, S.-H.; et al. Frequency and phenotypic characteristics of RPE65 mutations in the Chinese population. Orphanet. J. Rare Dis. 2021, 16, 174. [Google Scholar] [CrossRef]
- Cideciyan, A.V. Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog. Retin. Eye Res. 2010, 29, 398–427. [Google Scholar] [CrossRef]
- Gardiner, K.L.; Cideciyan, A.V.; Swider, M.; Dufour, V.L.; Sumaroka, A.; Komáromy, A.M.; Hauswirth, W.W.; Iwabe, S.; Jacobson, S.G.; Beltran, W.A.; et al. Long-Term structural outcomes of late-stage RPE65 gene therapy. Mol. Ther. 2020, 28, 266–278. [Google Scholar] [CrossRef]
- Maguire, A.M.; Bennett, J.; Aleman, E.M.; Leroy, B.P.; Aleman, T.S. Clinical Perspective: Treating RPE65-Associated Retinal Dystrophy. Mol. Ther. 2021, 29, 442–463. [Google Scholar] [CrossRef]
- National Library of Medicine. National Center for Biotechnology Information. Available online: https://www.ncbi.nlm.nih.gov/medgen/892932 (accessed on 30 October 2022).
- RETINITIS PIGMENTOSA 87 WITH CHOROIDAL INVOLVEMENT; RP87. Available online: https://www.omim.org/entry/618697 (accessed on 30 October 2022).
- Hull, S.; Mukherjee, R.; Holder, G.E.; Moore, A.T.; Webster, A.R. The clinical features of retinal disease due to a dominant mutation in RPE65. Mol. Vis. 2016, 22, 626–635. [Google Scholar]
- Jauregui, R.; Park, K.S.; Tsang, S.H. Two-year progression analysis of RPE65 autosomal dominant retinitis pigmentosa. Ophthalmic. Genet. 2018, 39, 544–549. [Google Scholar] [CrossRef]
- Jaissle, G.B.; May, C.A.; van de Pavert, S.A.; Wenzel, A.; Claes-May, E.; Giessl, A.; Szurman, P.; Wolfrum, U.; Wijnholds, J.; Fischer, M.D.; et al. Bone spicule pigment formation in retinitis pigmentosa: Insights from a mouse model. Graefe’s Arch. Clin. Exp. Ophthalmol. 2010, 248, 1063–1070. [Google Scholar] [CrossRef]
- Milam, A.H.; Li, Z.Y.; Fariss, R.N. Histopathology of the human retina in retinitis pigmentosa. Prog. Retin. Eye Res. 1998, 17, 175–205. [Google Scholar] [PubMed]
- Li, Z.Y.; Possin, D.E.; Milam, A.H. Histopathology of bone spicule pigmentation in retinitis pigmentosa. Ophthalmology 1995, 102, 805–816. [Google Scholar] [CrossRef] [PubMed]
- To, K.W.; Adamian, M.; Jakobiec, F.A.; Berson, E.L. Clinical and histopathologic findings in clumped pigmentary retinal degeneration. Arch. Ophthalmol. 1996, 114, 950–955. [Google Scholar]
- Shin, Y.; Moiseyev, G.; Chakraborty, D.; Ma, J.-X. A Dominant Mutation in Rpe65, D477G, Delays Dark Adaptation and Disturbs the Visual Cycle in the Mutant Knock-In Mice. Am. J. Pathol. 2017, 187, 517–527. [Google Scholar] [CrossRef][Green Version]
- Körner, A.; Pawelek, J. Mammalian tyrosinase catalyzes three reactions in the biosynthesis of melanin. Science 1982, 217, 1163–1165. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, M.; Fitzpatrick, T.B. On the nature of the pigment in retinal pigment epithelium. Science 1957, 126, 449–450. [Google Scholar] [CrossRef]
- Reinisalo, M.; Putula, J.; Mannermaa, E.; Urtti, A.; Honkakoski, P. Regulation of the human tyrosinase gene in retinal pigment epithelium cells: The significance of transcription factor orthodenticle homeobox 2 and its polymorphic binding site. Mol. Vis. 2012, 18, 38–54. [Google Scholar]
- Biesemeier, A.; Kreppel, F.; Kochanek, S.; Schraermeyer, U. The classical pathway of melanogenesis is not essential for melanin synthesis in the adult retinal pigment epithelium. Cell Tissue Res. 2010, 339, 551–560. [Google Scholar] [CrossRef]
- Liu, Y.; Ye, F.; Li, Q.; Tamiya, S.; Darling, D.S.; Kaplan, H.J.; Dean, D.C. Zeb1 represses Mitf and regulates pigment synthesis, cell proliferation, and epithelial morphology. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5080–5088. [Google Scholar] [CrossRef]
- Schraermeyer, U.; Kopitz, J.; Peters, S.; Henke-Fahle, S.; Blitgen-Heinecke, P.; Kokkinou, D.; Schwarz, T.; Bartz-Schmidt, K.-U. Tyrosinase biosynthesis in adult mammalian retinal pigment epithelial cells. Exp. Eye Res. 2006, 83, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Lamah, T.; Kokkinou, D.; Bartz-Schmidt, K.-U.; Schraermeyer, U. Melanin protects choroidal blood vessels against light toxicity. Z. Naturforsch. C J. Biosci. 2006, 61, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Żądto, A.; Ito, S.; Sarna, M.; Wakamatsu, K.; Mokrzyński, K.; Sarna, T. The role of hydrogen peroxide and singlet oxygen in the photodegradation of melanin. Photochem. Photobiol. Sci. 2020, 19, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Bowne, S.J.; Humphries, M.M.; Sullivan, L.S.; Kenna, P.F.; Tam, L.C.S.; Kiang, A.S.; Campbell, M.; Weinstock, G.M.; Koboldt, D.C.; Ding, L.; et al. A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur. J. Hum. Genet. 2011, 19, 1074–1081. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bjeloš, M.; Ćurić, A.; Rak, B.; Bušić, M.; Kuzmanović Elabjer, B. The First Homozygote Mutation c.499G>T (Asp167Tyr) in the RPE65 Gene Encoding Retinoid Isomerohydrolase Causing Retinal Dystrophy. Curr. Issues Mol. Biol. 2022, 44, 6397-6403. https://doi.org/10.3390/cimb44120436
Bjeloš M, Ćurić A, Rak B, Bušić M, Kuzmanović Elabjer B. The First Homozygote Mutation c.499G>T (Asp167Tyr) in the RPE65 Gene Encoding Retinoid Isomerohydrolase Causing Retinal Dystrophy. Current Issues in Molecular Biology. 2022; 44(12):6397-6403. https://doi.org/10.3390/cimb44120436
Chicago/Turabian StyleBjeloš, Mirjana, Ana Ćurić, Benedict Rak, Mladen Bušić, and Biljana Kuzmanović Elabjer. 2022. "The First Homozygote Mutation c.499G>T (Asp167Tyr) in the RPE65 Gene Encoding Retinoid Isomerohydrolase Causing Retinal Dystrophy" Current Issues in Molecular Biology 44, no. 12: 6397-6403. https://doi.org/10.3390/cimb44120436
APA StyleBjeloš, M., Ćurić, A., Rak, B., Bušić, M., & Kuzmanović Elabjer, B. (2022). The First Homozygote Mutation c.499G>T (Asp167Tyr) in the RPE65 Gene Encoding Retinoid Isomerohydrolase Causing Retinal Dystrophy. Current Issues in Molecular Biology, 44(12), 6397-6403. https://doi.org/10.3390/cimb44120436